

Abstract
Introduction
The genetic causes of limb-girdle muscular dystrophy (LGMD) have been studied in numerous countries, but such investigations have been limited in Egypt.
Methods
A cohort of 30 families with suspected LGMD from Assiut, Egypt was studied using immunohistochemistry, homozygosity mapping, Sanger sequencing, and whole exome sequencing.
Results
Six families were confirmed to have pathogenic mutations, 4 in SGCA and 2 in DMD. Of these, 3 families harbored a single nonsense mutation in SGCA, suggesting that this may be a common mutation in Assiut, Egypt originating from a founder effect.
Discussion
The Assiut region in Egypt appears to share at least several of the common LGMD genes found in other parts of the world. It is notable that 4 of the 6 mutations were ascertained via whole exome sequencing, even though it was the last approach adopted, illustrating the power of this technique for identifying causative mutations for muscular dystrophies.
Keywords: Founder effect, Homozygosity mapping, Limb-girdle muscular dystrophy, Sarcoglycanopathy, Whole exome sequencing
Introduction
Limb girdle muscular dystrophy (LGMD) is a category of a group of rare, heterogeneous, genetically inherited muscle diseases. LGMD causes progressive proximal muscle weakness and a range of histological abnormalities, with variable rates of progression and degrees of severity. Numerous genes have been associated with different forms of LGMD1. Inheritance patterns differentiate the two major subtypes: LGMD type 1 (LGMD1) is inherited in a dominant fashion and LGMD type 2 (LGMD2) follows a recessive pattern. To date, 8 dominant forms (LGMD1A-H) and 23 recessive forms (LGMD2A-W) have been described2,3. Onset of symptoms may occur at nearly any age, from childhood through adolescence into adulthood; the only exception is infancy, which by definition would indicate the presence of a congenital muscular dystrophy. Mutations in known genes can be found in approximately 45–46% of cases of neuromuscular disorders4 and LGMD5. Clinical genetic testing is not available for some associated genes, especially the less common ones, and access to such testing is often limited, due primarily to the costs involved. This makes it difficult for many affected families to obtain a definitive genetic diagnosis.
In this study, we recruited and analyzed 30 families of Egyptian descent, each of which has one or more individuals with the LGMD phenotype. Immunohistochemistry, homozygosity mapping, Sanger sequencing, and exome sequencing contributed to the identification of causative mutations in known genes for 6 of the families. Four of the families harbored mutations in SGCA, while 2 others were found to have cases of dystrophinopathy.
Materials and Methods
Patient recruitment and clinical diagnosis
Affected individuals with the clinical diagnosis of LGMD and informative relatives from 30 families were enrolled in an institutionally-approved research protocol at Assiut University, Egypt. The families were numbered 1267–1285, 1357–1364, and 1410–1412. Results of clinical evaluations were available for the study, including medical histories, family histories, physical examination, and serum creatine kinase (CK) levels.
Muscle histology
Medial gastrocnemius muscle biopsies were performed on selected affected individuals. This muscle was selected as it displayed a lesser degree of weakness in the subjects and would thus be less likely to display excessive fibrosis. The tissue samples were snap frozen and stored at −80°C. Hematoxylin and eosin staining was performed using standard protocols on samples representing families 1268, 1271, 1273, 1275, 1276, 1278, 1280, and 1282. Immunohistochemistry was performed using antibodies to dystrophin (DYS-1, 2, 3), α-, β-, γ- and δ- sarcoglycan, dysferlin, and α- and β-dystroglycan on muscle biopsy samples from subjects 1271-1, 1273-1, 1275-1, 1276-1, 1278-1, 1280-1, and 1282-1. Staining for DYS-1, 2, 3, and α-, β-, γ- and δ- sarcoglycan was performed on muscle biopsy samples from subjects 12704, 1272-1, 1274-1, 1277-1, 1279-1, 1283-1, and 1285-1. Novocastra NCL-a-SARC antibody was used to stain for α-sarcoglycan.
DNA extraction
Peripheral blood samples were obtained from affected individuals and informative relatives in the enrolled families. Genomic DNA was extracted from the blood samples using the QIAamp DNA Blood Midi Kit (Qiagen), then quantified using a NanoDrop 1000 Spectrophotometer (Fisher Scientific).
Homozygosity mapping
DNA samples from affected individuals in families known or suspected to be consanguineous (1267–1269, 1271–1275, 1277, 1280, 1284, 1357, 1358, 1360–1363) were selected for genotyping at 10,204 single nucleotide polymorphisms (SNPs) using the GeneChip Human Mapping 10K 2.0 Array (Affymetrix), and homozygosity mapping was performed as previously described6.
Whole exome sequencing
The Genomics Platform at the Broad Institute was used to perform whole exome sequencing of DNA samples representing selected subjects for whom immunohistochemistry and homozygosity mapping did not suggest the associated gene harboring the causative mutations. The Agilent Sure-Select Human All Exon v2.0, 44Mb baited target and the Broad’s in-solution hybrid selection process were used to target exons in genomic DNA. At least 250 ng of DNA with concentrations of at least 2ng/μl were submitted for each sample. The exome-sequencing pipeline included sample plating, library preparation (2- plexing of samples per hybridization), hybrid capture, sequencing (76bp paired reads), sample identification quality control check, and data storage. The hybrid selection libraries cover >80% of targets at 20× or more, with a mean target coverage of >80×. The exome sequencing data was de-multiplexed and each sample’s sequence data were aggregated into a single Picard BAM file. Exome sequencing data was processed through a pipeline based on Picard (https://github.com/broadinstitute/picard), using base quality score recalibration and local realignment at known insertions and deletions. The BWA aligner (https://github.com/lh3/bwa) mapped reads to the human genome build 37 (hg19) reference sequence. Single nucleotide polymorphisms (SNPs) and insertions/deletions (indels) were jointly called across all samples using the Genome Analysis Toolkit (GATK) HaplotypeCaller package version 3.3. Default filters were applied to SNP and indel calls using the GATK’s Variant Quality Score Recalibration (VQSR) approach. The variants were annotated using Variant Effect Predictor. The variant call set was uploaded on to xBrowse (https://atgu.mgh.harvard.edu/xbrowse/) and an analysis limited to the candidate gene list was performed using the various inheritance patterns. The main report contains variants restricted to nonsense, frameshift, essential splice site and missense variants and filtered on variant site and genotype quality. The appendix listing each gene contains all variants discovered regardless of annotation and quality. Species conservation and predicted pathogenicity of these variants was examined using SIFT7, PolyPhen-28, Mutation taster9, and FATHMM10.
Sanger sequencing
Sanger sequencing was performed for a number of candidate genes and mutations ascertained via muscle immunohistochemistry, homozygosity mapping, and whole exome sequencing. Sequencing was first performed in an affected family member for confirmation and then specific mutations were sequenced in other family members to track cosegregation patterns. Coding regions of SGCA, SGCB, SGCG, SGCD, and FKRP were amplified via PCR using standard oligonucleotide primers11. Amplicons were assessed via agarose gel electrophoresis, then purified by treating 5 μl of PCR product with 2μl of Exonuclease and Shrimp Alkaline Phosphatase mix (Exo-SAP-IT; Affymetrix) and submitted to the Molecular Genetics Core Facility at Boston Children’s Hospital and the Interdisciplinary Center for Biotechnology Research (ICBR) at the University of Florida for sequencing. Sequence data were analyzed using Sequencher v.5.2.3 (GeneCodes Corporation, Ann Arbor, Michigan).
Cloning of PCR products
The SGCA exon harboring the compound heterozygous NM_000023.2 c.739G>A and c.746T>C mutations (see Results Section below) was amplified from genomic DNA samples representing family 1280, then cloned into the pGEM®-T Easy Vector (Promega) following the manufacturer’s protocol. Five colonies from each sample were picked and multiplied. Plasmid DNA was extracted from these colonies and sequenced using universal M13 forward and reverse primers.
Results
Clinical features
All affected individuals were determined to have a clinical diagnosis consistent with LGMD. However, due to the lack of availability of local clinical genetic testing, the potential diagnosis of dystrophinopathy could not be excluded in all families prior to enrollment, especially those who did not have affected females and whose inheritance patterns did not indicate male-to-male transmission. Detailed clinical features of the probands for whom causative mutations were identified are described in Supplemental Table 1
Muscle histology
Hematoxylin and eosin staining confirmed dystrophic pathology in muscle samples from the selected affected individuals. The most notable immunohistochemical finding was absent α-sarcoglycan staining in an affected individual from family 1276 (Figure 1), prompting Sanger sequencing of the entire SGCA gene in the probands of this family. Of note, α-sarcoglycan staining was present albeit patchy in 1280-1, thus Sanger sequencing for SGCA was not performed prior to exome sequencing (Figure 1). Other immunohistochemical abnormalities were observed but Sanger sequencing did not confirm primary causative mutations for these other findings (data not shown).
Figure 1.
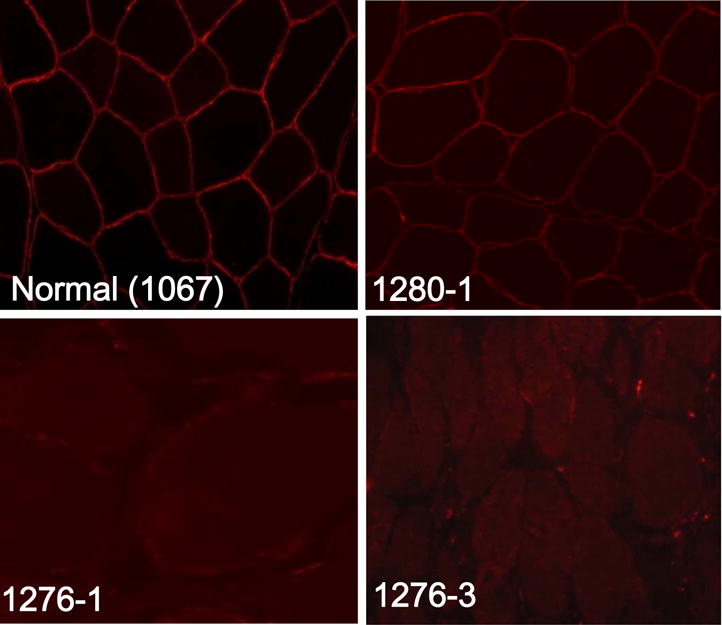
Immunohistochemistry with α–sarcoglycan antibodies (NCL-a-SARC, Novocastra) revealed the presence of α–sarcoglycan protein in control muscle sections (1067), but not in muscle sections from affected individuals 1276-1 or 1276-3; patchy staining was noted in 1280–1.
Homozygosity mapping
Due to the selection of only one affected individual for genotyping in each family, numerous candidate causative genes were identified on homozygosity mapping. Among these, SGCA was found to harbor pathogenic mutations on Sanger sequencing for family 1269 (see below). Sequencing of candidate genes for other families did not yield causative mutations (data not shown).
Whole exome sequencing
Affected individuals from families 1273–1275, 1277–1278, 1280–1284 were selected for whole exome sequencing based on a lack of plausible candidate causative mutations after the analyses noted above. This analysis identified likely mutations in 4 families: a homozygous NM_000023.2 c.574C>T; NP_000014.1 p.Arg192Ter nonsense mutation in SGCA for family 1274, a hemizygous NM_4006.2 c.8333G>A; NP_003997.1 p.Arg2778His missense mutation in the dystrophin (DMD) gene for family 1277, compound heterozygous NM_000023.2 c.739G>A; NP_000014.1 p.Val247Met and NM_000023.2 c.746T>C; NP_000014.1 p.Leu249Pro missense mutations in SGCA for family 1280, and a hemizygous NM_4006.2 c.7523delC; NP_003997.1 p.Glu2508Gly fs Ter2 frameshift mutation in DMD for family 1281.
Sanger sequencing and cloning
Sanger sequencing confirmed the homozygous SGCA NM_000023.2 c.574C>T; NP_000014.1 p.Arg192Ter nonsense mutation in family 1274 that was identified on whole exome sequencing, and identified the same homozygous mutation in families 1269 and 1276 (Supplemental Figure 1). Sanger sequencing also confirmed the mutations in families 1277, 1280, and 1281 found on whole exome sequencing (Supplemental Figure 1). Co-segregation was confirmed in all 6 families with identified mutations (Figures 2 and Supplemental Figure 1). Screening for the SGCA NM_000023.2 c.574C>T; NP_000014.1 p.Arg192Ter mutation in all remaining families demonstrated no additional affected kindreds (Supplemental Figure 2). Cloning of SGCA exon 6 amplified from the genomic DNA of 1280-1, 1280-2, and 1280-3 confirmed that the two mutations were in trans (Supplemental Figure 1).
Figure 2.
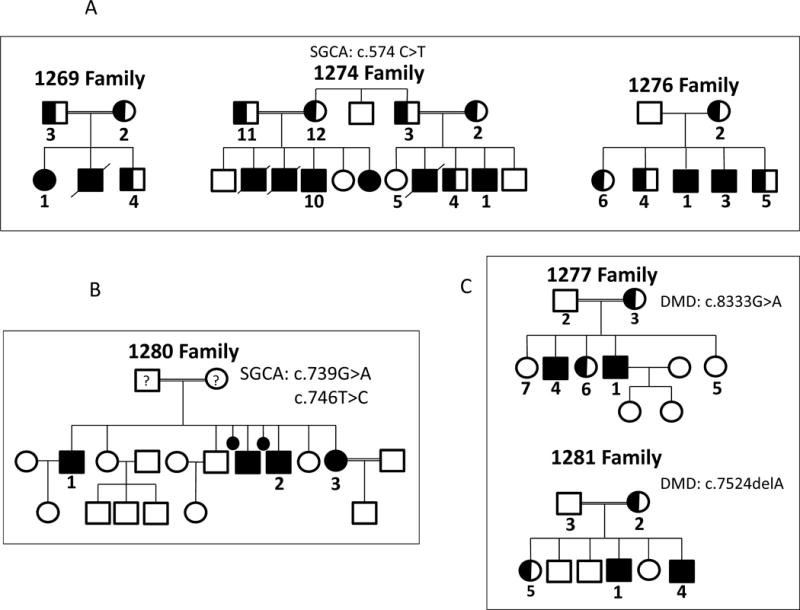
(A) Pedigrees for the three families who harbor the SGCA c.574C>T mutation. (B) Pedigree of family 1280 showed 3 affected siblings 1280-1, 1280-2, and 1280-3 who have compound heterozygous SGCA c.739G>A and c.746T>C mutations. Genotypes of parents of proband (1280-1) are unknown and hence represented with ‘?’. (C) Pedigree of family 1277 with two affected brothers harboring a hemizygous c.8333G>A DMD mutation, and pedigree of family 1281 with two affected brothers harboring a hemizygous c.7523delA DMD mutation. Filled circles squares represent affected individuals confirmed to have homozygous, compound heterozygous, or hemizygous mutations. Half-filled circles and squares represent heterozygous carriers. Unfilled circles and squares represent unaffected individuals.
Discussion
The analysis of these families spanned the years 2010–2015, a period of rapid advances in genomic technologies. As a result, several techniques helped identify the associated genes in various families, including muscle immunohistochemistry, homozygosity mapping, and whole exome sequencing. This hybrid approach illustrates how yields can be optimized by relying on complementary genetic approaches. It is notable that whole exome sequencing ascertained confirmed mutations in 4 of the 6 families in this cohort, including the compound heterozygous SGCA mutations in the setting of intact, albeit patchy α-staining for family 1280. Residual α-sarcoglycan staining in the setting of SGCA mutations has been described previously12,13. Other candidate genes that were sequenced due to results of immunohistochemistry and homozygosity mapping did not yield causative mutations, illustrating the power of whole exome sequencing in this setting. Given that all mutations identified were in previously described muscular dystrophy genes, the equivalent results could have been obtained via broad spectrum Sanger sequencing. However, the rapidly declining cost of exome sequencing suggests that this newer technology will play an increasingly important role in screening such cohorts. Analysis continues for the remaining 24 families for whom causative mutations have not been identified to date. One potential strategy for further analysis is comparative genomic hybridization, but that is beyond the scope of the current study.
Collectively, the sarcoglycanopathies (LGMD2C, LGMD2D, LGMD2E, LGMD2F), caused by mutations in SGCG14,15, SGCA16, SGCB17, and SGCD18 Respectively, comprise some of the more common subtypes of LGMD. They are especially common in Brazil, where they account for as much as 68% of cases of LGMD219. Other common forms of LGMD include: LGMD2A, caused by mutations in CAPN320; LGMD2B, a dysferlinopathy caused by mutations in DYSF21,22; LGMD2I, caused by mutations in FKRP23,24; and LGMD2L, caused by mutations in ANO525,26. A recent review mapped the geographical distribution and prevalence of LGMD throughout the world, based on currently published data, and did not report data from Egypt27. In the same year, dysferlinopathy was reported to be a common cause of LGMD in Egypt28. As the current cohort does not appear to harbor pathogenic mutations in DYSF, further studies on a broader geographic scale will be needed to help delineate the genetic epidemiology of LGMD in Egypt.
The tetrameric sarcoglycan complex is composed of the 4 sarcoglycans expressed in muscle (α- sarcoglycan, β-sarcoglycan, γ-sarcoglycan, and δ-sarcoglycan) and is in turn part of a larger macromolecular complex of proteins called the dystrophin-associated protein complex (DAPC). The DAPC contributes to the stability of the plasma membrane cytoskeleton, maintains sarcolemmal integrity, and ensures transduction during muscle contraction29. Mutations in SGCG were reported as early as 1995 in Northern Africa, though not including Egypt14. LGMD2D is caused by mutations in SGCA. SGCA c.229C>T, p.R77C in exon 3 is a founder mutation from the Magdalen Islands in the Gulf of St. Lawrence. It is the most common SGCA mutation reported internationally30. The same mutation was also reported as a founder mutation in several LGMD2D patients from Finland31 and also from Europe and Brazil27. A homozygous SGCA c. 157G>A mutation in exon 2 was found in two Tunisian siblings32. The current study identified a homozygous SGCA NM_000023.2 c.574C>T; NP_000014.1 p.Arg192Ter nonsense mutation in 3 unrelated families among the 30 that were screened. The frequency of this mutation suggests a possible founder effect. More than 70 pathogenic SGCA mutations have been described to date33. The SGCA c.574 C>T mutation has been reported previously, in a sporadic family with two affected siblings from Albania34, and appears to be the only previously published report of this mutation. Genetic diagnosis in LGMD along with other genetic disorders will become increasingly important in the coming years as new molecular therapies will target specific gene defects and even specific mutation types. For example, several studies suggest that LGMD2D may be amenable to gene therapy approaches35–37.
There is considerable phenotypic overlap between LGMDs and the dystrophinopathies, the latter caused by mutations in DMD38,39. As more LGMD genes were discovered, patients who had previously been diagnosed with Duchenne muscular dystrophy were sometimes found to have LGMD instead40. Sarcoglycanopathies may cause either a severe Duchenne-like phenotype or a milder Becker-like phenotype. The two may often be distinguished by the inheritance pattern, though this may be difficult to discern in a sporadic male case. Cardiac complications are essentially universal in Duchenne muscular dystrophy, whereas they are less consistently present in the sarcoglycanopathies; as the cardiac complications arise in the later stages of disease, this clinical feature may not help distinguish between the two in the early stages of disease41. The two families in the current report found to have DMD mutations had clinical features that resembled those of LGMD, and have milder phenotypes, suggesting that they are most likely to have Becker muscular dystrophy (1277-1 had positive dystrophin staining on muscle biopsy and was able to father children, while 1281-1 has a normal CK and remains ambulatory). A study of Duchenne muscular dystrophy in Egypt using multiplex PCR found that 78 (51.3%) of 152 subjects showed out-of-frame deletions in the DMD gene42. The DMD NM_4006.2 c.8333G>A mutation found in family 1277 does not appear to have been published before, but is reported online for a patient with a Becker muscular dystrophy phenotype from Valentia, Spain (http://dmd.nl). The arginine amino acid residue at this locus is very well conserved among several species, and the DMD c.8333G>A mutation co-segregated with the phenotype in family 1277. The DMD NM_4006.2 c.7523delA mutation in family 1281 also co-segregates with the phenotype among the other family members. This mutation does not appear to have been published previously.
It is notable that both 1277 and 1281 families are consanguineous, but harbor causal mutations in DMD, which is X-linked. This is an interesting example of how consanguinity may be misleading in genetic analyses. Similarly, family 1280, also consanguineous, harbors SGCA mutations that are compound heterozygous. Thus, it is important to remember that genetic diseases in consanguineous families may not always be caused by homozygous recessive mutations.
This study shows that mutations in SGCA are a common cause of LGMD in Assiut, Egypt, along with the DYSF mutations reported previously. The moderate size of this clinic-based cohort makes it premature to draw more general conclusions about the overall genetic epidemiology of LGMD in Egypt; however, the data presented here provide useful information that will complement future studies. The SGCA NM_000023.2 c.574C>T; NP_000014.1 p.Arg192Ter mutation is likely due to a founder effect, and is expected to be present in other affected families. These results also illustrate the difficulty in distinguishing dystrophinopathies from LGMDs without access to genetic testing or muscle pathology resources.
Supplementary Material
Acknowledgments
The authors would like to thank the participants for enrolling in the study, along with Marielle Thorne for assistance with sample processing. This project was funded by NIH R01 NS080929 (HMR and PBK), NIH R01 GM104371 (DGM), Muscular Dystrophy Association Grant 186796 (PBK), the Bernard F. and Alva B. Gimble Foundation (LMK), and the Department of Pediatrics at the University of Florida College of Medicine (HMR, MJ, KAC and PBK). Sanger DNA sequencing was performed at the IDDRC Molecular Genetics Core Facility at Boston Children’s Hospital and the Interdisciplinary Center for Biotechnology Research (ICBR) Sanger Sequencing Core at the University of Florida. Exome sequencing was supported by Medical Sequencing Program Grant U54HG003067 from the National Human Genome Research Institute.
Abbreviations
- CK
creatine kinase
- DMD
dystrophin
- LGMD
limb-girdle muscular dystrophy
- SGCA
α-sarcoglycan
- SGCB
β-sarcoglycan
- SGCD
δ-sarcoglycan
- SGCG
γ-sarcoglycan
References
- 1.Mitsuhashi S, Kang PB. Update on the genetics of limb girdle muscular dystrophy. Seminars in pediatric neurology. 2012;19(4):211–218. doi: 10.1016/j.spen.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 2.England JD. Making sense of the muscular dystrophies: diagnosis and treatment guideline for limb-girdle muscular dystrophy. Muscle Nerve. 2014;50(5):721–722. doi: 10.1002/mus.24459. [DOI] [PubMed] [Google Scholar]
- 3.Nigro V, Savarese M. Genetic basis of limb-girdle muscular dystrophies: the 2014 update. Acta myologica: myopathies and cardiomyopathies: official journal of the Mediterranean Society of Myology/edited by the Gaetano Conte Academy for the study of striated muscle diseases. 2014;33(1):1–12. [PMC free article] [PubMed] [Google Scholar]
- 4.Ankala A, da Silva C, Gualandi F, Ferlini A, Bean LJ, Collins C, et al. A comprehensive genomic approach for neuromuscular diseases gives a high diagnostic yield. Annals of neurology. 2015;77(2):206–214. doi: 10.1002/ana.24303. [DOI] [PubMed] [Google Scholar]
- 5.Ghaoui R, Cooper ST, Lek M, Jones K, Corbett A, Reddel SW, et al. Use of Whole-Exome Sequencing for Diagnosis of Limb-Girdle Muscular Dystrophy: Outcomes and Lessons Learned. JAMA neurology. 2015;72(12):1424–1432. doi: 10.1001/jamaneurol.2015.2274. [DOI] [PubMed] [Google Scholar]
- 6.Boyden SE, Salih MA, Duncan AR, White AJ, Estrella EA, Burgess SL, et al. Efficient identification of novel mutations in patients with limb girdle muscular dystrophy. Neurogenetics. 2010;11(4):449–455. doi: 10.1007/s10048-010-0250-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nature protocols. 2009;4(7):1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 8.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nature methods. 2010;7(4):248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nature methods. 2014;11(4):361–362. doi: 10.1038/nmeth.2890. [DOI] [PubMed] [Google Scholar]
- 10.Shihab HA, Gough J, Cooper DN, Stenson PD, Barker GL, Edwards KJ, et al. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Human mutation. 2013;34(1):57–65. doi: 10.1002/humu.22225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bennett RR, Schneider HE, Estrella E, Burgess S, Cheng AS, Barrett C, et al. Automated DNA mutation detection using universal conditions direct sequencing: application to ten muscular dystrophy genes. BMC genetics. 2009;10:66. doi: 10.1186/1471-2156-10-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moreira ES, Vainzof M, Suzuki OT, Pavanello RC, Zatz M, Passos-Bueno MR. Genotype- phenotype correlations in 35 Brazilian families with sarcoglycanopathies including the description of three novel mutations. Journal of medical genetics. 2003;40(2):E12. doi: 10.1136/jmg.40.2.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vainzof M, Moreira ES, Canovas M, Anderson LV, Pavanello RC, Passos-Bueno MR, et al. Partial alpha-sarcoglycan deficiency with retention of the dystrophin-glycoprotein complex in a LGMD2D family. Muscle Nerve. 2000;23(6):984–988. doi: 10.1002/(sici)1097-4598(200006)23:6<984::aid-mus24>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 14.Noguchi S, McNally EM, Ben Othmane K, Hagiwara Y, Mizuno Y, Yoshida M, et al. Mutations in the dystrophin-associated protein gamma-sarcoglycan in chromosome 13 muscular dystrophy. Science. 1995;270(5237):819–822. doi: 10.1126/science.270.5237.819. [DOI] [PubMed] [Google Scholar]
- 15.McNally EM, Passos-Bueno MR, Bonnemann CG, Vainzof M, de Sa Moreira E, Lidov HG, et al. Mild and severe muscular dystrophy caused by a single gamma-sarcoglycan mutation. American journal of human genetics. 1996;59(5):1040–1047. [PMC free article] [PubMed] [Google Scholar]
- 16.Piccolo F, Roberds SL, Jeanpierre M, Leturcq F, Azibi K, Beldjord C, et al. Primary adhalinopathy: a common cause of autosomal recessive muscular dystrophy of variable severity. Nature genetics. 1995;10(2):243–245. doi: 10.1038/ng0695-243. [DOI] [PubMed] [Google Scholar]
- 17.Bonnemann CG, Modi R, Noguchi S, Mizuno Y, Yoshida M, Gussoni E, et al. Beta- sarcoglycan (A3b) mutations cause autosomal recessive muscular dystrophy with loss of the sarcoglycan complex. Nature genetics. 1995;11(3):266–273. doi: 10.1038/ng1195-266. [DOI] [PubMed] [Google Scholar]
- 18.Nigro V, de Sa Moreira E, Piluso G, Vainzof M, Belsito A, Politano L, et al. Autosomal recessive limb-girdle muscular dystrophy, LGMD2F, is caused by a mutation in the delta- sarcoglycan gene. Nature genetics. 1996;14(2):195–198. doi: 10.1038/ng1096-195. [DOI] [PubMed] [Google Scholar]
- 19.Vainzof M, Passos-Bueno MR, Pavanello RC, Marie SK, Oliveira AS, Zatz M. Sarcoglycanopathies are responsible for 68% of severe autosomal recessive limb-girdle muscular dystrophy in the Brazilian population. Journal of the neurological sciences. 1999;164(1):44–49. doi: 10.1016/s0022-510x(99)00040-4. [DOI] [PubMed] [Google Scholar]
- 20.Richard I, Brenguier L, Dincer P, Roudaut C, Bady B, Burgunder JM, et al. Multiple independent molecular etiology for limb-girdle muscular dystrophy type 2A patients from various geographical origins. American journal of human genetics. 1997;60(5):1128–1138. [PMC free article] [PubMed] [Google Scholar]
- 21.Bashir R, Britton S, Strachan T, Keers S, Vafiadaki E, Lako M, et al. A gene related to Caenorhabditis elegans spermatogenesis factor fer-1 is mutated in limb-girdle muscular dystrophy type 2B. Nature genetics. 1998;20(1):37–42. doi: 10.1038/1689. [DOI] [PubMed] [Google Scholar]
- 22.Liu J, Aoki M, Illa I, Wu C, Fardeau M, Angelini C, et al. Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nature genetics. 1998;20(1):31–36. doi: 10.1038/1682. [DOI] [PubMed] [Google Scholar]
- 23.Kang PB, Feener CA, Estrella E, Thorne M, White AJ, Darras BT, et al. LGMD2I in a North American population. BMC musculoskeletal disorders. 2007;8:115. doi: 10.1186/1471-2474-8-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brockington M, Yuva Y, Prandini P, Brown SC, Torelli S, Benson MA, et al. Mutations in the fukutin-related protein gene (FKRP) identify limb girdle muscular dystrophy 2I as a milder allelic variant of congenital muscular dystrophy MDC1C. Human molecular genetics. 2001;10(25):2851–2859. doi: 10.1093/hmg/10.25.2851. [DOI] [PubMed] [Google Scholar]
- 25.Sarkozy A, Hicks D, Hudson J, Laval SH, Barresi R, Hilton-Jones D, et al. ANO5 gene analysis in a large cohort of patients with anoctaminopathy: confirmation of male prevalence and high occurrence of the common exon 5 gene mutation. Human mutation. 2013;34(8):1111–1118. doi: 10.1002/humu.22342. [DOI] [PubMed] [Google Scholar]
- 26.Bolduc V, Marlow G, Boycott KM, Saleki K, Inoue H, Kroon J, et al. Recessive mutations in the putative calcium-activated chloride channel Anoctamin 5 cause proximal LGMD2L and distal MMD3 muscular dystrophies. American journal of human genetics. 2010;86(2):213–221. doi: 10.1016/j.ajhg.2009.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mahmood OA, Jiang XM. Limb-girdle muscular dystrophies: where next after six decades from the first proposal (Review) Molecular medicine reports. 2014;9(5):1515–1532. doi: 10.3892/mmr.2014.2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fahmy NA, Abd Elhady A, Abd El-Naser A, Ashour S, Etribi A, Nonaka I, et al. G.P.288: Dysferlinopathy in Egypt: Clinical, Pathological and Genetic characteristics. Neuromuscular Disord. 2014;24(9–10):903–904. [Google Scholar]
- 29.Filosto M, Scarpelli M, Padovani A. eLS. John Wiley & Sons, Ltd; 2001. Molecular Genetics of Limb-Girdle Muscular Dystrophies. [Google Scholar]
- 30.Tetreault M, Srour M, Allyson J, Thiffault I, Loisel L, Robitaille Y, et al. Founder mutation for alpha-sarcoglycan-LGMD2D in a Magdalen Islands Acadian cluster. The Canadian journal of neurological sciences Le journal canadien des sciences neurologiques. 2011;38(5):747–752. doi: 10.1017/s0317167100054135. [DOI] [PubMed] [Google Scholar]
- 31.Hackman P, Juvonen V, Sarparanta J, Penttinen M, Aarimaa T, Uusitalo M, et al. Enrichment of the R77C alpha-sarcoglycan gene mutation in Finnish LGMD2D patients. Muscle Nerve. 2005;31(2):199–204. doi: 10.1002/mus.20267. [DOI] [PubMed] [Google Scholar]
- 32.Fendri K, Kefi M, Hentati F, Amouri R. Genetic heterogeneity within a consanguineous family involving the LGMD 2D and the LGMD 2C genes. Neuromuscular Disord. 2006;16(5):316–320. doi: 10.1016/j.nmd.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 33.Diniz G, Tosun Yildirim H, Akinci G, Hazan F, Ozturk A, Yararbas K, et al. Sarcolemmal alpha and gamma sarcoglycan protein deficiencies in Turkish siblings with a novel missense mutation in the alpha sarcoglycan gene. Pediatr Neurol. 2014;50(6):640–647. doi: 10.1016/j.pediatrneurol.2013.12.024. [DOI] [PubMed] [Google Scholar]
- 34.Babameto-Laku A, Tabaku M, Tashko V, Cikuli M, Mokini V. The First Case of Primary Alpha-Sarcoglycanopathy Identified in Albania, in Two Siblings with Homozygous Alpha- Sarcoglycan Mutation. Genet Counsel. 2011;22(4):377–383. [PubMed] [Google Scholar]
- 35.Pacak CA, Walter GA, Gaidosh G, Bryant N, Lewis MA, Germain S, et al. Long-term skeletal muscle protection after gene transfer in a mouse model of LGMD-2D. Molecular therapy: the journal of the American Society of Gene Therapy. 2007;15(10):1775–1781. doi: 10.1038/sj.mt.6300246. [DOI] [PubMed] [Google Scholar]
- 36.Mendell JR, Rodino-Klapac LR, Rosales XQ, Coley BD, Galloway G, Lewis S, et al. Sustained alpha-sarcoglycan gene expression after gene transfer in limb-girdle muscular dystrophy, type 2D. Annals of neurology. 2010;68(5):629–638. doi: 10.1002/ana.22251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mendell JR, Rodino-Klapac LR, Rosales-Quintero X, Kota J, Coley BD, Galloway G, et al. Limb-girdle muscular dystrophy type 2D gene therapy restores alpha-sarcoglycan and associated proteins. Annals of neurology. 2009;66(3):290–297. doi: 10.1002/ana.21732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Piccolo F, Moore SA, Mathews KD, Campbell KP. Limb-girdle muscular dystrophies. Advances in neurology. 2002;88:273–291. [PubMed] [Google Scholar]
- 39.Monaco AP, Neve RL, Colletti-Feener C, Bertelson CJ, Kurnit DM, Kunkel LM. Isolation of candidate cDNAs for portions of the Duchenne muscular dystrophy gene. Nature. 1986;323(6089):646–650. doi: 10.1038/323646a0. [DOI] [PubMed] [Google Scholar]
- 40.Schwartz M, Hertz JM, Sveen ML, Vissing J. LGMD2I presenting with a characteristic Duchenne or Becker muscular dystrophy phenotype. Neurology. 2005;64(9):1635–1637. doi: 10.1212/01.WNL.0000157654.59374.E5. [DOI] [PubMed] [Google Scholar]
- 41.Al-Zaidy S, Rodino-Klapac L, Mendell JR. Gene therapy for muscular dystrophy: moving the field forward. Pediatr Neurol. 2014;51(5):607–618. doi: 10.1016/j.pediatrneurol.2014.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Elhawary NA, Shawky RM, Hashem N. Frameshift deletion mechanisms in Egyptian Duchenne and Becker muscular dystrophy families. Molecules and cells. 2004;18(2):141–149. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.