

Abstract
Ratiometric sensors generally couple binding events or chemical reactions at a distal site to changes in the fluorescence of a core fluorophore scaffold. However, such approaches are often hindered by spectral overlap of the product and reactant species. We provide a strategy to design ratiometric sensors that display dramatic spectral shifts by leveraging the chemoselective reactivity of novel functional groups inserted within fluorophore scaffolds. As a proof-of-principle, fluorophores containing a borinate (RF620) or silanediol (SiOH2R) functionality at the bridging position of the xanthene ring system are developed as endogenous H2O2 sensors. Both these fluorophores display far-red to near-infrared excitation and emission prior to reaction. Upon oxidation by H2O2 both sensors are chemically converted to tetramethylrhodamine, producing significant (≥66 nm) blue-shifts in excitation and emission maxima. This work provides a new concept for the development of ratiometric probes.
Keywords: bioorthogonal reaction, fluorescent probes, reactive oxygen species, sensors, signal transduction
Graphical abstract
Ratiometric sensors: A novel method for designing ratiometric sensors termed chemoselective alteration of fluorophore scaffolds (CAFS) is disclosed. Two proof-of-concept hydrogen peroxide sensors with large excitation and emission shifts (≥ 66 nm) were obtained using this approach.
Strategies that are often employed for the development of ratiometric probes involve the appendage of species capable of intramolecular charge transfer (ICT) or fluorescence resonance energy transfer (FRET) to existing fluorophores (Scheme 1).[1] These types of sensors have found applications in the detection of small molecules[1,2] in complex biological systems. Despite the advantages of these approaches, challenges in ratiometric sensor design remain. For example, the sensitivity of current approaches can suffer from spectral overlap of product and reactant species. Furthermore, the rational design of probes that undergo changes in both excitation and emission in response to analyte detection remains difficult. To address these issues, we set out to develop a design approach in which a novel functional group capable of undergoing a chemoselective reaction with an analyte is placed directly within a fluorophore scaffold. We envisioned that reaction with a target analyte would then chemically alter the identity of the fluorophore producing robust changes in both excitation and emission maxima of the product (Scheme 1). We term this approach Chemoselective Alteration of Fluorophore Scaffolds (CAFS).
Scheme 1.
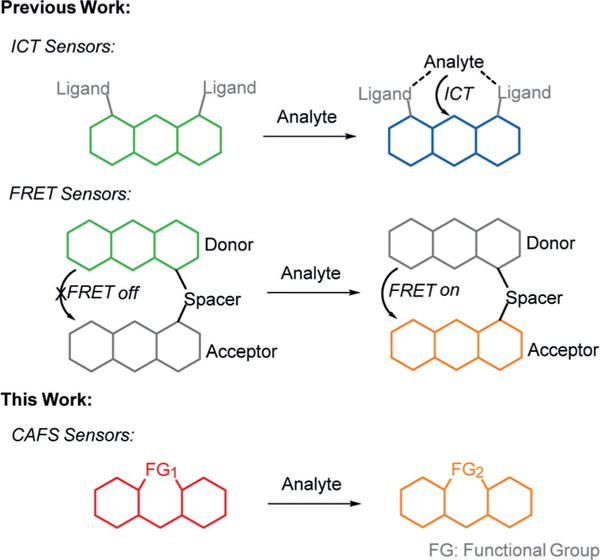
A comparison between common ratiometric sensor designs and the CAFS approach.
For the initial demonstration of CAFS we chose the xanthene scaffold since this structure has found extensive application due to its relative brightness (ε × ϕ) and photostability.[3] Recent work has shown that introduction of new atoms or groups at the bridging position of the xanthene ring can produce significant effects on the photophysical properties of the resulting fluorophores.[4] For example, replacing the oxygen atom of tetramethylrhodamine (TMR) with dimethyl silicon results in a new fluorophore SiR (Figure 1) with a 90 nm red-shift of the emission maxima while maintaining the brightness of the parent scaffold.[4b] Indeed, even longer emission shifts can be achieved by incorporating phosphorus[4a] or sulfur[4d] at the analogues position. Interestingly, our studies have shown that a simple hydrolysis of a bridging phosphinate ethyl ester can yield a 37 nm blue-shift in the emission of the corresponding fluorophore.[5] This experimental observation led us to ask whether chemoselective reactions of novel functional groups in xanthene-based fluorophores could be utilized to afford ratiometric chemodosimeters displaying large shifts in both excitation and emission.
Figure 1.
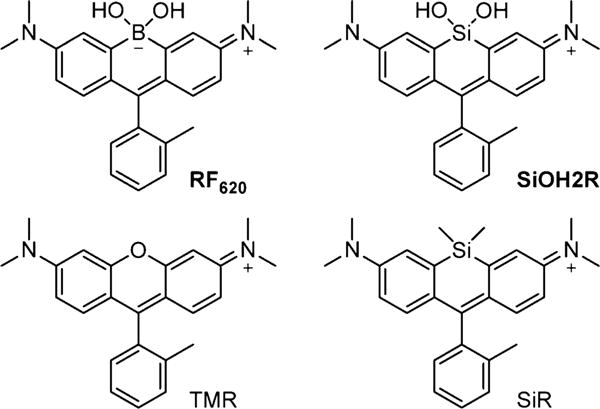
Structures of RF620, SiOH2R, TMR, and SiR.
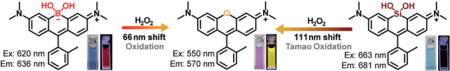
To explore this idea, we introduced a borinate functionality into the xanthene scaffold with the goal of leveraging the selective chemical reactivity of this group to afford a ratiometric sensor for H2O2, termed Rachael Fluor620 (RF620) (Figure 1 and Scheme 2). Hydrogen peroxide is a reactive oxygen species (ROS) and is now recognized as a critical signaling molecule involved in the reversible oxidation of proteins.[6] Thus, there is a need to evaluate the amount of H2O2 in living cells in order to understand protein function and cellular redox mechanisms. Leveraging the chemoselectivity of the borinate functionality in RF620, we envisioned the ability to generate TMR in situ upon reaction with H2O2. Under physiological conditions (10 mM PBS, pH 7.4, with 0.1% DMSO), RF620 exhibited excitation and emission maxima at 620 and 636 nm, respectively. The overlap of TMR emission and RF620 excitation provide the possibility for future FRET applications in settings in which these fluorophores are held in close proximity. Importantly, this corresponds to a 70 nm (excitation) and 66 nm (emission) red-shift relative to TMR. In addition, RF620 (ε = 109000 M−1 cm−1, ϕ = 0.36) was 1.7-fold brighter than TMR (ε = 74500 M−1 cm−1, ϕ = 0.31) under the same buffer conditions (Figure 2). Analysis of RF620 fluorescence as a function of pH revealed a quenching of fluorescence with decreasing pH, with an observed midpoint of 4.36 (Figure S1 in the Supporting Information). Nonetheless, RF620 remained highly fluorescent under physiological conditions (pH 7.4). We also observed an interaction of RF620 with polyols such as fructose and glucose (Figures S2 and S3). Upon binding, the RF620–fructose complex displayed a red-shift in its excitation (638 nm) and emission (654 nm). The disassociation constant was determined to be 14 mM (Figure S2). Similar results were obtained for a borinate-containing xanthene fluorophore reported during the preparation of this manuscript.[7]
Scheme 2.
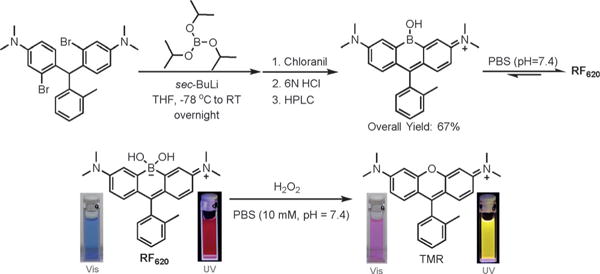
Synthetic scheme for RF620 and its absorbance and fluorescence change upon oxidation by H2O2 under physiological conditions.
Figure 2.
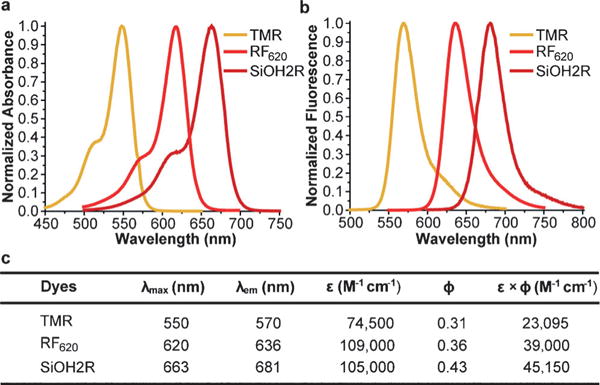
Comparison of the photophysical properties of TMR, RF620, and SiOH2R. All experiments were conducted in PBS (10 mM, pH 7.4 with 0.1% DMSO). a) Absorption spectra. b) Emission spectra. c) Summary of photophysical parameters.
In the presence of H2O2 the formation of TMR was observed, corresponding to 70 and 66 nm blue-shifts in excitation and emission maxima, respectively (Scheme 2). Kinetic studies revealed that the rate of fluorescence quenching of RF620 was 7.96 ± 0.22 M−1 s−1, 53-fold faster than the rate of TMR formation (0.15 ± 0.003 M−1 s−1) under physiological conditions (Figure S4). These data indicate a stepwise oxidation and the existence of a relatively stable intermediate. UV/Vis experiments clearly showed the appearance of a species with absorbance at 580 nm during the course of the reaction (Figure S5). The structural identity of this intermediate is currently under investigation. Importantly, the chemoselectivity of RF620 was confirmed by assessing the formation of TMR in the presence of a panel of ROS including H2O2, superoxide, tert-butyl hydrogen peroxide (TBHP), HOCl, NO, ·OtBu, and ·OH. These experiments yielded a robust 1720-fold increase in the TMR/RF620 emission ratio after 3 hours incubation with H2O2, while all other ROS species tested did not significantly influence the TMR/RF620 ratio (Figure S6). Incubation of HeLa cells with 10 μM RF620 and subsequent addition of 80 μM H2O2 resulted in a dramatic, 870-fold increase in the TMR/RF620 emission ratio (Figure S7). Furthermore, stimulation of endogenous H2O2 production in HeLa cells with phorbol 12-myristate 13-acetate (PMA)[8] or human epidermal growth factor (EGF)[9] resulted in 16- and 14-fold increases in the TMR/RF620 ratio, respectively (Figure 3). TMR generation was abolished in the presence of ebselen, a known H2O2 scavenger,[10] validating the selectivity of RF620 in cells (Figure 3). Colocalization experiments indicated that TMR fluorescence was generated in the mitochondria (Figure S8, Pearson’s coefficient = 0.93). In addition, no cellular toxicity was observed from RF620 at the concentrations used in these experiments (Figure S9). Taken together, the RF620 probe provides the initial proof-of-principle for the ability of CAFS to afford robust ratiometric sensors.
Figure 3.
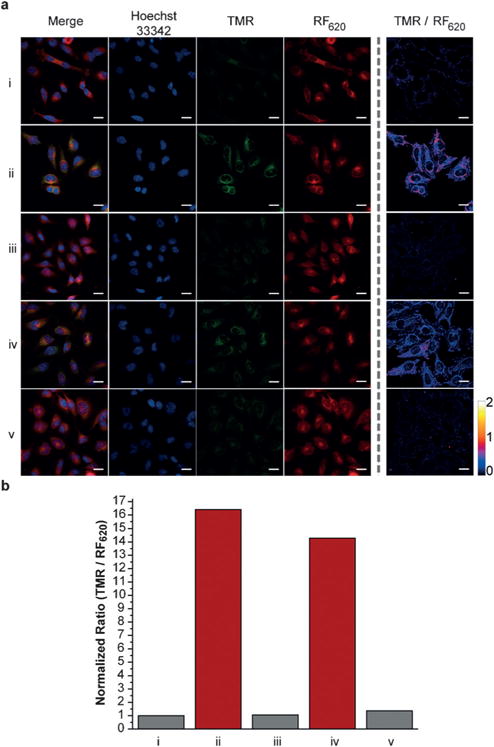
RF620 can detect endogenous production of H2O2 in living cells. a) Confocal fluorescence microscopy imaging of living HeLa cells incubated with 10 μM RF620 for 30 min, washed (3×) and incubated with i. blank, ii. PMA (1 μgmL−1), iii. PMA (1 μgmL−1) with ebselen (5 μm), iv. EGF (500 ngmL−1) or v. EGF (500 ngmL−1) with ebselen (5 μm) for 90 min. b) Comparison of the normalized ratio between TMR and RF620 emission. Scale bar: 25 μm.
Building upon these results, we next asked whether we could further increase the shifts in excitation and emission wavelength upon reaction with H2O2 using the CAFS approach. The Tamao oxidation has been well-established as a strategy to liberate hydroxy functionalities masked with a silane group.[11] Upon reaction with an oxidative reagent like H2O2 or peroxyacids, the C–Si bond is oxidized to a C–O bond. Importantly, computational calculations indicated that a xanthene-based dye containing a silanediol functional group, termed SiOH2R (Figure 1), would display a red-shifted absorption relative to RF620 (Figure S10). We hypothesized that CAFS might be employed with SiOH2R, in order to yield a ratiometric H2O2 probe with an increased excitation and emission shift upon reaction with H2O2 (Scheme 3). Indeed, the resulting SiOH2R fluorophore displayed excitation and emission maxima at 663 and 681 nm, respectively (Figure 2). Moreover, SiOH2R retained the brightness of the parent TMR scaffold (Figure 2), no appreciable binding to polyols was observed (Figure S11), and no significant change in fluorescence upon varying the pH from 3 to 8 was observed (Figure S12). Upon oxidation, SiOH2R displayed a dramatic color change from cyan to pink and a fluorescence emission change from far-red to yellow (Scheme 3 and Figure S13). Gratifyingly, the shift in excitation and emission wavelengths were 113 and 111 nm, respectively. While investigating the kinetics of this reaction, we observed an increase in reaction rate with increasing pH. The apparent second-order rate constant for TMR formation increased linearly from 0.01 M−1 s−1 at pH 7.0 to 0.22 M−1 s−1 at pH 8.5 (Figure S14). This result is consistent with the mechanism of the Tamao oxidation, which proceeds through a hypervalent silicone intermediate.[12] To demonstrate the necessity of a reactive functional group in CAFS, a control experiment was conducted by exposing SiR (Figure 1) to H2O2. No TMR formation was observed in this experiment (Figure S15), indicating the requirement for specially tuned reactive functional groups in CAFS. The chemoselectivity of the silanediol was demonstrated by incubation with a panel of ROS, these experiments demonstrated the selectivity of SiOH2R for H2O2 (Figure S16).
Scheme 3.
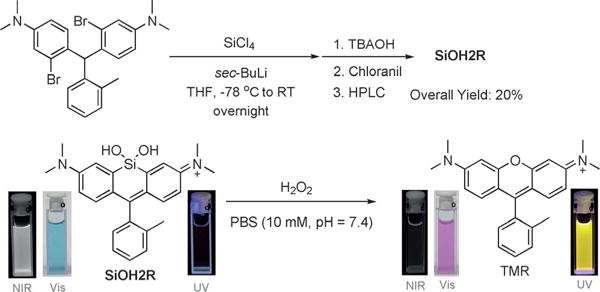
Synthetic scheme for SiOH2R and its color and fluorescence change upon Tamao oxidation with H2O2 under physiological conditions.
Confident in the sensing mechanism of SiOH2R, we further investigated its usefulness in living systems. For these experiments, we found that serum starvation decreased background signal and allowed for increased resolution of TMR formation from SiOH2R. HeLa cells incubated with exogenous H2O2 (80 μM) yielded a 5000-fold increase in the TMR/SiOH2R emission ratio, clearly demonstrating the ability to detect H2O2 (Figure S17). To investigate the ability to detect endogenous H2O2, HeLa cells were stimulated with PMA. These experiments yielded a clear 4.26-fold increase in the TMR/SiOH2R emission ratio (Figure 4). Colocalization experiments indicated that TMR fluorescence was observed in the mitochondria (Figure S18, Pearson’s coefficient = 0.94). Importantly, ebselen abolished TMR generation in these experiments (Figure 4) and no cell toxicity was observed at the concentrations of SiOH2R used in these experiments (Figure S19). Taken together, SiOH2R displays a relatively large shift in both excitation and emission upon reaction with H2O2, providing evidence for the ability to rationally tune ratiometric sensors using the CAFS approach.
Figure 4.
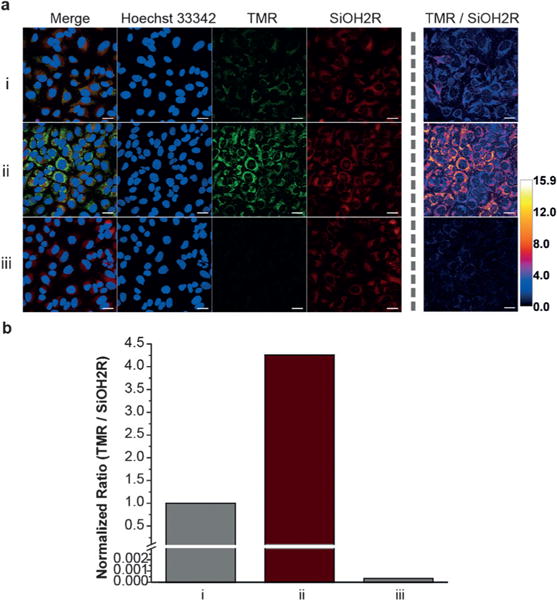
SiOH2R can detect endogenous H2O2 in living cells. a) Serum starved HeLa cells were incubated with i. 0 μgmL−1 PMA 30 min, ii. 1 μgmL−1 PMA 30 min, and iii. 1 μgmL−1 PMA 30 min followed by washing and addition of 5 μM ebselen. Cells were washed (3×) and further incubated with 10 μm SiOH2R for 1 h. c) Comparison of the normalized ratio between TMR and SiOH2R emission. Scale bar: 25 μm.
In conclusion, we have demonstrated the ability of CAFS to yield robust and tunable ratiometric sensors. Two proof-of-concept H2O2 sensors (RF620 and SiOH2R) were designed and validated using this method. The relatively large excitation and emission shifts generated by these probes allows the signal of the product to be easily distinguished from reactant. In addition, to the best of our knowledge, we have demonstrated the first example of the Tamao oxidation for the detection of a biologically relevant signaling molecule. We envision utilizing CAFS to afford multiplexable sensors with orthogonal selectivity to biologically important species.
Supplementary Material
Acknowledgments
We acknowledge Prof. Martha Morton, the Research Instrumentation/NMR facility, and the Nebraska Center for Mass Spectrometry for assistance with characterization of new compounds. We also thank the Morrison Microscopy Core Facility for assistance with confocal fluorescence microscopy as well as Prof. Edward Harris for use of cell culture equipment. Calculations were performed with resources at the University of Nebraska-Lincoln Holland Computing Center. We thank Prof. James Takacs and Veronica Shoba for helpful conversations. This work was funded by the NIH (R35GM119751), the Center for Nanohybrid Functional Materials (NSF EPS-1004094), and the University of Nebraska-Lincoln. The content of this work is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Supporting information for this article can be found under: http://dx.doi.org/10.1002/anie.201612628.
Conflict of interest
The authors declare no conflict of interest.
Contributor Information
Xinqi Zhou, Department of Chemistry, University of Nebraska – Lincoln, Lincoln, NE 68588 (USA).
Lauren Lesiak, Department of Chemistry, University of Nebraska – Lincoln, Lincoln, NE 68588 (USA).
Rui Lai, Department of Chemistry, University of Nebraska – Lincoln, Lincoln, NE 68588 (USA); Nebraska Center for Materials and Nanoscience, University of Nebraska – Lincoln, Lincoln, NE 68588 (USA).
Dr. Jon R. Beck, Department of Chemistry, University of Nebraska – Lincoln, Lincoln, NE 68588 (USA)
Dr. Jia Zhao, Department of Chemistry, University of Nebraska – Lincoln, Lincoln, NE 68588 (USA)
Prof. Dr. Christian G. Elowsky, Department of Agronomy and Horticulture, University of Nebraska – Lincoln, Lincoln, NE 68588 (USA)
Prof. Dr. Hui Li, Department of Chemistry, University of Nebraska – Lincoln, Lincoln, NE 68588 (USA) Nebraska Center for Materials and Nanoscience, University of Nebraska – Lincoln, Lincoln, NE 68588 (USA).
Prof. Dr. Cliff I. Stains, Department of Chemistry, University of Nebraska – Lincoln, Lincoln, NE 68588 (USA)
References
- 1.Lee MH, Kim JS, Sessler JL. Chem Soc Rev. 2015;44:4185–4191. doi: 10.1039/c4cs00280f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chan J, Dodani SC, Chang CJ. Nat Chem. 2012;4:973–984. doi: 10.1038/nchem.1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lavis LD, Raines RT. ACS Chem Biol. 2008;3:142–155. doi: 10.1021/cb700248m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Chai X, Cui X, Wang B, Yang F, Cai Y, Wu Q, Wang T. Chem Eur J. 2015;21:16754–16758. doi: 10.1002/chem.201502921. [DOI] [PubMed] [Google Scholar]; b) Fu MY, Xiao Y, Qian XH, Zhao DF, Xu YF. Chem Commun. 2008:1780–1782. doi: 10.1039/b718544h. [DOI] [PubMed] [Google Scholar]; c) Grimm JB, Gruber TD, Ortiz G, Brown TA, Lavis LD. Bioconjugate Chem. 2016;27:474–480. doi: 10.1021/acs.bioconjchem.5b00566. [DOI] [PubMed] [Google Scholar]; d) Liu J, Sung YQ, Zhang HX, Shi HP, Shi YW, Guo W. ACS Appl Mater Interfaces. 2016;8:22953–22962. doi: 10.1021/acsami.6b08338. [DOI] [PubMed] [Google Scholar]
- 5.Zhou X, Lai R, Beck JR, Li H, Stains CI. Chem Commun. 2016;52:12290–12293. doi: 10.1039/c6cc05717a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.D’Autreaux B, Toledano MB. Nat Rev Mol Cell Biol. 2007;8:813–824. doi: 10.1038/nrm2256. [DOI] [PubMed] [Google Scholar]
- 7.Shimomura N, Egawa Y, Miki R, Fujihara T, Ishimaru Y, Seki T. Org Biomol Chem. 2016;14:10031–10036. doi: 10.1039/c6ob01695b. [DOI] [PubMed] [Google Scholar]
- 8.Xu J, Zhang Y, Yu H, Gao XD, Shao SJ. Anal Chem. 2016;88:1455–1461. doi: 10.1021/acs.analchem.5b04424. [DOI] [PubMed] [Google Scholar]
- 9.Ermakova YG, Bilan DS, Matlashov ME, Mishina NM, Markvicheva KN, Subach OM, Subach FV, Bogeski I, Hoth M, Enikolopov G, Belousov VV. Nat Commun. 2014;5:5222. doi: 10.1038/ncomms6222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Müller A, Cadenas E, Graf P, Sies H. Biochem Pharmacol. 1984;33:3235–3239. doi: 10.1016/0006-2952(84)90083-2. [DOI] [PubMed] [Google Scholar]
- 11.a) Spletstoser JT, Zacuto MJ, Leighton JL. Org Lett. 2008;10:5593–5596. doi: 10.1021/ol802489w. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Barrett AGM, Head J, Smith ML, Stock NS, White AJP, Williams DJ. J Org Chem. 1999;64:6005–6018. [Google Scholar]
- 12.Mader MM, Norrby PO. Chem Eur J. 2002;8:5043–5048. doi: 10.1002/1521-3765(20021104)8:21<5043::AID-CHEM5043>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.