

Abstract
Dry eye disorders are a significant health problem for which limited therapeutic options are available. CFTR is a major prosecretory chloride channel at the ocular surface. We previously identified, by high-throughput screening, aminophenyl-1,3,5-triazine CFTRact-K089 (1) that activated CFTR with EC50 ≈ 250 nM, which when delivered topically increased tear fluid secretion in mice and showed efficacy in an experimental dry eye model. Here, functional analysis of aminophenyl-1,3,5-triazine analogs elucidated structure–activity relationships for CFTR activation and identified substantially more potent analogs than 1. The most potent compound, 12, fully activated CFTR chloride conductance with EC50 ≈ 30 nM, without causing cAMP or calcium elevation. 12 was rapidly metabolized by hepatic microsomes, which supports its topical use. Single topical administration of 25 pmol of 12 increased tear volume in wild-type mice with sustained action for 8 h and was without effect in CFTR-deficient mice. Topically delivered 12 may be efficacious in human dry eye diseases.
INTRODUCTION
CFTR (cystic fibrosis transmembrane conductance regulator) is a cAMP-gated chloride channel with prosecretory activity in the airways, gastrointestinal organs, testis, and exocrine glands.1–4 Loss-of-function mutations in CFTR cause cystic fibrosis, and inappropriate activation of CFTR causes certain secretory diarrheas such as cholera.5–7 CFTR is an important drug target for which major advances have been made in the development of potentiators and correctors of mutant CFTRs in cystic fibrosis, with two drugs having received FDA approval.8 Activators of wild-type CFTR have potential clinical indications for prosecretory therapy of constipation and dry eye disorders and possibly for disorders of the liver, pancreas, and airways,9–12 and CFTR inhibitors are under development for certain secretory diarrheas and polycystic kidney disease.13,14
We previously identified, by high-throughput screening, small-molecule activators of wild-type CFTR that function by a direct activation mechanism.12,15 One class of phenylquinoxalinone CFTR activators showed efficacy following oral administration in experimental mouse models of constipation.9,10 A second class of CFTR activators, which includes the aminophenyl-1,3,5-triazine CFTRact-K089 (compound 1, Figure 1A), activated ocular surface CFTR activity in mice and increased tear fluid secretion after topical delivery.12 In a mouse model of aqueous-deficient dry eye produced by lacrimal gland excision, topical delivery of 1 three times daily corrected defective tear fluid secretion and prevented ocular surface clinical signs. An ideal dry eye therapeutic would produce sustained tear fluid secretion with once or twice daily dosing.
Figure 1.
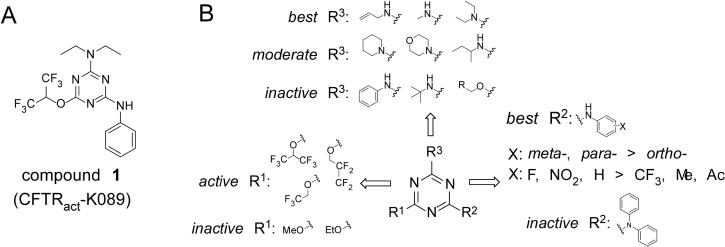
Structure–activity analysis of aminophenyl-1,3,5-triazine CFTR activators. (A) Chemical structure of 1. (B) Preliminary SAR analysis based on commercial analogs (see Supporting Information Table 1 for data on all commercial analogs).
Here, we report structure–activity relationship studies of aminophenyl-1,3,5-triazine CFTR activators with a focus on dry eye applications. Dry eye is a heterogeneous group of disorders associated with reduced tear volume and tear fluid hyperosmolarity, resulting in ocular surface inflammation, eye discomfort, and visual disturbance. Dry eye is a major health problem in an aging population, with five million patients in the U.S. age 50 and over.16,17 The only approved dry eye drugs, topical cyclosporine and lifitegrast, act by an anti-inflammatory mechanism.18,19 CFTR activators would be first-in-class dry eye therapeutics that function by a prosecretory mechanism. Synthesis and characterization of aminophenyl-1,3,5-triazines here produced potential lead candidates with substantially improved potency and in vivo prosecretory efficacy compared to 1, as well as compounds with greater metabolic stability for potential treatment of nonocular conditions.
CHEMISTRY
General Synthesis of Aminophenyl-1,3,5-triazine Analogs
The synthesis of aminophenyl-1,3,5-triazine analogs was accomplished by serial substitution of cyanuric chloride using fluorinated alkoxides, diethylamine, and anilines (Scheme 1). Substitution of cyanuric chloride with 1,1,1,3,3,3-hexafluoroisopropanol or 2,2,3,3-tetrafluoro-1-propanol afforded 2 and 3, respectively. Amination of 2 and 3 using diethylamine afforded intermediates 4 and 5. Refluxing of 4 and 5 with aniline afforded 1 and 7a, respectively. Refluxing m- or p-F, Cl, NO2, CO2H-substituted anilines with 4 and 5 afforded aminophenyl-1,3,5-triazines 6a–g and 7b–g, respectively. 5 was refluxed with 6- or 5-aminoindazole, 6-aminobenzoxazole, and 6-aminobenzothiazole under basic condition to afford 6h–k.
Scheme 1. Synthesis of N,N-Diethyl-1,3,5-triazine Amine Analogsa.
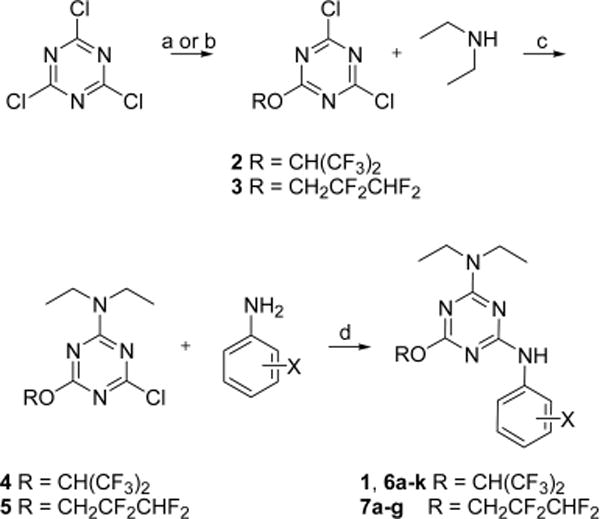
aReagents and conditions: (a) (CF3)2CHOH, K2CO3, THF, −80 °C, 62% for 2; (b) CHF2CF2CH2OH, K2CO3, THF, −80 °C, 68% for 3; (c) Et2NH, DIPEA, THF, 0 °C, 55%; (d) DIPEA, THF, reflux, 25–81%.
N-Methyl-1,3,5-triazine amine analogs 11 and 12 were synthesized as shown in Scheme 2. Methylamine was substituted with cyanuric chloride to afford N-methyl-4,6-dichloro-1,3,5-triazine-2-amine 8 in 75% yield. With 8 in hand, substitution with 1,1,1,3,3,3-hexafluoroisopropanol or 2,2,3,3-tetrafluoro-1-propanol afforded 9 and 10, respectively. Aniline substitution on 9 and 10 afforded N-methyl-N′-phenyl-1,3,5-triazine diamine analogs 11 and 12, respectively.
Scheme 2. Synthesis of N-Methyl-N′-phenyl-1,3,5-triazine Diamine Analogsa.
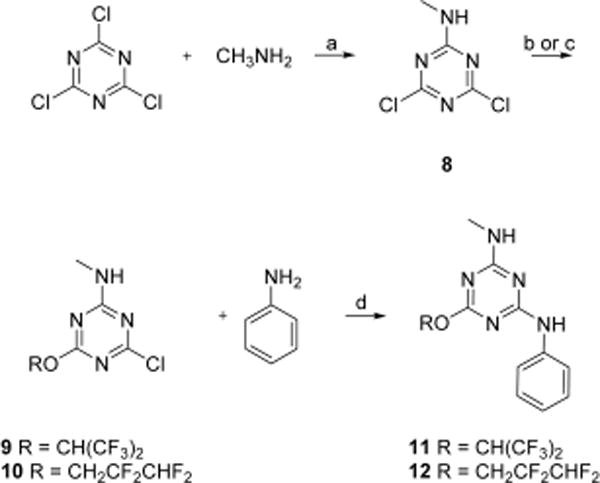
aReagents and conditions: (a) DIPEA, THF, 0 °C, 75%; (b) (CF3)2CHOH, K2CO3, THF, 0 °C, 43% for 9; (c) CHF2CF2CH2OH, K2CO3, THF, 0 °C, 66% for 10; (d) DIPEA, THF, reflux, 74% for 12; 68% for 11.
RESULTS
Preliminary Structure–Activity Analysis of Commercially Available Analogs
Initial evaluation of compound potency was done using a cell-based plate reader assay of CFTR function. Figure 1B summarizes preliminary SAR analysis from functional assays on 91 commercially available analogs of 1 (data for all commercial compounds provided in Supporting Information Table 1). The commercial 1,3,5-triazine analogs tested here were trisubstituted at the 2-, 4-, 6-positions with various substituents, including alkoxide, aniline (aminophenyl), and primary or secondary amines. SAR analysis revealed that trisubstitued-1,3,5-triazines whose three substituents contain a fluorinated alkoxide (R1), an aniline (R2), and an alkylamine (R3) gave the best potency (compounds K032, K059, and K089, Supporting Information Table 1). Analogs with two-alkoxide substituents or nonfluorinated alkoxide greatly reduced potency (K010–018, K023). Further, ortho-substituted aniline (R2) reduced potency as compared to meta- and para-substitution (K007, K032, K059). Electron-donating groups including methoxy, acetyl, trifluoromethoxy (K001, K024–028, K079) reduced activity as compared to electron-withdrawing group such as nitro as in K032. Though we focused on anilines here, benzylamine and substituted benzylamines in place of anilines on R2 may be interesting to test. Finally, sterically bulky alkylamines (R3) including cyclic, tert-butyl- and isobutylamine reduced potency (K029, K056, K060–065). Short alkylamines such as methylamine and diethylamine gave greatest potency.
Because the initial SAR data from commercial analogs were limited and confounded by simultaneous variations in R1, R2, and R3, we synthesized targeted trisubstituted-1,3,5-triazine analogs. EC50 values for CFTR activation for the synthesized compounds are summarized in Charts 1–3. Active compounds produced full activation of CFTR, as seen by the similar efficacy produced by maximal forskolin (20 μM) but with different EC50 values.
Chart 1.
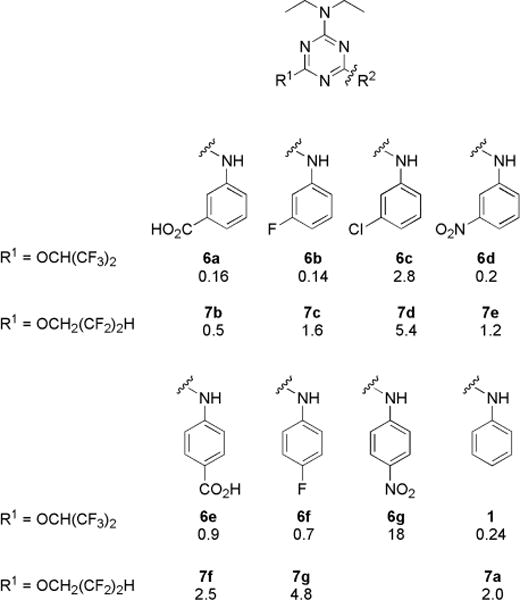
Structures of N,N-Diethyl-N′-aryl-1,3,5-triazine Diamine Analogs and Their EC50 (μM)
Chart 3.
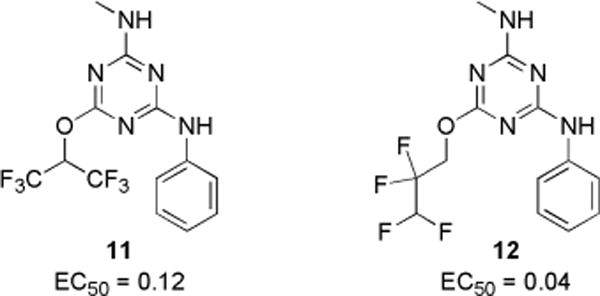
N-Methyl-N′-phenyl-1,3,5-triazine Diamine Analogs and Their EC50 (μM)
Modifications of Substituted Anilines and Fluorinated Alkoxides
Since both fluorinated alkoxide (R1) and short alkylamine (R3) appeared to be crucial for activity, we first prepared N-aryl-1,3,5-triazine amines with R3 as diethylamine and explored the activity of two fluorinated alkoxides, 1,1,1,3,3,3-hexafluoro-2-propanol and 2,2,3,3-tetrafluoro-1-propanol. As shown in Chart 1, hexafluoroisopropanol (1) was more potent than tetrafluoropropanol (7a). With various aryl (R2) substituent, hexafluoroisopropanol analogs (6a–f) were ~3- to 8-fold more potent than corresponding tetrafluoropropanol analogs (7b–g). To investigate aniline substituents (R2), meta- and para-substituted aniline analogs with electron-neutral and electron-withdrawing groups were prepared. In general, meta-substituted anilines (6a and 6b) were more active than para-substituted aniline analogs (6e and 6f). Among meta-substituted anilines, F- (6b), NO2- (6d), and CO2H-aniline (6a) had comparable or slightly better potency than non-substituted aniline (1), but chloroaniline (6c) had significantly reduced potency. From these findings we hypothesized that hydrogen bond donor or acceptor substituents are important for potency; we therefore synthesized more analogs with variations in R2.
Modifications of Bicyclic Anilines
To further investigate aniline substituents, compounds were synthesized with bicyclic anilines at the R2 position with diethylamine (R3) and hexafluoroisopropanol (R1), including 6- or 5-aminoindazole (6h and 6i), 6-aminobenzoxazole (6j), and 6-aminobenzothiazole (6k). As shown in Chart 2, the indazole analog 6h was ~3-fold more potent than its regioisomer 6i; however, both were less potent than 1. Notably, replacement of indazole (6h) by benzoxazole (6j) gave greater potency than 1; further, replacement of benzoxazole (6j) by benzothiazole (6k) increased potency by ~3-fold to EC50 ≈ 50 nM. Analogs with electroneutral (6i) or hydrogen bond donor (6h) substituents on the meta-position were less potent than the nonsubstituted aniline analog (1), and analogs with hydrogen bond acceptor substituents, 6j and 6k, showed significantly greater potency than 1. These results support the importance of hydrogen bond acceptor substituents at the aniline meta-position.
Chart 2.
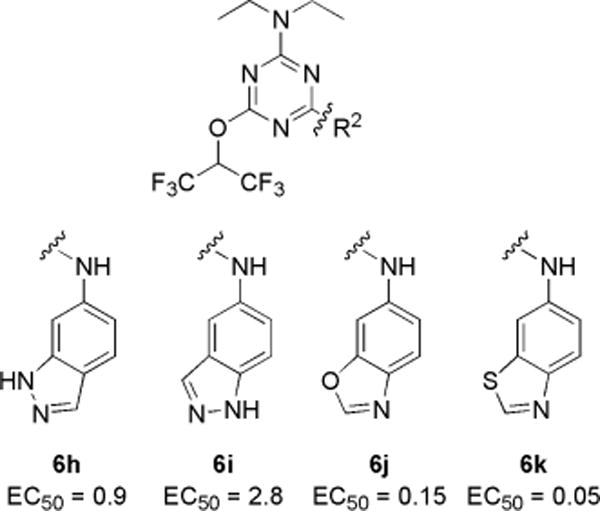
Substitution with Bicyclic Anilines and Their EC50 (μM)
Modifications with N-Methylamine
Finally, N,N-diethylamine at R3 was replaced by methylamine with two fluorinated alkoxides (Chart 3). Notably, N-methyl-2,2,3,3-tetrafluoro-1-propanoxy-1,3,5-triazine amine 12 had EC50 of ~50-fold better than diethylamine analog 6a. Interestingly, for diethylamine (R3) analogs, hexafluoropropanol (1) is more potent than tetrafluoropropanol (7a), whereas for methylamine (R3) analogs, tetrafluoropropanol analog 12 was ~3-fold more potent than hexafluoropropanol analog 11. Compounds that showed greater potency than 1 from the SAR analysis were chosen for biological characterization and additional efficacy studies.
Biology
In Vitro Characterization of Aminophenyl-1,3,5-triazines
Short-circuit current measurements were done on CFTR-expressing FRT cells using a transepithelial chloride gradient and following permeabilization of the cell basolateral membrane such that current provides a quantitative, linear measure of CFTR chloride conductance. Representative data in Figure 2A for 6k and 12 show a small increase in current with addition of a low concentration of forskolin, followed by concentration-dependent increases in current following compound additions. The compounds produced full CFTR activation, as little further increase in current was seen following a maximal concentration of forskolin. EC50 values are 30 nM for 6k and 31 nM for 12 (Figure 2B).
Figure 2.
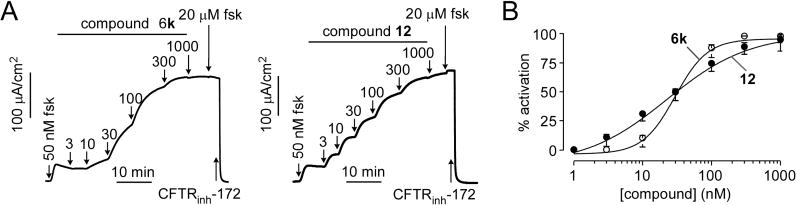
Short-circuit current measurement of CFTR activation by 6k and 12. (A) Measurements done in FRT cells expressing human wild-type CFTR showing responses to indicated concentrations of forskolin, 6k, or 12 and 10 μM CFTR inhibitor CFTRinh-172. (B) Concentration-dependent activation of CFTR (mean ± SEM, n = 3).
Possible off-target effects on cell signaling and relevant non-CFTR ion channels were investigated. 6k and 12 at 10 μM did not elevate cellular cAMP (Figure 3A), increase cytoplasmic calcium, or inhibit the ATP-stimulated elevation in cytoplasmic calcium (Figure 3B). The effects of 6k and 12 on TMEM16A and non-TMEM16A calcium-activated chloride channel (CaCCs) were investigated in FRT-expressing TMEM16A and in HT-29 cells, respectively. Both cell types were transfected with a cytoplasmic YFP-based halide sensor. Upon CaCC activation, extracellular iodide addition results in CaCC-facilitated iodide influx, as detected by fluorescence quenching of the cytoplasmic YFP halide sensor. 6k and 12 at 10 μM neither inhibited nor activated CaCCs in TMEM16A-expressing FRT cells (Figure 3C) or HT-29 cells (Figure 3D). To further investigate compound specificity, the major ion transport pathways in human bronchial epithelial (HBE) cells were studied by short-circuit current. HBE cells were sequentially treated with amiloride to inhibit the epithelial sodium channel (ENaC), with forskolin to activate CFTR, CFTRinh-172 to inhibit CFTR, and with ATP to activate CaCC, which produced the anticipated changes in short-circuit current in control cells (Figure 3E). Pretreatment of cells for 10 min with 10 μM 6k or 12 did not alter short-circuit current responses to ENaC, maximal forskolin, or ATP, indicating that the compounds do not inhibit ENaC, CaCC, or the supporting epithelial cell transporters necessary to generate current (NKCC1, epithelial K+ channels, Na+/K+ pump). Finally, as found in FRT cells expressing wild-type CFTR, 12 did not activate CFTR in HBE cells in the absence of basal CFTR phosphorylation produced by forskolin (Figure 3F, top), whereas 0.1 μM 12 fully activated CFTR following basal CFTR phosphorylation by 100 nM forskolin (Figure 3F, bottom).
Figure 3.
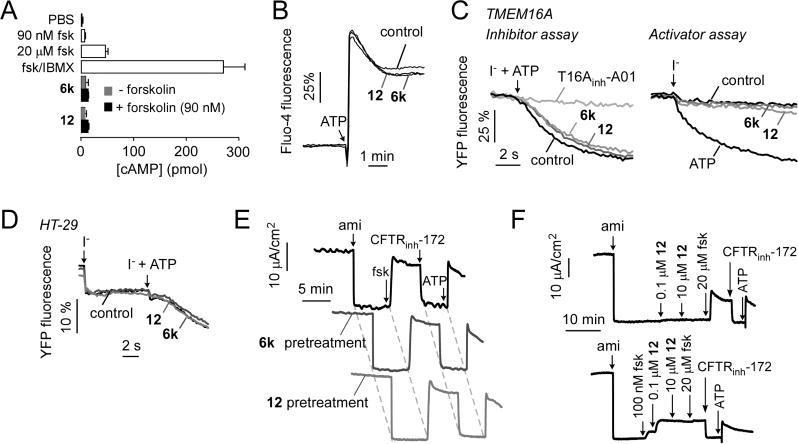
Characterization of 6k and 12. (A) Cellular cAMP in FRT cells following incubation for 10 min with 10 μM 6k or 12, without or with 90 nM forskolin (fsk), as well as forskolin alone (90 nM and 20 μM) and forskolin (20 μM) + IBMX (100 μM) (mean ± SEM, n = 4). (B) Cytoplasmic calcium measured by Fluo-4 fluorescence. FRT cells were pretreated for 5 min with 10 μM 6k or 12 (or control), with 100 μM ATP added as a calcium agonist as indicated. (C) TMEM16A activity measured in FRT cells expressing YFP showing no inhibition (left, iodide + ATP addition) or activation (right, iodide addition) by 10 μM 6k or 12. (D) CaCC activity measured in HT-29 cells expressing YFP showing no activation (iodide addition) or inhibition (iodide + ATP addition) by 10 μM 6k or 12. (E) Short circuit current in primary cultures of human bronchial epithelial cells in response to agonists and inhibitors that target key ion transport processes: 20 μM amiloride (ami); 20 μM forskolin (fsk); 10 μM CFTRinh-172; 100 μM ATP. Experiments were done following a 10 min preincubation with 10 μM 6k or 12. (F) CFTR activation by indicated concentration of 12 without forskolin (top) and after 100 nM forskolin (bottom) addition. Studies in parts B–F are representative of two to four separate sets of experiments.
Pharmacological Properties and in Vivo Efficacy
Compounds were further studied for their solution and metabolic stability and cellular toxicity. Figure 4A shows that 6k and 12 did not produce significant toxicity using an Alamar Blue assay, with 33% DMSO as positive control. When dissolved at 100 μM in an ophthalmic vehicle containing 0.3% CMC in Ringer’s solution, 12 remained chemically stable for 24 h at 40 °C as assayed by LC/MS (data not shown). To assess metabolic stability, compounds at 5 μM were incubated with rat hepatic microsomes in the presence of NADPH. Figure 4B shows the kinetics of compound metabolism and LC/MS profiles of 12. 1 and 12, which contain nonsubstituted anilines, were metabolized rapidly with <40% of the original compounds remaining at 15 min, whereas 6j and 6k, which contain bicyclic anilines, were metabolized slowly with ~60% of the original compounds remaining at 60 min. The benzoxazole-containing analog 6j showed greater metabolic stability than benzothiazole analog 6k, which may be due to relative lability of the sulfide in benzothiazole versus the oxygen in benzoxazole. Low metabolic stability is desirable for a dry eye therapeutic to minimize systemic exposure following potential absorption, whereas high metabolic stability is desirable for liver and lung applications of CFTR activators where systemic exposure and organ accumulation are needed. Though possible off-target effects related to ion channels were investigated here, more extensive screening for off-target activity is indicated, particularly for liver and lung applications where systemic exposure is required.
Figure 4.
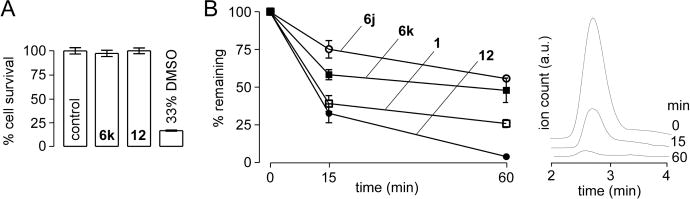
Compound pharmacology. (A) Cytotoxicity was measured by Alamar Blue assay in FRT cells incubated for 8 h with 10 μM 6k or 12, with 33% DMSO as positive control (mean ± SEM, n = 8). (B) In vitro metabolic stability. Compounds at 5 μM were incubated for indicated times with 1 mg/mL hepatic microsomes in the presence of NADPH and parent compound assayed by LC/MS (mean ± SEM, n = 3). LC/MS profile of 12 is shown on the right with elution time on the x-axis for incubation times of 0, 15, and 60 min.
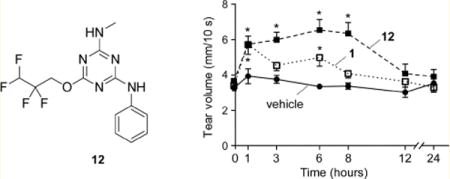
Measurements of tear fluid volume were done following a single ophthalmic dosing of test compound in 2.5 μL of an ophthalmic vehicle or vehicle alone as control. Compound 12 was selected following preliminary testing of compound efficacy at 6 h. At 100 μM (250 pmol) 12 produced a sustained increase in tear volume for 8 h that returned to baseline over 12–24 h, with improved kinetics compared to 1 (Figure 5A). As evidence for compound specificity, 12 did not increase tear volume in CF mice, which lack functional CFTR (Figure 5B). A dose–response study was done to determine the minimal amount of 12 that produced sustained tear fluid secretion. Figure 5C shows similar activity of 12 at doses down to 25 pmol with maximal tear volume at 1–3 h and sustained effect to 8 h.
Figure 5.
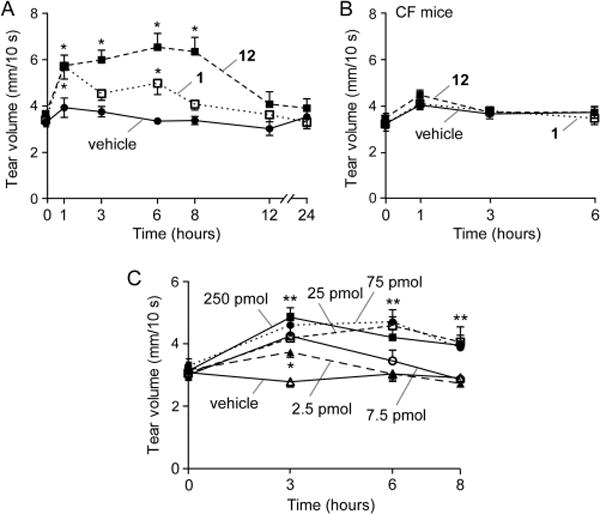
Tear fluid volume in mice following ocular delivery of 1 or 12. (A) Tear volume was measured just before and at the indicated times after single ocular delivery of vehicle, 1 (250 pmol), or 12 (250 pmol) in a 2.5 μL volume. (B) Study as in part A but in CF mice lacking functional CFTR. (C) Single dose study as in part A with different amounts of 12. Data are reported as the mean ± SEM, 5 mice, 10 eyes per condition, (*) p < 0.05, (**) p < 0.01 compared to vehicle control.
DISCUSSION
The structure–activity study here identified analogs of 1 with substantially greater CFTR activation potency, with the most potent compound 12 having EC50 ≈ 30 nM, CFTR selectivity, and sustained in vivo efficacy in increasing tear volume for at least 8 h in mice. CFTR selectivity was demonstrated in cellular studies and by the lack of increase in tear fluid volume in CFTR-deficient mice. 12 was chemically stable, including when formulated in a standard ophthalmic vehicle. Minimal systemic exposure of 12 is predicted based on the small amount needed to stimulate tear secretion and its rapid predicted hepatic metabolism. Further studies supporting its clinical development will be needed, such as ocular tissue pharmacokinetics, long-term toxicity, and patient tolerability, which are beyond the scope of this medicinal chemistry study. Of note, synthesized analogs 6j and 6k had substantially greater in vitro metabolic stability than 1 or 12 and therefore have potential utility in therapy of liver and lung diseases where systemic exposure is necessary.
Various biological activities have been reported for the 1,3,5-triazines scaffold. Atrazine, a chlorotriazine with two alkylamines substituent, is a widely used herbicide.20 Some N,N-diaminobenzyl-N-isopropyl-1,3,5-triazines were found to have anti-tubulin activities in vitro and in vivo.21 In addition, triazines containing bulky piperazinyl and cyclohexylamino substitutions were reported to have cathepsin inhibition activity.22 The substituents in these biologically active triazines are very different from those that conferred CFTR activation in this study. Structure–activity analysis of commercial and synthesized aminophenyl-1,3,5-triazines determined that three specific substituents (alkoxide, alkylamine, and phenylamine) on the triazine are needed for CFTR activity. For each of the substituents, (i) fluorinated alkoxides showed superior activity than nonfluorinated alkoxides, (ii) short and linear alkylamines are crucial for activity while bulky amines greatly reduced activity, and (iii) phenylamines containing electron-withdrawing groups, including nitro, carboxylic acid, or fluoride, in the meta-position increased activity. Of note, compound 12 has druglike properties, including favorable molecular weight (331 Da), topological polar surface area (70.3 Å2), and cLogP (4.51), the last much improved compared to cLogP of 5.99 for 1.
CFTR is a compelling target for dry eye therapy, as it is expressed in corneal and conjunctival epithelia at the ocular surface23–26 where it functions as a prosecretory chloride channel.27,28 While CFTR is largely inactive under basal conditions, when activated, it has the capacity to drive secretion of tear fluid onto the ocular surface. By analogy, CFTR is largely inactive in the intestine under basal conditions, but when activated by bacterial enterotoxins as in cholera, it can promote massive secretion of fluid into the intestinal lumen. As topically delivered CFTR activators act primary on ocular surface epithelia, they are predicted to be efficacious in dry eye produced by multiple etiologies, including disorders targeting lacrimal glands such as Sjogren’s syndrome. It is noted, however, that a pure prosecretory therapeutic replaces primarily the aqueous compartment of the tear film, which also contains lipid and mucin components. This potential limitation also applies to antiabsorptive therapeutics in development targeting the ENaC sodium channel.29 Notwithstanding this potential limitation, the demonstration of efficacy in rodent models30 and the known pathogenesis of dry eye in which tear film hyperosmolality produces downstream pathology31,32 support the utility of CFTR activator therapy in human dry eye.
In conclusion, CFTR activator therapy is predicted to have broad clinical indications for prosecretory therapy of dry eye caused by a variety of etiologies, including “idiopathic” dry eye. Because of their unique mechanism of action, CFTR activators are combinable with approved immunosuppressants and possible future therapeutics such as ENaC inhibitors. Compared with immunosuppressant drugs, CFTR activator therapy rescues an early, initiating event in dry eye pathogenesis in an etiology-agnostic manner. The aminophenyl-1,3,5-triazines synthesized and tested herein are candidate lead compounds for further preclinical development.
EXPERIMENTAL DETAILS AND DATA
General Synthesis Procedures
All solvents and chemicals were used as purchased without further purification. Reaction progress was monitored on Merck precoated silica gel plates (with fluorescence at 254 nm) using ethyl acetate/n-hexane as solvent system. Column chromatography was done with Fluka silica gel 60 (230–400 mesh). Proton (1H) and carbon (13C) NMR spectra were recorded on a Bruker Avance 300 or 500 (500.13 MHz for 1H; 125.76 MHz for 13C) using CDCl3, CD3OD, acetone-d6, or DMSO-d6 as solvents. Chemical shifts are given in parts per million (ppm). HRMS was performed using a hybrid quadrupole Orbitrap mass analyzer, QExactive (Thermo, Bremen, Germany), with an electrospray ionization source. The mass resolution was set as 70 000 at m/z 200, and the mass accuracy was less than 3 ppm. Purity of all final compounds was 95% or higher. Purities were determined by low-resolution mass spectrometry using a LC/MS system (Waters 2695 HPLC with Micromass ZQ). LC was done on a Xterra MS C18 column (2.1 mm × 100 mm, 3.5 μm) with 0.2 mL/min water/acetonitrile (containing 0.1% formic acid), 16 min linear gradient, 5–95% acetonitrile. UV absorbance was detected at 254 nm. Below we report synthesis and analytic data for intermediates and representative final compounds 1 and 12. Analytic data for 6a–k, 7a–g, and 11 are reported in Supporting Information.
N,N-Diethyl-6-[(1,1,1,3,3,3-hexafluoropropan-2-yl)oxy]-N′-phenyl-1,3,5-triazine-2,4-diamine (1)
To a solution of 4 (500 mg, 1.42 mmol) in THF (30 mL) were added diisopropylethylamine (0.49 mL, 2.83 mmol) and aniline (0.15 mL, 1.7 mmol). The mixture was refluxed for 16 h. After cooling to room temperature, the reaction mixture was extracted with EtOAc, washed with brine, and dried over MgSO4. The solvent was removed under reduced pressure and purified by flash chromatography (1:10 EtOAc/Hex) to afford 1 as a white powder (280 mg, 48%). 1H NMR (300 MHz, CDCl3): δ = 7.61 (d, 2H, J = 7.8 Hz), 7.55 (brs, 1H), 7.35 (t, 1H, J = 7.5 Hz), 7.11 (t, 1H, J = 7.4 Hz), 6.37 (sep, 1H, J = 6.2 Hz), 3.69–3.58 (m, 4H), 1.29–1.20 (m, 6H). 13C NMR (75 MHz, CDCl3): δ = 168.5, 165.1, 165.0, 138.4, 128.7, 123.4, 122.7, 120.3, 119.0, 68.3 (t, 34 Hz), 42.4, 42.0, 12.9, 12.8. HRMS (ESI): m/z calculated for C16H18F6N5O [M + H+]: 410.1410. Found: 410.1411.
Analogs 6a–k and 7a–g were prepared as described for 1, with various substituted anilines and 4 or 5.
2,4-Dichloro-6-[(1,1,1,3,3,3-hexafluoropropan-2-yl)oxy]-1,3,5-triazine (2)
To a solution of cyanuric chloride (2 g, 10.84 mmol) in THF (50 mL) at −80 °C was added potassium carbonate (3.0 g, 21.74 mmol). A solution of 1,1,1,3,3,3-hexafluoro-2-propanol (1.36 mL, 10.30 mmol) in THF (20 mL) was then added dropwise over 30 min. The mixture was stirred for additional 30 min at −80 °C and quenched with water. The reaction mixture was diluted with EtOAc and washed with 1 N HCl solution. The organic layer was washed with brine and dried over MgSO4. The solvent was removed under reduced pressure to afford a white solid (1.5 g, 62%). The residue was used for further reaction without purification. 1H NMR (300 MHz, CDCl3): δ = 6.25 (sep, 1H, J = 5.7 Hz).
2,4-Dichloro-6-[(2,2,3,3-tetrafluoropropan)oxy]-1,3,5-triazine (3)
To a solution of cyanuric chloride (2 g, 10.84 mmol) in THF (50 mL) at −80 °C was added potassium carbonate (3 g, 21.74 mmol). A solution of 2,2,3,3-tetrafluoropropanol (1.2 mL, 10.3 mmol) in THF (20 mL) was then added dropwise over 30 min. The mixture was stirred for additional 30 min at −80 °C and quenched with water. The reaction mixture was diluted with EtOAc, and the organic layer was washed with brine and dried over MgSO4. The solvent was removed under reduced pressure to afford a white solid (1.3 g, 68%). The residue was used for further reaction without purification. 1H NMR (300 MHz, CDCl3): δ = 6.01 (tt, 1H, J = 4.0, 53 Hz), 4.90–4.79 (m, 2H).
4-Chloro-N,N-diethyl-6-[(1,1,1,3,3,3-hexafluoropropan-2-yl)-oxy]-1,3,5-triazin-2-amine (4)
To a solution of 2 (1.6 g, 5.06 mmol) in THF (40 mL) were added diisopropylethylamine (1.06 mL, 6.08 mmol) and diethylamine (0.5 mL, 5.0 mmol) at 0 °C. The mixture was stirred for 1 h at 0 °C, then quenched with water. The reaction mixture was extracted with EtOAc, washed with water and brine, and dried over MgSO4. The solvent was removed under reduced pressure to afford a white solid (950 mg, 55%). 1H NMR (300 MHz, CDCl3): δ = 6.25 (sex, 1H, J = 6.0 Hz), 3.79–3.53 (m, 4H), 1.25–1.16 (m, 6H).
4-Chloro-N,N-diethyl-6-[(2,2,3,3,-tetrafluoropropan)oxy]-1,3,5-triazin-2-amine (5)
To a solution of 3 (1.3 g, 5 mmol) in THF (30 mL) were added diisopropylethylamine (1 mL, 6 mmol) and diethylamine (0.5 mL, 5.0 mmol) at 0 °C. The mixture was stirred for 1 h at 0 °C, then quenched with water. The reaction mixture was extracted with EtOAc, washed with water and brine, and dried over MgSO4. The solvent was removed under reduced pressure to afford yellow oil (900 mg, 57%). 1H NMR (300 MHz, CDCl3): δ = 6.02 (td, 1H, J = 1.6, 4.7 Hz), 4.70 (dt, 2H, J = 1.5, 12.2 Hz), 3.65–3.55 (m, 4H), 1.19 (t, 6H, J = 7.1 Hz).
4,6-Dichloro-N-methyl-1,3,5-triazine-2-amine (8)
To a solution of cyanuric chloride (5 g, 27.3 mmol) in acetone (30 mL) at 0 °C was added methylamine (13.7 mL, 2 M in THF) dropwise over 30 min. The mixture was stirred for 1 h at 0 °C, and then crushed ice was added. The resulting precipitate was filtered and washed with water to afford 8 as a white solid (3.7 g, 75%), which was used for further reaction without purification. 1H NMR (300 MHz, CDCl3) δ = 7.02 (brs, 1H), 3.10 (dd, 3H, J = 1.2, 5.1 Hz); 13C NMR (75 MHz, CDCl3) δ = 171.0, 169.4, 166.2, 28.3.
4-Chloro-6-((1,1,1,3,3,3-hexafluoropropan-2-yl)oxy)-N-methyl-1,3,5-triazin-2-amine (9)
To a solution of 8 (2 g, 11.17 mmol) in THF (40 mL) at 0 °C were added potassium carbonate (11.56 g, 83.8 mmol) and 1,1,1,3,3,3-hexafluoro-2-propanol (1.4 mL, 13.4 mmol). The mixture was stirred overnight at ambient temperature. The reaction mixture was diluted with EtOAc, and then organic layer was washed with brine and dried over MgSO4. The solvent was removed under reduced pressure. A white solid was obtained (1.4 g, 43%). 1H NMR (300 MHz, CDCl3) δ = 7.02 (brs, 1H), 6.27 (quint, 1H, J = 5.7 Hz), 3.11 (d, 3H, J = 5.1 Hz).
4-Chloro-6-[(2,2,3,3-tetrafluoropropan)oxy]-N-methyl-1,3,5-triazine-2-amine (10)
To a solution of 8 (630 mg, 3.52 mmol) in THF (30 mL) at 0 °C was added potassium carbonate (970 mg, 7.04 mmol). A solution of 2,2,3,3-tetrafluoropropanol (0.31 mL, 3.52 mmol) in THF (20 mL) was added dropwise over 30 min. The mixture was stirred for additional 30 min at 0 °C, then quenched with water. The reaction mixture was diluted with EtOAc, and the organic layer was washed with brine and dried over MgSO4. Solvent was removed under reduced pressure to afford a white solid (640 mg, 66%). The residue was used for further reaction without purification. 1H NMR (300 MHz, CDCl3): δ = 6.98–5.79 (m, 1H), 4.87–4.69 (m, 2H), 3.05 (m, 3H).
N-Methyl-N′-phenyl-6-(2,2,3,3-tetrafluoropropoxy)-1,3,5-triazine-2,4-diamine (12)
To a solution of 10 (70 mg, 0.26 mmol) in THF (3 mL) were added diisopropylethylamine (0.09 mL, 0.51 mmol) and aniline (0.03 mL, 0.31 mmol). The mixture was refluxed for 16 h. After cooling to room temperature, the reaction mixture was extracted with EtOAc, washed with brine, and dried over MgSO4. Solvent was removed under reduced pressure and purified by flash chromatography (1:2 EtOAc/Hex) to afford 12 as a white powder (63 mg, 74%). 1H NMR (300 MHz, CDCl3): δ = 7.61 (d, 2H, J = 7.8 Hz), 7.55 (brs, 1H), 7.35 (t, 1H, J = 7.5 Hz), 7.11 (t, 1H, J = 7.4 Hz), 6.37 (sep, 1H, J = 6.2 Hz), 3.69–3.58 (m, 4H), 1.29–1.20 (m, 6H). 13C NMR (75 MHz, CDCl3): δ = 168.5, 165.1, 165.0, 138.4, 128.7, 123.4, 122.7, 120.3, 119.0, 68.3 (t, 34 Hz), 42.4, 42.0, 12.9, 12.8. HRMS (ESI): m/z calculated for C13H14F4N5O [M + H+]: 332.1129. Found: 332.1127.
Mice
Wild-type and CF (homozygous ΔF508-CFTR mutant) mice in a CD1 genetic background were bred at the University of California San Francisco (UCSF) Animal Facility. Mice aged 8–12 weeks (25–35 g) were used. Animal protocols were approved by the UCSF Institutional Animal Care and Use Committee and were in compliance with the ARVO Statement for the Use of Animals in Ophthalmic and Visual Research.
Cell Culture
Fischer rat thyroid (FRT) cells stably coexpressing human wild-type CFTR or TMEM16A and the halide-sensitive yellow fluorescent protein (YFP)-H148Q were cultured as described.15,33 HT-29 expressing YFP was cultured as described.34 Primary cultures of human bronchial epithelial (HBE) cells were maintained at an air–liquid interface as described previously.35
Plate Reader Assays of Chloride Channel Function
CFTR activity was assayed as described.15 FRT cells coexpressing YFP and wild-type CFTR were washed with phosphate-buffered saline (PBS) and then incubated for 10 min with test compounds in PBS containing 125 nM forskolin. I− influx was measured in a plate reader with initial baseline read for 2 s and then for 12 s after rapid addition of an I−-containing solution. TMEM16A activity was assayed similarly as described33 using FRT cells coexpressing YFP and TMEM16A. Activity of non-TMEM16A CaCC was assayed as described34 in HT-29 cells expressing YFP. In each assay initial rates of I− influx were computed as a linear measure of channel function.
Short-Circuit Current Measurement
Short-circuit current measurements were done as described.9 Briefly, Snapwell (Corning Costar, Corning, NY) inserts containing CFTR-expressing FRT cells or human bronchial epithelial cells were mounted in Ussing chambers (Physiological Instruments, San Diego, CA). For FRT cells the hemichambers were filled with 5 mL of HCO3−-buffered solution (basolateral) and half-Cl− solution (apical), and the basolateral membrane was permeabilized with 250 μg/mL amphotericin B. Symmetrical HCO3−-buffered solutions were used for human bronchial epithelial cells. Solutions were bubbled with 95% O2/5% CO2 and maintained at 37 °C, and short-circuit current was measured on a DVC-1000 voltage clamp (World Precision Instruments Inc., Sarasota, FL) using Ag/AgCl electrodes and 3 M KCl agar bridges.
cAMP and Ca2+ Assays
FRT cells expressing wild-type CFTR were cultured in 12-well plates. After washing with PBS, cells were incubated for 10 min with compounds in the absence or presence of 90 nM forskolin and then lysed. Lysates were assayed for cAMP content using a cAMP immunoassay kit (parameter cAMP immunoassay kit, R&D Systems, Minneapolis, MN). For cytoplasmic calcium measurements, FRT cells in 96-well black-walled microplates were loaded with Fluo-4 NW (Invitrogen, Carlsbad, CA) and Fluo-4 fluorescence was measured with a plate reader equipped with syringe pumps and monochromators.
Cytotoxicity
FRT cells were cultured overnight in black 96-well Costar microplates and incubated with test compounds at up to 10 μM (the maximum solubility in PBS) for 8 h. Cytotoxicity was measured by Alamar Blue assay (Invitrogen, Carlsbad, CA).
Tear Volume
Tear volume was measured using phenol red threads (Zone-Quick, Oasis Medical, Glendora, CA) as described.36 Threads were placed for 10 s in the lateral canthi of isofluorane-anesthetized mice using jewelers’ forceps. Tear volume was determined from the length of thread wetting as visualized under a dissecting microscope. Serial measurements were done to evaluate compound pharmacodynamics after topical application of a single, 2.5 μL drop of ophthalmic formulation containing 0–100 μM 1 or 12 in Ringer’s solution with 0.3% carboxymethylcellulose (high viscosity, VWR, Radnor, PA), 0.015% benzalkonium chloride preservative, and 1% DMSO. Both eyes in each mouse received the same treatment to control for potential contralateral effects of a given treatment.
In Vitro Metabolic Stability
Compounds (each 5 μM) were incubated for specified times at 37 °C with rat liver microsomes (1 mg/mL; Sigma-Aldrich) in potassium phosphate buffer containing 1 mM NADPH, as described.14 Ice-cold EtOAc was then added, and the mixture was centrifuged for 15 min at 3000 rpm. The supernatant was analyzed by low-resolution mass spectrometry using a LC/MS system (Waters 2695 HPLC with Micromass ZQ). LC was done on a Xterra MS C18 column (2.1 mm × 100 mm, 3.5 μm) with 0.2 mL/min water/acetonitrile (containing 0.1% formic acid), 12 min linear gradient, 5–95% acetonitrile. UV absorbance was detected at 254 nm.
Statistics
Statistical analysis was performed using Prism 5 GraphPad Software package (San Diego, CA). Serial tear volume measurements were analyzed by two-way ANOVA with Dunnett post hoc analysis. Data are presented as the mean ± standard error of the mean (SEM), and statistical significance was set at p < 0.01 (*).
Supplementary Material
Acknowledgments
This work was supported by NIH Grants DK72517, EY13574, DK099803, EB00415, DK35124, and DK101373 and by NIH/NCATS UCSF-CTSI Grant UL1 TR000004.
ABBREVIATIONS
- CFTR
cystic fibrosis transmembrane conductance regulator
- cAMP
cyclic adenosine monophosphate
- YFP
yellow fluorescent protein
- CF
cystic fibrosis
- FRT
Fischer rat thyroid
- LC/MS
liquid chromatography/mass spectrometry
- fsk
forskolin
- SAR
structure–activity relationship
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.6b01792.
Analytical data of synthetic compounds 6a–k, 7a–g, and 11; Table 1 showing chemical structures and data (PDF) Molecular formula strings and some data (CSV)
ORCID
Sujin Lee: 0000-0003-3345-2073
Notes
The authors declare the following competing financial interest(s): Drs. Levin and Verkman are named co-inventors on a provisional patent application on CFTR activators for dry eye therapy filed December 2015, whose rights are owned by the University of California.
References
- 1.Verkman AS, Galietta LJ. Chloride channels as drug targets. Nat Rev Drug Discovery. 2009;8:153–171. doi: 10.1038/nrd2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schmidt BZ, Haaf JB, Leal T, Noel S. Cystic fibrosis transmembrane conductance regulator modulators in cystic fibrosis: current perspectives. Clin Pharmacol: Adv Appl. 2016;8:127–140. doi: 10.2147/CPAA.S100759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thiagarajah JR, Donowitz M, Verkman AS. Secretory diarrhea: mechanisms and emerging therapies. Nat Rev Gastroenterol Hepatol. 2015;12:446–457. doi: 10.1038/nrgastro.2015.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ong T, Ramsey BW. New rherapeutic approaches to modulate and correct cystic fibrosis transmembrane conductance regulator. Pediatr Clin North Am. 2016;63:751–764. doi: 10.1016/j.pcl.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chao AC, de Sauvage FJ, Dong YJ, Wagner JA, Goeddel DV, Gardner P. Activation of intestinal CFTR Cl-channel by heat-stable enterotoxin and guanylin via cAMP-dependent protein kinase. EMBO J. 1994;13:1065–1072. doi: 10.1002/j.1460-2075.1994.tb06355.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moon C, Zhang W, Sundaram N, Yarlagadda S, Reddy VS, Arora K, Helmrath MA, Naren AP. Drug-induced secretory diarrhea: A role for CFTR. Pharmacol Res. 2015;102:107–112. doi: 10.1016/j.phrs.2015.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Field M. Mechanisms of action of cholera and Escherichia coli enterotoxins. Am J Clin Nutr. 1979;32:189–196. doi: 10.1093/ajcn/32.1.189. [DOI] [PubMed] [Google Scholar]
- 8.Solomon GM, Marshall SG, Ramsey BW, Rowe SM. Breakthrough therapies: Cystic fibrosis (CF) potentiators and correctors. Pediatr Pulmonol. 2015;50:S3–S13. doi: 10.1002/ppul.23240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cil O, Phuan PW, Lee S, Tan J, Haggie PM, Levin MH, Sun L, Thiagarajah JR, Ma T, Verkman AS. CFTR activator increases intestinal fluid secretion and normalizes stool output in a mouse model of constipation. Cell Mol Gastroenterol Hepatol. 2016;2:317–327. doi: 10.1016/j.jcmgh.2015.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cil O, Phuan PW, Son JH, Zhu JS, Ku CK, Tabib NA, Teuthorn AP, Ferrera L, Zachos NC, Lin R, Galietta LJ, Donowitz M, Kurth MJ, Verkman AS. Phenylquinoxalinone CFTR activator as potential prosecretory therapy for constipation. Transl Res. 2016 Oct 14; doi: 10.1016/j.trsl.2016.10.003. [Online early access] Published Online. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Solomon GM, Raju SV, Dransfield MT, Rowe SM. Therapeutic approaches to acquired cystic fibrosis transmembrane conductance regulator dysfunction in chronic bronchitis. Ann Am Thorac Soc. 2016;13(Suppl 2):S169–S176. doi: 10.1513/AnnalsATS.201509-601KV. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Flores AM, Casey SD, Felix CM, Phuan PW, Verkman AS, Levin MH. Small-molecule CFTR activators increase tear secretion and prevent experimental dry eye disease. FASEB J. 2016;30:1789–1797. doi: 10.1096/fj.201500180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cil O, Phuan PW, Gillespie AM, Lee S, Tradtrantip L, Yin J, Tse M, Zachos NC, Lin R, Donowitz M, Verkman AS. Benzopyrimido-pyrrolo-oxazine-dione CFTR inhibitor (R)-BPO-27 for antisecretory therapy of diarrheas caused by bacterial enterotoxins. FASEB J. 2016 Nov 8; doi: 10.1096/fj.201600891R. [Online early access] Published Online. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Snyder DS, Tradtrantip L, Yao C, Kurth MJ, Verkman AS. Potent, metabolically stable benzopyrimido-pyrrolo-oxazine-dione (BPO) CFTR inhibitors for polycystic kidney disease. J Med Chem. 2011;54:5468–5477. doi: 10.1021/jm200505e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ma T, Vetrivel L, Yang H, Pedemonte N, Zegarra-Moran O, Galietta LJ, Verkman AS. High-affinity activators of CFTR chloride conductance identified by high-throughput screening. J Biol Chem. 2002;277:37235–37241. doi: 10.1074/jbc.M205932200. [DOI] [PubMed] [Google Scholar]
- 16.Schaumberg DA, Dana R, Buring JE, Sullivan DA. Prevalence of dry eye disease among US men: estimates from the Physicians’ Health Studies. Arch Ophthalmol. 2009;127:763–768. doi: 10.1001/archophthalmol.2009.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schaumberg DA, Sullivan DA, Buring JE, Dana MR. Prevalence of dry eye syndrome among US women. Am J Ophthalmol. 2003;136:318–326. doi: 10.1016/s0002-9394(03)00218-6. [DOI] [PubMed] [Google Scholar]
- 18.Alves M, Fonseca EC, Alves MF, Malki LT, Arruda GV, Reinach PS, Rocha EM. Dry eye disease treatment: a systematic review of published trials and critical appraisal of therapeutic strategies. Ocul Surf. 2013;11:181–192. doi: 10.1016/j.jtos.2013.02.002. [DOI] [PubMed] [Google Scholar]
- 19.Sheppard JD, Torkildsen GL, Lonsdale JD, D’Ambrosio FA, Jr, McLaurin EB, Eiferman RA, Kennedy KS, Semba CP, OPUS-1 Study Group Lifitegrast ophthalmic solution 5.0% for treatment of dry eye disease: results of the OPUS-1 phase 3 study. Ophthalmology. 2014;121:475–483. doi: 10.1016/j.ophtha.2013.09.015. [DOI] [PubMed] [Google Scholar]
- 20.Jowa L, Howd R. Should atrazine and related chlorotriazines be considered carcinogenic for human health risk assessment? J Environ Sci Health Part C Environ Carcinog Ecotoxicol Rev. 2011;29:91–144. doi: 10.1080/10590501.2011.577681. [DOI] [PubMed] [Google Scholar]
- 21.Moon H-S, Jacobson EM, Khersonsky SM, Luzung MR, Walsh DP, Xiong W, Lee JW, Parikh PB, Lam JC, Kang T-W, Rosania GR, Schier AF, Chang Y-T. A novel microtubule destabilizing entity from orthogonal synthesis of triazine library and zebrafish embryo screening. J Am Chem Soc. 2002;124:11608–11609. doi: 10.1021/ja026720i. [DOI] [PubMed] [Google Scholar]
- 22.Plebanek E, Chevrier F, Roy V, Garenne T, Lecaille F, Warszycki D, Bojarski AJ, Lalmanach G, Agrofoglio LA. Straightforward synthesis of 2,4,6-trisubstituted 1,3,5-triazine compounds targeting cysteine cathepsins K and S. Eur J Med Chem. 2016;121:12–20. doi: 10.1016/j.ejmech.2016.05.009. [DOI] [PubMed] [Google Scholar]
- 23.Kompella UB, Kim KJ, Lee VH. Active chloride transport in the pigmented rabbit conjunctiva. Curr Eye Res. 1993;12:1041–1048. doi: 10.3109/02713689309033501. [DOI] [PubMed] [Google Scholar]
- 24.Turner HC, Bernstein A, Candia OA. Presence of CFTR in the conjunctival epithelium. Curr Eye Res. 2002;24:182–187. doi: 10.1076/ceyr.24.3.182.8297. [DOI] [PubMed] [Google Scholar]
- 25.Al-Nakkash L, Reinach PS. Activation of a CFTR-mediated chloride current in a rabbit corneal epithelial cell line. Invest Ophthalmol Visual Sci. 2001;42:2364–2370. [PubMed] [Google Scholar]
- 26.Shiue MH, Gukasyan HJ, Kim KJ, Loo DD, Lee VH. Characterization of cyclic AMP-regulated chloride conductance in the pigmented rabbit conjunctival epithelial cells. Can J Physiol Pharmacol. 2002;80:533–540. doi: 10.1139/y02-066. [DOI] [PubMed] [Google Scholar]
- 27.Levin MH, Verkman AS. CFTR-regulated chloride transport at the ocular surface in living mice measured by potential differences. Invest Ophthalmol Visual Sci. 2005;46:1428–1434. doi: 10.1167/iovs.04-1314. [DOI] [PubMed] [Google Scholar]
- 28.Levin MH, Kim JK, Hu J, Verkman AS. Potential difference measurements of ocular surface Na+ absorption analyzed using an electrokinetic model. Invest Ophthalmol Visual Sci. 2006;47:306–316. doi: 10.1167/iovs.05-1082. [DOI] [PubMed] [Google Scholar]
- 29.Yu D, Thelin WR, Rogers TD, Stutts MJ, Randell SH, Grubb BR, Boucher RC. Regional differences in rat conjunctival ion transport activities. Am J Physiol Cell Physiol. 2012;303:C767–780. doi: 10.1152/ajpcell.00195.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thelin WR, Johnson MR, Hirsh AJ, Kublin CL, Zoukhri DJ. Effect of topically applied epithelial sodium channel inhibitors on tear production in normal mice and in mice with induced aqueous tear deficiency. J Ocul Pharmacol Ther. 2012;28:433–438. doi: 10.1089/jop.2011.0157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gilbard JP, Carter J, Sang D, Refojo M, Hanninen L, Kenyon KR. Morphologic effect of hyperosmolarity on rabbit corneal epithelium. Ophthalmology. 1984;91:1205–1212. doi: 10.1016/s0161-6420(84)34163-x. [DOI] [PubMed] [Google Scholar]
- 32.Liu H, Begley C, Chen M, Bradley A, Bonanno J, McNamara N, Nelson J, Simpson T. A link between tear instability and hyperosmolarity in dry eye. Invest Ophthalmol Visual Sci. 2009;50:3671–3679. doi: 10.1167/iovs.08-2689. [DOI] [PubMed] [Google Scholar]
- 33.Namkung W, Phuan PW, Verkman AS. TMEM16A inhibitors reveal TMEM16A as a minor component of calcium-activated chloride channel conductance in airway and intestinal epithelial cells. J Biol Chem. 2011;286:2365–2374. doi: 10.1074/jbc.M110.175109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.De La Fuente R, Namkung W, Mills A, Verkman AS. Small-molecule screen identifies inhibitors of a human intestinal calcium-activated chloride channel. Mol Pharmacol. 2008;73:758–768. doi: 10.1124/mol.107.043208. [DOI] [PubMed] [Google Scholar]
- 35.Haggie PM, Phuan PW, Tan JA, Zlock L, Finkbeiner WE, Verkman AS. Inhibitors of pendrin anion exchange identified in a small molecule screen increase airway surface liquid volume in cystic fibrosis. FASEB J. 2016;30:2187–2197. doi: 10.1096/fj.201600223R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stewart P, Chen Z, Farley W, Olmos L, Pflugfelder SC. Effect of experimental dry eye on tear sodium concentration in the mouse. Eye Contact Lens. 2005;31:175–178. doi: 10.1097/01.icl.0000161705.19602.c9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.