

Abstract
Biotinylated mono and bi- antennary di/trisaccharides were synthesized to evaluate their ability to capture Escherichia coli strains that express pilus types with different receptor specificities. The synthesized biotinylated di/trisaccharides contain Galα1-4Gal, GalNHAcα1-4Gal, Galα1-4GalNHAc, Galα1-4Galβ1-4Glc and GalNHAcα1-4Galβ1-4Glc as carbohydrate epitopes. These biotinylated oligosaccharides were immobilized on streptavidin coated magnetic beads, incubated with different strains of live E. coli. Capturing ability was assessed using a luciferase assay which detects bacterial ATP. The trisaccharides containing Galα1-4Galβ1-4Glc and the disaccharides containing Galα1-4Gal as the epitopes exhibited strong capturing ability for uropathogenic E.coli strains with the pap pilus genotype, including CFT073, J96 and J96 pilE. The same ligands failed to capture E.coli strains with fim, prs, or foc genotypes. Uropathogenic CFT073 was also captured moderately by biantennary disaccharide containing GalNHAc moiety at the reducing end; however, other saccharides containing GalNHAc at the non-reducing end did not capture the CFT073 strain. These synthetic glycoconjugates could potentially be adapted as rapid diagnostic agents to differentiate between different E. coli pathovars.
Keywords: Glycosides, biotin, E. coli, glycoconjugates, diagnostic agents
Introduction
Escherichia coli strains range in virulence from essentially harmless to deadly pathogens with extremely low infectious doses. While all E. coli share the same basic core genome, pathogenic strains have additional genes that encode the virulence factors that allow them to cause disease. [1, 2] Virulence factors fall into general categories, including adhesins and toxins. Adhesins allow bacteria to bind to specific human tissues or organs and establish infection at that site. The presence of E. coli anywhere other than the large intestine results in human disease, and some E. coli can cause disease even when localized to the large intestine.
Pili are extracellular bacterial structures associated with adhesion. E. coli produce a variety of pilus types, including common type 1 (encoded in the fim locus) which bind α-mannosides and P or pyelonephritis-associated pili (encoded in the pap locus) which bind Gal(αl-4)Gal expressed by the neutral glycolipid globotriaosylceramide (Gb3), while other variants bind to other neutral glycolipids, Gb4 and Gb5. The S pili (encoded in the prs locus) bind α N-acetyl neuraminic acid attached to galactose ligands. The F1C fimbriae (encoded in the foc locus) binds to GalNAcβ1-4Gal residues.[3, 4] Pili share a common structure, for example, type 1 fimbriae are made of repeating FimA subunits coupled to a short tip fibrillum structure containing the adhesion FimH, via adaptor proteins, FimF and FimG.[5, 6] FimH is the only subunit that possesses a mannose binding site and it recognizes α-mannoside present on glycoproteins.[4] These different pilus types allow E. coli to attach preferentially to different tissues and organs, and are associated with different human diseases, for example: Type 1 pili producing E. coli are associated with cystitis, sepsis and meningitis. Type P pili producing E. coli strains are associated with pyelonephritis and those that produce S/F1C pili are associated with urinary tract infections, sepsis and meningitis. [4]
Differentiating pathogenic E. coli from non-pathogenic strains presents a diagnostic challenge because there are no rapid tests that can broadly discriminate between pathogenic E. coli and harmless E. coli. Stereotyping, or the identification of lipopolysaccharide and flagellar proteins on the E. coli cell-surface, has utility in some cases such as serotype O157:H7 E. coli, infamously associated with hemolytic uremic syndrome (HUS). However, serotyping has limited utility, as evidenced by the 2011 outbreak of HUS in Germany caused by serotype O104:H4, where more than 20,000 people were sickened and 800 developed life-threatening complications.[7] In that outbreak, testing clinical samples and food for the presence of O157:H7 caused a considerable delay in the identification of the actual agent of this outbreak. Nucleic acid-based tests such as PCR can be used to differentiate between E. coli pathovars, but these are expensive and can only be performed in resource-rich settings. Other common techniques used to detect pathogenic E. coli strains can be too time-consuming, such as bacterial culturing, or too expensive and perishable, to be used for point of care diagnostics.[8]
Since the ability of pathogenic E. coli to cause disease is dependent on the ability to bind to human tissues, development of a panel of synthetic ligands that mimic the human host receptors used by various pathogenic forms of E. coli could aid in the rapid diagnosis of pathogenic and non-pathogenic E.coli. The recognition site of a typical carbohydrate ligand bound by E. coli strains is often localized to the terminal disaccharide or trisaccharide moiety of the oligosaccharides.[9] Therefore, synthesis of chemically defined oligosaccharides could be used to detect or capture E. coli strains. Carbohydrates are stable, robust, selective, amenable to scale up, and adaptable to existing biosensor platforms making them attractive candidates to be exploited for diagnostic and therapeutic purposes.[8, 10]
Monosaccharides typically interact weakly with proteins, usually in the millimolar to micromolar range. The weak binding of carbohydrates to their complementary proteins is circumvented through the use of multiple interactions. Carbohydrates are densely displayed on the mammalian cell surface on lipid rafts, and either carbohydrate binding proteins expressed by pathogens typically possess multiple binding sites, or a single pathogen will display multiple proteins with a single carbohydrate binding site. The strong binding achieved is due to the so-called cluster effect, which is highly prevalent in nature and has inspired the design of multivalent inhibitors to block protein–carbohydrate interactions.[11-13] A variety of diverse scaffolds have been formulated for multivalent ligand presentation, these include dendrimers, polymers, gold-nanoparticles and magnetic beads/nanoparticles[11, 14-17] and these glycomaterials have been used to bind and detect bacteria. In this study, streptavidin-coated magnetic beads are used for multiple presentations of biotinylated oligosaccharides to determine carbohydrate binding specificity of different E. coli strains. Attaching the biotinylated oligosaccharides to streptavidin-coated magnetic beads is advantageous for several reasons. First, it is relatively straightforward to conjugate biotinylated oligosaccharides to streptavidin-coated beads without the need to optimize conditions. Second, multiple glycans can be displayed from streptavidin-coated platforms because a single streptavidin molecule binds four biotin molecules. Third, streptavidin-coated magnetic beads and microplates are commercially available for high throughput screening of pathogens or toxins that bind to biotinylated compounds. We describe here our synthesis of biotinylated oligosaccharides and their ability to capture specific classes of pathogenic E. coli strains using streptavidin-coated magnetic beads.
Results and Discussion
I. Synthesis of glycoconjugates
Our approach towards the synthesis of glycoconjugates was to first synthesize azide-terminated di/trisaccharides followed by 1, 3 dipolar cycloaddition (“click” reaction) with alkyne bearing monomeric and dimeric scaffolds as shown in Scheme 1 and 2. The protected biotinylated oligosaccharides were readily deprotected using Zemplén conditions to afford the glycoconjugates. Syntheses of the scaffolds have been reported previously by our group.[18, 19],[20] Synthesis of the glycoconjugates containing Gal-α(1,4)-Gal and Gal-α(1,4)-GalNHAc oligosaccharide have been described below.
Scheme 1. Representation of the synthesized biotinylated glycoconjugates.
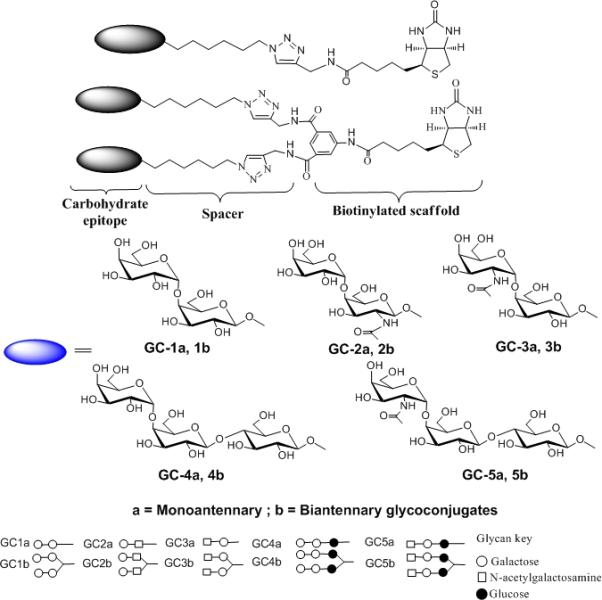
The compounds comprise of di or trisaccharides attached to a biotinylated monomeric and dimeric scaffold via a 6-carbon spacer. The black eclipse represents the carbohydrate epitope.
Scheme 2. Synthesis of GC-1a and GC-1b Reagents and conditions.
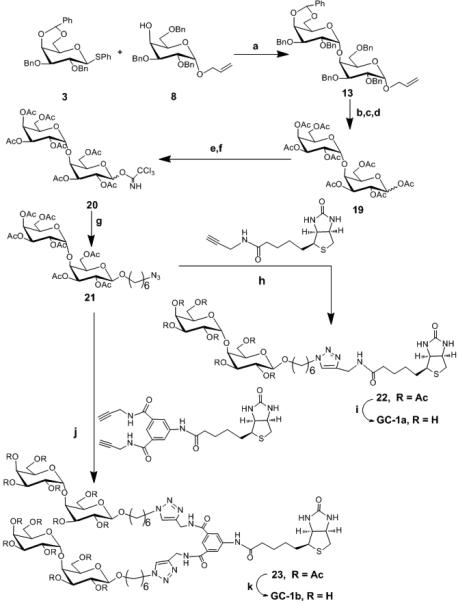
a) CH2Cl2, AgOTf, p-NO2PhSCl, TTBP, −78°C, 60% b) PdCl2, NaOAc, AcOH, H2O, rt, 54% c) Pd(OH)2, H2, EtOAc / EtOH, rt , 67%, d) Ac2O, pyridine, DMAP, 0°C to rt , 57% e) H2NNH2.HOAc, THF, rt, 63% f) K2CO3, CH2Cl2, Cl3CCN, rt, 77% g) CH2Cl2, HO(CH2)6Cl, TMSOTf, −30°C to rt, 55% h) CuSO4.5H2O, C6H7O6Na, THF/H2O, 70% i) MeOH, NaOMe, rt, quantitative j) CuSO4.5H2O, C6H7O6Na, THF/H2O, 70% k) MeOH, NaOMe, rt quantitative.
Synthesis of glycoconjugate GC1a and 1b
While synthesis of Gal-α(1,4)-Gal linkages has been reported, efficient and reproducible yields are still sometimes problematic. Our attempted glycosylation reactions to generate Gal-α(1,4)-Gal linkages are shown in Table 1. Synthesis of Gal-α(1,4)-Gal was first attempted using per-benzyl trichloroacetimidate 1 as a donor and 8 as an acceptor, however, only a trace amount of the desired product was obtained (entry a, Table 1). The low yield was attributed to the instability of the donor as TLC analysis revealed rapid decomposition of donor. Therefore, we decreased the number of benzyl protecting groups to improve the stability of the donor. Accordingly, we synthesized donor 2; unfortunately, this change did not improve the stability or the yields (entry b, Table 1). Glycosylation reaction between donor 2 and acceptor 9 also gave trace amount of α-linked product 14 (entry c, Table 1). Next, we replaced the leaving group, trichloroacetimidate, with thioglycoside. While it is well known that the per-benzyl and benzylidene protected thioglycosides are readily prepared and stable;[21, 22] unfortunately the coupling reaction between per-benzyl thioglycoside donor 4 and acceptor 8 did not furnish the desired product. However, the conformationally-restrained benzylidene thioglycoside 3 gave a reasonable yield when reacted with 8 (entry d, Table 1) and this glycosylation reaction was used to make Gal-α(1,4)-Gal containing glycoconjugates. Surprisingly, trace amount of 14 was obtained when 3 was reacted with 9 (entry e, Table 1). We attributed the result to the spacer; presumably the chlorine in acceptor 9 is interfering with the reagents used to activate the thioglycoside or the linker is affecting the coupling efficiency. All thioglycoside donors were activated using para-nitrobenzenesulfenyl chloride in conjunction with silver trifluromethanesulfonate (AgOTf) according to the protocol developed by Crich et al.[23] Once we identified the appropriate condition for efficient coupling, we synthesized Gal-α(1,4)-Gal containing glycoconjugates (GC-1a and GC-1b) as shown in Scheme 2. Briefly, the allyl group of compound 13 was removed using PdCl2 and NaOAc in the presence of acetic acid and water. The resulting intermediate was subjected to reductive hydrogenation reaction followed by global acetylation reaction to furnish compound 19. Treatment of compound 19 with hydrazine acetate in THF cleaved the anomeric acetate protecting group of 19 to afford a hemiacetal product which was converted to a trichloroacetimidate donor 20 using trichloroacetonitrile in the presence of potassium carbonate. A glycosylation reaction between Compound 20 and 6-azido-hexanol in the presence of TMSOTf as a promoter resulted in the formation of compound 21. The coupled product was confirmed by 1H ( H1 = 5.01 δ, J = 3.6 Hz, H1’ = 4.48 δ, J = 8.0 Hz) and 13C NMR ( C1 = 101.2 δ, C1’ = 99.4 δ) and 1H-13C HSQC NMR experiment. Subsequent coupling of compound 21 with alkyne bearing monomeric and dimeric scaffolds afforded compounds 22 and 23, respectively. Deprotection of compounds 22 and 23 under Zemplén conditions furnished GC-1a and GC-1b respectively in excellent yield.
Table 1.
Attempted glycosylation reactions.

|

|
The imidate donors were activated with TMSOTf in CH2Cl2 at −30°C and thioglycoside donors were activated with AgOTf, PhClNO4S in CH2Cl2 at 78 °C. Experimental details are given in the experimental section or supplementary material.
Isolated yields.
Synthesis of glycoconjugate GC-2a and 2b
For the synthesis of Gal-α(1,4)-GalNHAc glycoconjugates, we used the imidate donor 6 instead of 3 for the coupling reaction because the coupling of 3 with acceptor 9 (entry e, Table 1) led to lower yields. The imidate donor 6 was first used for glycosylation reaction with 9 to form the α-glycosidic bond (entry h, table 1). Encouraged by the stability of the donor and the glycosylation reaction outcome we made an acceptor similar to 9 but with an azide protecting group on carbon-2 of the acceptor. For the synthesis of 10, we used the α-linked imidate donor 7, which was synthesized from acetylated and azide protected galactosamine using DBU as a base. Activation of 7 with TMSOTf in the presence of 6-chlorohexanol as an aglycon acceptor gave us mainly β-product 17 (entry j, Table 1) in good yield presumably via SN2 reaction pathway. The β-glycosidic linkage was confirmed by 1HNMR (H1 = 4.5, J1,2 = 8.0 Hz) and 13CNMR (C1 = 102.3 ppm). We note here that the β-glycoside could be obtained by employing 2-phthalimido (NPhth) and N-trichloroacetamide as participating protecting groups.[24, 25] These groups can decrease the reactivity of glycosyl donors and acceptors.[26, 27] Thus, we obtained the desired β linkage using appropriate reaction conditions without having to resort to a participating group at the 2 position.
As indicated on Scheme 3, 17 was subjected to deacetylation reaction followed by 4, 6-O-benzylidation and benzylation reactions to obtain acceptor 10 through regioselective reductive cleavage of the benzylidene acetal of 24. As expected, glycosylation reaction between 6 and 10 afforded 16 in decent yield (entry i, table 1). The α-linkage was confirmed by 1H NMR (H1 = 5.12 ppm, J1,2 = 3.6 Hz and 13C NMR ( C1 = 100.2 ppm). Reduction of the azide group using AcSH to the N-acetyl group was followed by nucleophilic substitution of the terminal chlorine with sodium azide to obtain 25 in reasonable yield. [28] Next, 1, 3 dipolar cycloaddition between 26 and the monomeric scaffold furnished compound 27. Although the click reaction worked well using the standard conditions (THF/water, copper sulfate and sodium ascrobate), deprotectation of the benzyl groups using hydrogenation and Birch reductions (sodium in liquid ammonia) from the biotinylated compound 27 to obtain GC-2a proved problematic despite numerous attempts (Scheme 3).While we attribute the failure of the hydrogenation reaction to the presence of sulfur in the biotin group; presumably sulfur is interfering with the metal-promoted catalytic hydrogenation. However we were surprised that the Birch reduction resulted in significant decomposition.
Scheme 3. Attempted synthesis of GC-2a and GC-2b.
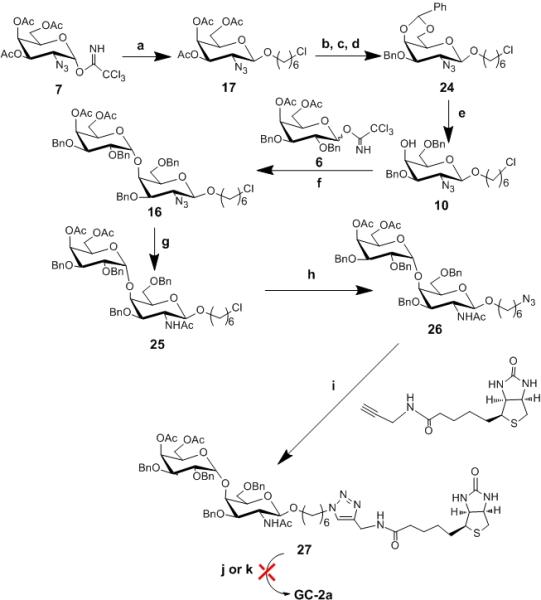
Reagents and conditions. a) CH2Cl2, TMSOTf, -30°C to rt, 90% b)NaOMe, MeOH, rt, 93% c) PhCH(OMe)2, p-TsOH, THF, 76% d) NaH, BnBr, THF, reflux, 62% e) NaCNBH3, HCl.Et2O, THF, 73% f) CH2Cl2, TMSOTf, −30°C, 62% g) AcSH, 46% h) DMF, NaN3, 90% l) CuSO4.5H2O, C6H7O6Na, THF/H2O, 70% j) i.MeOH, NaOMe, rt ii. Pd (OH)2, H2, EtOH/EtOAc, k) Na, NH3
The problem of deprotection with the biotinylated compound 27 was circumvented by removing the benzyl groups before performing 1, 3 dipolar cycloaddition reaction as shown in Scheme 4. To this end, the benzyl groups were removed using Pd/C and hydrogen at ambient temperature after converting azide to N-acetyl group. This was followed by global acetylation and nucleophilic substitution of the terminal chlorine with an azide functionality using sodium azide to obtain 28. Finally, 1, 3 dipolar cycloaddition of 28 with the monomeric and dimeric scaffolds using standard 1, 3 dipolar cycloaddition conditions , followed by removal of acetate groups afforded us GC-2a and GC-2b in reasonable yield (Scheme 4).
Scheme 4. Synthesis of GC-2a and GC-2b.
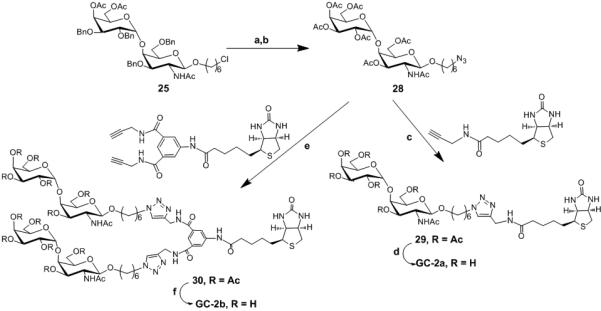
Reagents and conditions: a) i. NaOMe, MeOH, rt ii. Pd(OH)2, EtOH, H2, over two steps 75% b) i. Ac2O, Pyridine, DMAP, 78% ii. DMF, NaN3, 90% c) CuSO4.5H2O, C6H7O6Na, t-BuOH/H2O, 70% d) MeOH, NaOMe, rt ,90% e) CuSO4.5H2O, C6H7O6Na, t-BuOH/H2O, 70% f) MeOH, NaOMe, rt, 90%
Synthesis of glycoconjugates GC-3a, b, GC-4a, b and GC-5a, b
Synthesis of GalNHAcα(1, 4)-Gal part of the glycoconjugates GC-3a and 3b was relatively straight forward due to the fact that azides are good non-participating protecting groups. Azide protected acetylated galactose donor[29] gave exclusively α-glycoside when treated with 8 as judged by 1HNMR (H1 = 4.88 ppm, J1,2 = 3.6 Hz ) and 13CNMR (C1 = 98.2 ppm). This was followed by standard 1, 3 dipolar cycloaddition to the alkyne bearing scaffolds and standard deprotection. (Please refer supporting information for details). Synthesis of the trisaccharide glycoconjugates containing Galα(1, 4)-Galα(1, 4)Glc and GalNHAcα(1, 4)-Galα(1, 4)-Glc) was simplified by synthesizing lactose derivative with six carbon linker as a glycosyl acceptor 11 (entry K, Table 1) compared to the previously reported synthesis.[18] 6 and 7 were used as glycosyl donors for the synthesis of Galα(1, 4)-Galα(1, 4)Glc (GC-4a and 4b) and GalNHAcα(1, 4)-Galα(1, 4)-Glc (GC-5a and GC-5b) respectively. Details of the glycosylation reactions towards the synthesis of the disaccharide and the trisaccharide glycoconjugates are described on Table 1 and in the Supporting Information.
Binding studies
Capturing of E. coli strains with the biantennary trisaccharide (GC-4b)
Properties of the E. coli strains from different pathovars used in this study are described in Table 2. ORN178 and the afimbrial mutant, ORN208, are derivatives of a K12 intestinal isolate, lacking pathogenic potential.[30] J96 and CFT073 were isolated from urinary tract infections and are members of the uropathogenic E. coli (UPEC) pathovar.[31, 32] J96 pilE is a mutant of J96 which is unable to produce mannose-binding pili.[33] Strain PT22Δtox is an E. coli O157:H7 clinical isolate deleted for Shiga toxin, and is a member of the enterohemorrhagic E. coli (EHEC) pathovar.[34] In a previous study,[8] mannose-coated magnetic beads captured only strains expressing Type 1 fim-encoded pili, as summarized in Table 2.
Table 2.
E. coli strains used in this study and in the previous study[8] describing capture by biotinylated α-mannoside glycoconjugates
| Strain | Pathovar | Pilus type | α Mannoside binding[8] | GC-4b (Galα1-4Galβ1-4Glc) binding |
|---|---|---|---|---|
| ORN178 | None | Fim | + | – |
| ORN208 | None | None | – | – |
| J96 | UPEC | Fim, Pap, Prs | + | + |
| J96 PilE | UPEC | Pap, Prs | – | + |
| CFT073 | UPEC | Fim, Pap, Foc | + | + |
| O157:H7 PT22Δtox | EHEC | None | – | – |
All strains were screened for binding to streptavidin-coated magnetic beads displaying biantennary trisaccharide ligand, GC-4b. E. coli strains were grown overnight on L-agar at room temperature, transferred to 37°C for 2-3 hours and harvested in PBS. Addition of beads coated with GC-4b to E. coli strain CFT073 induced aggregation within minutes and the aggregation was visible to the naked eye and under a light microscope (Figure 1A). In contrast, strain ORN178 (Figure 1B), when incubated with beads coated with GC-4b showed no aggregation. Next, we used ESEM (Environmental Scanning Electron Microscope) to confirm the results. As seen in Figure 1C, significant aggregation was observed when GC-4b coated beads were incubated with CFT073 strain, and at high magnification, individual bacteria were seen. In contrast, little aggregation was observed when ORN178 was used. (Figure 1D). These experiments demonstrate that aggregation is highly dependent on the bacterial strain.
Figure 1. Bacterial-induced aggregation of GC-4b coated beads.

CFT073 (A and B) and ORN178 (C and D) were incubated with GC-4b coated beads. A) Visual aggregation of the beads incubated with CFT073 was observed, as documented by light microscopy. B) In ESEM CFT073 bacteria were seen bound to the beads (arrows). C) No aggregation was observed for beads incubated with ORN178. D) In ESEM no bacteria are seen bound to the beads incubated with ORN178.
To quantify the binding efficiency, capture of E. coli strains was assessed using BacTiter-Glo™ assay substrate.[8] (Figure 2) In this particular assay, the enzyme luciferase oxidizes luciferin, which in turn produces light in a reaction dependent on ATP produced by the metabolically active E. coli. The amount of light produced is equivalent to the amount of ATP present. The light produced is quantified by a luminometer (Thermo Labsystem Luminoskan Ascent).
Figure 2. Binding of E. coli strains to the biantennary Pk trisaccharide (GC-4b).

Error bars represent mean ± SEM of three independent experiments. Statistical analysis using a student t-test was performed comparing the binding of each E. coli strain to GC-4b (*P < 0.05).
E. coli strains producing type P fimbriae (CFT073, J96, and J96 pilE) bound to the trisaccharide containing Galα1-4Galβ1-4Glc (GC-4b). While CFT073 showed strong binding to the ligand, J96 and J96 pilE exhibited moderate binding. The other strains, which lack P fimbriae expression failed to bind to GC-4b. The binding of type P fimbriae is consistent with previous reports, as it is known to produce pili that bind to Galα1-4Gal (galabiose) containing glycolipids.[3, 4] However there is statistically significant difference in binding to GC-4b among the type P expressing strains. This may be because CFT073 and the other type P expressing strains produce different PapG variants (PapG I, II and III) which are known to exhibit subtle differences in binding to galabiose containing glycolipids.[4] Alternatively pilus expression is highly regulated and not all bacteria in a population are piliated.
The capturing efficiency of the magnetic beads bearing GC-4b was also determined by comparing the ATP production, determined by the aforementioned luciferase assay, of the bacteria captured on beads to the activity of a known amount of bacteria added to the microplate wells. GC-4b bearing magnetic beads captured 34.0% of CFT0T3 and 17.0% of both J96 and J96 pilE when each strain was added at around 5×106 cells per 100 μl. CFT073 is captured more efficiently than the other Pap-producing strains.
Capturing of CFT073 strain with the panel of biotinylated glycoconjugates
Binding of CFT073 to a panel of oligosaccharides was also assessed. Binding to monoantennary trisaccharide (GC-4a), Galα1-4Gal monoantennary (GC-1a) and biantennary disaccharides (GC-1b) was similar to biantennary Pk-trisaccharide, GC-4b (Figure 3). This indicates that the digalactose units are the critical determinants for binding to CFT073. It is also important to note here that although multivalency is essential for enhanced binding, magnetic beads displaying GC-4a or GC-4b exhibited almost the same capturing efficiency of CFT073. The trisaccharide GC-4a and GC-4b magnetic beads captured 38.0% and 35.6% CFT073 respectively, while the disaccharide beads displaying GC-1a and GC-1b captured 29.4% and 33.0% of CFT073 (Figure 3). Typically, protein-glycan interactions are rather weak and strong binding is achieved through avidity, or the ability to simultaneously engage multiple glycans However, the requirements to achieve avidity are very different for different glycan binding proteins. [35] For example, Shiga toxin, which is approximately 5 nanometers in diameter has 15 glycan binding sites, spaced approximately 1-2 nanometers apart. At least four tightly spaced glycans are needed for efficient binding. [36] Glycan spacing is less critical for influenza binding, since influenza virus is much larger (~130 nanometers) and has more glycan binding sites, with approximately 300 copies of the trimeric sialic acid binding protein, hemagglutinin per virus. The distance between sialic acid binding sites on the hemagglutinin trimer is approximately 5 nanometers. In the case of CFT073 strain, the Pap pili are 2-7 nm in width and roughly 1000 nm in length. Each pilus has one glycan binding site, and a single bacterial cell can display hundreds of pili. Both Shiga toxin and influenza are rigid structures and the glycan display must be compatible with the spacing of the glycan binding sites. In marked contrast, the long and flexible bacterial pili can bend to accommodate the glycan spacing. Thus, it is not unexpected that the bacteria bound equally well to the dimeric and monomeric ligands. In this case, the number of glycans displayed from the monoantennary scaffold is sufficient to promote maximal binding when 106 bacteria are present. No binding was observed to beads coated with biotinylated forms of fetuin, a bovine serum protein with extensive sialic acid-rich glycosylation, or to biotinylated forms of heparin, a highly sulfated glycosaminoglycan (data not shown).
Figure 3. Capturing of CFT073 with the panel of glycans.

Error bars represent mean ±standard error of three independent experiments. Statistical analysis using a student t-test was performed comparing the binding and capturing ability of each ligand; * no statistical differences between the group (P > 0.05), ** significantly reduced binding compared to * group (P < 0.05).
Glycans with GalNHAc at the terminal end did not bind or capture CFT073. This implies that the GalNHAc at the terminal end interferes with binding, suggesting that the terminal galactose is critical for recognition to this strain. Similarly, none of the other E. coli strains bound to the GalNHAc containing oligosaccharides (data not shown). However, the biantennary disaccharide with the GalNHAc (GC-2b) at the reducing end showed intermediate capturing ability, while the monoantennary disaccharide (GC-2a) with GalNHAc the reducing end was somewhat less effective at binding to CFT073. These results suggest that the penultimate galactose residue also plays a role in recognition, but is less critical than the terminal galactose sugar.
In summary, our results show that binding of pathogenic bacteria to the synthetic oligosaccharides was observed after incubation of E. coli strains with carbohydrate-presenting magnetic beads. The oligosaccharides demonstrated binding specificity of E. coli strains expressing the P pilus type. The current study demonstrates that magnetic beads coated with oligosaccharides of various specificities can be considered for development of detection devices that are responsive to the presence of specific pathogens.
Experimental section
Synthesis
Synthesis of the biotinylated glycoconjugates is given in the Supporting Information
Microbiology
Reagents
Streptavidin-coated magnetic beads (Dynabeads® M-280 streptavidin, 2.8 μm in diameter) were purchased from Invitrogen (Carlsbad, CA). BacTiter-Glo™ substrate was purchased from Promega (Madison, WI). Fetuin (Sigma) was biotinylated using EZ-Link® Sulfo-NHS-LC-Biotin (Thermo Scientific). Heparin (Sigma) was purchased in the biotinylated form.
Bacterial cultures and cell recovery assay
LB agar plates were streaked with each E. coli strain (ORN208, ORN178, J96, J96 pilE, CFT073 and PT22Δtox) and incubated overnight at room temperature and transferred to 37°C for 2-3hrs. Bacterial colonies were harvested and placed in PBS buffer (3 ml). Appropriate dilutions were made to obtain an OD600 measurement of approximately 0.8 (OD600 of 1.0 is approximately 108 cells mL−1), and approximately 5.0×106 bacterial cells were incubated with conjugated magnetic beads for all the assays. BacTiter-Glo™ assay was used to quantify viable cells. In this assay the enzyme luciferase oxidizes luciferin, which in turn produces light in a reaction dependent on ATP, an indicator of metabolically active bacterial cells. The amount of light produced is equivalent to the amount of ATP present. The light produced was quantified by a luminometer (Thermo labsystem Luminoskan Ascent 96-microwell plate reader).
Conjugation of biotinylated glycans to magnetic beads
Conjugation of the biotinylated oligosaccharides was carried out as previously described.[8] Briefly, 25 μl of magnetic beads (1 ml, 10 mg) was aliquoted into a 1.5 ml microcentrifuge tube. Prior to incubation with the biotinylated ligands, the beads were washed three times with PBS by placing the tubes in a magnet. The beads were then incubated with the biotinylated ligands (1000 pmol of ligand per 1 mg of beads) on an orbital shaker for 1 hr at room temperature. The glycan-coated magnetic beads were isolated using a standard magnet and washed three times with PBS to ensure complete removal of the unbound ligands.
ESEM images
E. coli strains CFT073 and ORN178 (107 CFU/ml) in PBS (pH 7.4) were incubated with beads bearing GC-4b at room temperature for 1 h on the orbital shaker. The tubes were placed over a magnet for 5 min and the PBS was carefully removed. The tubes were washed with sterile deionized water (500 μl) and the beads were suspended in water (500 μl) to generate the final SEM samples. Droplets from each sample were placed on a gold disc and allowed to dry in vacuo. The gold disc was sputtered with gold and Images were captured by using a Phillips XL30 ESEM.
Ability of the glycomagnetic beads to capture E. coli strains
Approximately 5.0×106 bacteria per 100 μl were prepared. Each E. coli strain was incubated with 25 μg of beads at room temperature for 1 hr on an orbital shaker. The beads were then placed over a magnet to remove the supernatants. The beads were washed with PBS three times and suspended in 100 μl of PBS. The bead suspension was transferred into 96-well microtiter plate and 100 μl of BacTiter-Glo® substrate was added to each well. Beads conjugated with ligands were used as a negative control. After adding BacTiter-Glo™ reagent, the microplate was placed in the luminometer. Before measuring the luminescence, the microplate was allowed to shake for 5 min followed by 5 min incubation at room temperature.
Supplementary Material
Acknowledgements
Financial support for this work was provided by NIAID (U01-AI075498 A.A.W.). and NSF (0845005 – S. S. I). S.S.I thanks Dr. Necati Kaval for the ESEM analysis.
References
- 1.Moriel DG, Rosini R, Seib LK, Serino L, Pizza M, Rappuoli R. mBio. 2012;3:e00118. doi: 10.1128/mBio.00118-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Croxen MA, Finlay BB. Nat.Rev.Microbiol. 2009;8:26. doi: 10.1038/nrmicro2265. [DOI] [PubMed] [Google Scholar]
- 3.Pieters JR. Medicinal Research Reviews. 2006;27:796. doi: 10.1002/med.20089. [DOI] [PubMed] [Google Scholar]
- 4.Mulvey AM. Cellular Microbiology. 2002;4:257. doi: 10.1046/j.1462-5822.2002.00193.x. [DOI] [PubMed] [Google Scholar]
- 5.Madison B, Ofek I, Clegg S, Abraham NS. Infec. Immun. 1994;62:843. doi: 10.1128/iai.62.3.843-848.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ofek I, Hasty LD, Abraham NS, Sharon N. Adv. Exp. Med. Biol. 2000;485:183. doi: 10.1007/0-306-46840-9_25. [DOI] [PubMed] [Google Scholar]
- 7.Nitschke M, Sayk F, Hartel C, Roseland TR, Hauswaldt S, Steinhoff J, Fellermann K, Derad I, Wellhoner P, Buning J, Tiemer B, Katalinic A, Jan Rupp J, Lehnert H, Solbach W, Knobloch K-MJ. JAMA. 2012;307:1046. doi: 10.1001/jama.2012.264. [DOI] [PubMed] [Google Scholar]
- 8.Hatch MD, Weiss AA, Kale RR, Iyer SS. ChemBioChem. 2008;9:2433. doi: 10.1002/cbic.200800188. [DOI] [PubMed] [Google Scholar]
- 9.Collins EB, Paulson CJ. Curr.Opin.Chem.Biol. 2004;8:617. doi: 10.1016/j.cbpa.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 10.Disney MD, Zheng J, Swager TM, Seeberger PH. J Am Chem Soc. 2004;126:13343. doi: 10.1021/ja047936i. [DOI] [PubMed] [Google Scholar]
- 11.Pieters JR. Org.Biomol.Chem. 2009;7:2013. doi: 10.1039/b901828j. [DOI] [PubMed] [Google Scholar]
- 12.Lundquist JJ, Toone JE. Chem.Rev. 2002;102:555. doi: 10.1021/cr000418f. [DOI] [PubMed] [Google Scholar]
- 13.Kiessling LL, Gestwicki JE, Strong LE. Curr Opin Chem Biol. 2000;4:696. doi: 10.1016/s1367-5931(00)00153-8. [DOI] [PubMed] [Google Scholar]
- 14.Chien YY, Jan MD, Adak AK, Tzeng HC, Lin YP, Chen YJ, Wang KT, Chen CT, Chen CC, Lin CC. Chembiochem. 2008;9:1100. doi: 10.1002/cbic.200700590. [DOI] [PubMed] [Google Scholar]
- 15.Boltje JT, Buskas T, Boons G-T. Nature Chemistry. 2009;1:611. doi: 10.1038/nchem.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pera NP, Kouki A, Haataja S, Branderhorst HM, Liskamp RM, Visser GM, Finne J, Pieters RJ. Org Biomol Chem. 8:2425. doi: 10.1039/c000819b. [DOI] [PubMed] [Google Scholar]
- 17.El-Boubbou K, Gruden C, Huang X. J Am Chem Soc. 2007;129:13392. doi: 10.1021/ja076086e. [DOI] [PubMed] [Google Scholar]
- 18.Kale RR, McGannon CM, Fuller-Schaefer C, Hatch DM, Flagler MJ, Gamage SD, Weiss AA, Iyer SS. Angew.Chem. Int. Ed. 2008;47:1265. doi: 10.1002/anie.200703680. [DOI] [PubMed] [Google Scholar]
- 19.Lewallen DM, Siler D, Iyer SS. ChemBioChem. 2009;10:1486. doi: 10.1002/cbic.200900211. [DOI] [PubMed] [Google Scholar]
- 20.Kale RR, Mukundan H, Price DN, Harris JF, Lewallen DM, Swanson BI, Schmidt JG, Iyer SS. JACS. 2008;130:8169. doi: 10.1021/ja800842v. [DOI] [PubMed] [Google Scholar]
- 21.Crich D. Accounts of Chemical Research. 2010;43:1143. doi: 10.1021/ar100035r. [DOI] [PubMed] [Google Scholar]
- 22.Deng S, Gangadharmath U, Chang CW. J Org Chem. 2006;71:5179. doi: 10.1021/jo060374w. [DOI] [PubMed] [Google Scholar]
- 23.Crich D, Cai F, Yang F. Carbohydrate Research. 2008;2008(343):1858–1862. 1858. doi: 10.1016/j.carres.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Palmacci RE, Seeberger HP. Tetrahedron. 2004;60:7755. [Google Scholar]
- 25.Horlacher T, Oberli AM, Werz BD, Krçck L, Bufali S, Rashmi Mishra R, Sobek J, Simons K, Hirashima M, Niki T, Seeberger HP. ChembioChem. 2010;11:1563. doi: 10.1002/cbic.201000020. [DOI] [PubMed] [Google Scholar]
- 26.Rele MS, Iyer SS, Baskaran S, Chaikof LE. JOC. 2004;69:9159. doi: 10.1021/jo049092r. [DOI] [PubMed] [Google Scholar]
- 27.Crich D, Dudkin V. JACS. 2001;123:6819. doi: 10.1021/ja010086b. [DOI] [PubMed] [Google Scholar]
- 28.Shangguan N, Katukojvala S, Greenberg R, Williams LJ. J Am Chem Soc. 2003;125:7754. doi: 10.1021/ja0294919. [DOI] [PubMed] [Google Scholar]
- 29.Koeller KM, Smith ME, Wong CH. Bioorg Med Chem. 2000;8:1017. doi: 10.1016/s0968-0896(00)00041-9. [DOI] [PubMed] [Google Scholar]
- 30.Harris SL, Spears PA, Havell EA, Hamrick TS, Horton JR, Orndorff PE. J Bacteriol. 2001;183:4099. doi: 10.1128/JB.183.13.4099-4102.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mobley HL, Green DM, Trifillis AL, Johnson DE, Chippendale GR, Lockatell CV, Jones BD, Warren JW. Infect Immun. 1990;58:1281. doi: 10.1128/iai.58.5.1281-1289.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Minshew BH, Jorgensen J, Counts GW, Falkow S. Infect Immun. 1978;20:50. doi: 10.1128/iai.20.1.50-54.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Keith BR, Maurer L, Spears PA, Orndorff PE. Infect Immun. 1986;53:693. doi: 10.1128/iai.53.3.693-696.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gamage SD, Patton AK, Hanson JF, Weiss AA. Infect Immun. 2004;72:7131. doi: 10.1128/IAI.72.12.7131-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kulkarni AA, Weiss AA, Iyer SS. Medicinal Research Reviews. 2010;30:327. doi: 10.1002/med.20196. [DOI] [PubMed] [Google Scholar]
- 36.Flagler MJ, Mahajan SS, Kulkarni AA, Iyer SS, Weiss AA. Biochemistry. 2010;49:1649. doi: 10.1021/bi902084y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.