

Abstract
Using transcriptional profiling of platelets from patients presenting with acute myocardial infarction, we identified myeloid-related protein-14 (MRP-14, also known as S100A9) as an acute myocardial infarction gene and reported that platelet MRP-14 binding to platelet CD36 regulates arterial thrombosis. However, whether MRP-14 plays a role in venous thrombosis is unknown. We subjected WT and Mrp-14–deficient (Mrp-14-/-) mice to experimental models of deep vein thrombosis (DVT) by stasis ligation or partial flow restriction (stenosis) of the inferior vena cava. Thrombus weight in response to stasis ligation or stenosis was reduced significantly in Mrp-14-/- mice compared with WT mice. The adoptive transfer of WT neutrophils or platelets, or the infusion of recombinant MRP-8/14, into Mrp-14-/- mice rescued the venous thrombosis defect in Mrp-14-/- mice, indicating that neutrophil- and platelet-derived MRP-14 directly regulate venous thrombogenesis. Stimulation of neutrophils with MRP-14 induced neutrophil extracellular trap (NET) formation, and NETs were reduced in venous thrombi harvested from Mrp-14-/- mice and in Mrp-14-/- neutrophils stimulated with ionomycin. Given prior evidence that MRP-14 also regulates arterial thrombosis, but not hemostasis (i.e., reduced bleeding risk), MRP-14 appears to be a particularly attractive molecular target for treating thrombotic cardiovascular diseases, including myocardial infarction, stroke, and venous thromboembolism.
Keywords: Cardiology, Vascular Biology
Introduction
Venous thromboembolism (VTE), including deep vein thrombosis (DVT), is the third-most common cause of death from cardiovascular disease after myocardial infarction and stroke (1). VTE is associated with a high case fatality rate, with 10% to 30% of patients dying within 1 month of diagnosis (2). Estimates suggest that 60,000–100,000 Americans die of DVT/pulmonary embolism (DVT/PE), which is the most common cause of death after elective surgery and pregnancy as well as the leading cause of preventable hospital deaths. Among people who have had a DVT, up to one-half will have long-term complications (post-thrombotic syndrome), such as swelling, pain, discoloration, and scaling in the affected limb (1, 2). Despite therapeutic anticoagulation, one-third of people with DVT/PE will have a recurrence within 10 years. Based on observations in animal models of DVT, there is a growing understanding of the importance of monocytes, neutrophils, and platelets cooperating to initiate and propagate venous thrombosis in vivo (3). However, the precise molecular mechanisms responsible for this cross talk are incompletely defined.
To gain insight into novel regulators of arterial inflammation and thrombosis, we leveraged platelet transcriptional profiling to examine platelet mRNA transcripts that are differentially expressed in patients with ST-segment elevation myocardial infarction (STEMI) compared with stable coronary artery disease (4). Myeloid-related protein-14 (MRP-14, also referred to as S100A9) was one of the strongest predictors of STEMI that arose from the transcriptional profiling analysis. MRP-14, a member of the S100 family of calcium-modulated proteins as well as a member of the alarmin or danger-associated molecular pattern molecules family, complexes with MRP-8 (also known as S100A8), and together, the MRP-8/14 heterodimer regulates myeloid cell function by modulating calcium signaling (5) and cytoskeletal reorganization (6), by operating as a chemoattractant (7), and by binding to cell-surface receptors, including CD36 (8), TLR-4 (9), and receptor for advanced glycation end products (RAGE) (10).
We recently reported an essential role for MRP-14 in arterial thrombosis, identifying a novel pathway of thrombosis that involves platelet MRP-14 and the platelet receptor CD36, without influence on tail bleeding time and other hemostatic parameters (11). Whether MRP-14 plays a role in venous thrombosis is unknown. In this study, we provide evidence that neutrophil-derived and platelet-derived MRP-14 directly modulate venous thrombosis.
Results
Deep vein thrombus formation is attenuated in Mrp-14-/- mice.
To investigate the role of MRP-14 in DVT, WT and Mrp-14-/- mice were subjected to inferior vena cava (IVC) ligation (stasis) (12) or partial flow restriction (stenosis) (3) on day 0. Thrombi were harvested on day 2, day 4, and day 12 after IVC ligation. In the stasis model, mean thrombus weight on day 2 in WT mice was 18.5 ± 3.1 mg; this was reduced significantly in Mrp-14-/- mice to 12.3 ± 6.6 mg (P = 0.013; Figure 1A). A similar reduction in thrombus weight was observed in the partial flow restriction (stenosis) model (WT 17.7 ± 8.7 vs. Mrp-14-/- 8.2 ± 7.9 mg, P = 0.042; Figure 1B). Stenosis-induced IVC thrombi were observed in 8 of 8 WT mice, whereas 3 of 8 Mrp-14-/- mice had no detectable thrombus.
Figure 1. Deep vein thrombus formation is attenuated in Mrp-14-/- mice.

Inferior vena cava (IVC) thrombus weight (mg) of individual mice after deep vein thrombosis (DVT) induction. (A) Two days after IVC ligation (stasis) in WT (n = 10) and Mrp-14-/- (n = 11) mice. Representative images of WT and Mrp-14-/- thrombi (n = 10–11). (B) Two days after IVC partial flow restriction (stenosis) in WT (n = 8) and Mrp-14-/- (n = 8) mice. (C) Four days after IVC ligation (stasis) in WT (n = 8) and Mrp-14-/- (n = 6) mice. (D) Twelve days after IVC ligation (stasis) in WT (n = 6) and Mrp-14-/- (n = 7) mice. (E) Representative image of hind limbs of WT (left) and Mrp-14-/- (right) mice 12 days after IVC ligation (stasis). Each dot represents 1 mouse. Data represent mean ± SD. P values were obtained by conducting unpaired, 2-tailed t test using Excel.
Thrombus growth was evident from day 2 to day 4 in both WT and Mrp-14-/- mice. However, while thrombus weight increased to 24.1 ± 6.7 mg in WT mice at day 4, it was increased to 19.4 ± 8.1 mg in Mrp-14-/- mice (P = 0.018; Figure 1C).
Deep vein thrombus resolution is enhanced in MRP-14-/- mice.
Next, we investigated whether MRP-14 also plays a role in decreasing thrombus burden over time (i.e., resolution of DVT). Mice that underwent stasis IVC ligation were therefore harvested on day 12. In WT mice, thrombus weight decreased from 24.1 ± 6.7 mg at day 4 to 13.6 ± 4.7 mg. In Mrp-14-/- mice, thrombus burden decreased from 19.4 ± 8.1 mg to 7.6 ± 4.3 mg, consistent with a significant 44% reduction in thrombus weight compared with WT mice (P = 0.032; Figure 1D). Remarkably, MRP-14-/- mice were protected from the development of severe hind limb edema on day 12 after IVC ligation (Figure 1E).
Unimpaired recruitment and accumulation of leukocytes in MRP-14-/- mice.
Platelets, neutrophils, and monocytes cooperate to initiate and propagate venous thrombosis in mice in vivo (3). MRP-14 is expressed in, and regulates the functions of neutrophils, monocytes, and platelets (11, 13). An essential role for MRP-8/14 (also known as S100A8/A9) in leukocyte recruitment in vivo is supported by evidence that Mrp-14-/- mice have diminished neutrophil and/or monocyte recruitment during tissue wound healing (6) and acute pancreatitis (14) as well as in a variety of models of vascular inflammation (e.g., atherosclerosis, vasculitis, and neointimal formation after mechanical injury) (13).
Therefore, we reasoned that the recruitment and accumulation of neutrophils and monocytes may be reduced in Mrp-14-/- thrombi, thereby providing a possible mechanism for reduced venous thrombogenesis. The accumulation of leukocytes in stasis-induced venous thrombi was examined on day 2, day 4, and day 14 after DVT induction. Immunohistochemistry using the neutrophil-specific marker 7/4 (Figure 2A) and the macrophage-specific marker Mac-3 (Figure 2B) demonstrated no significant difference in inflammatory cell accumulation at any time point. Absence of MRP-14 did not affect neutrophil (Figure 2C, day 2: WT vs. Mrp-14-/-, P = 0.821; Figure 2D, day 4: P = 0.859; and Figure 2E, day 12: P = 0.855) or macrophage (Figure 2F, day 2: WT vs. Mrp-14-/-, P = 0.197; Figure 2G, day 4: P = 0.103; and Figure 2H day 12: P = 0.596) accumulation at any time point.
Figure 2. The accumulation of inflammatory cells is unimpaired in Mrp-14-/- thrombi.

Immunohistochemistry staining of neutrophils (7/4-positive cells) and monocytes/macrophages (Mac-3–positive cells) in thrombi harvested at day 2, day 4, and day 12 after inferior vena cava (IVC) ligation. Representative photomicrographs of neutrophils (A) and macrophages (B) in WT and Mrp-14-/- thrombi harvested on day 4 after stasis IVC ligation. Quantification of neutrophils (C–E) and monocytes/macrophages (F–H) in WT and Mrp-14-/- thrombi harvested on day 2 (C and F), day 4 (D and G), and day 12 (E and H) after stasis IVC ligation. Data represent mean ± SD. Scale bars: 200 μm (top; original magnification, ×10); 100 μm (middle; original magnification, ×20); 50 μm (bottom; original magnification, ×40). P values were obtained by conducting unpaired, 2-tailed t test using Excel.
In addition to immunohistochemical analysis, we also quantified inflammatory cell number using automated complete blood count analysis of day 2 thrombi that were easily dispersible. Total white blood cell count and constituent neutrophils, lymphocytes, and monocytes, as well as cell counts normalized to thrombus weight, were similar in WT and Mrp-14-/- thrombi (Table 1).
Table 1. Cellular composition of IVC thrombi.

Adoptive transfer of WT neutrophils or platelets rescued the defect of DVT in Mrp-14-/- mice.
Although we provided evidence that deficiency of Mrp-14 is associated with reduced deep venous thrombus formation, we utilized mice with global rather than tissue-specific deficiency of Mrp-14, thereby limiting our ability to identify definitively the precise cellular source of MRP-14 that is critical for venous thrombus formation. To address this issue, we performed adoptive transfer or transfusion experiments using WT and Mrp-14-/- donor neutrophils, peripheral blood mononuclear cells (PBMCs), and platelets transferred into Mrp-14-/- recipient mice prior to stasis-induced DVT induction. Mrp-14-/- recipient mice that received Mrp-14-/- donor neutrophils formed venous thrombi that weighed 12.8 ± 5.1 mg (Figure 3A), similar to those in Mrp-14-/- mice without transfer (12.3 ± 6.6 mg; Figure 1A). Strikingly, the weight of thrombi in Mrp-14-/- mice receiving WT donor neutrophils increased significantly to 20.4 ± 5.1 mg (P = 0.047), restoring thrombi to the weight observed in WT mice (18.5 ± 3.1 mg; Figure 1A). In contrast, adoptive transfer of WT donor PBMCs in Mrp-14-/- mice did not significantly increase thrombus weight (WT donor PBMCs: 16.5 ± 10.4 vs. Mrp-14-/- donor PBMCs: 10.6 ± 9.2, P = 0.222; Figure 3B). In addition, we also investigated the effect of the adoptive transfer of platelets (i.e., transfusion) on thrombus weight. Mrp-14-/- mice that received Mrp-14-/- donor platelets formed thrombi weighing 5.8 ± 4.6 mg (Figure 3C). Thrombus weight was increased significantly in Mrp-14-/- recipient mice that received WT donor platelets to 13.1 ± 8.7 mg (P = 0.045), although transfer of WT platelets did not completely restore the thrombi weights to those observed in WT mice (18.5 ± 3.1 mg; Figure 1A). Taken together, these adoptive transfer experiments indicate that both neutrophil and platelet MRP-14 contribute to thrombus formation.
Figure 3. Adoptive transfer of WT neutrophils or platelets rescues the defect of thrombus formation in Mrp-14-/- mice.

Thrombus weight (mg) of individual Mrp-14-/- mice (recipient) that received WT or Mrp-14-/- neutrophils (A), PBMCs (B), or platelets (C) before recipient mice were subjected to inferior vena cava (IVC) ligation (stasis). Thrombi were harvested 2 days after DVT induction. Each dot represents 1 mouse. Data represent mean ± SD. P values were obtained from unpaired, 2-tailed t test using Excel.
Role of intracellular versus extracellular MRP-8/14 in thrombosis.
In our prior report (11), we identified a molecular pathway of arterial thrombosis involving platelet-secreted MRP-14 (i.e., extracellular) and its binding to the platelet receptor CD36. Our observations that transfusion of WT platelets or infusion of purified MRP-8/14 into Mrp-14-/- mice shortened the prolonged carotid artery occlusion time of Mrp-14-/- mice strongly suggested that extracellular, rather than intracellular, MRP-14 is largely responsible for MRP-14 action in arterial thrombosis. In the current study, adoptive transfer of WT neutrophils corrected, and platelets partially corrected, the venous thrombotic defect in Mrp-14-/- mice, indicating that neutrophil and/or platelet MRP-8/14 content regulates venous thrombosis. To determine whether extracellular MRP-8/14 action modulates venous thrombosis, we infused purified recombinant human MRP-8/14 (0.08 μg/g mouse) or buffer control into Mrp-14-/- mice (Figure 4). Infusion of purified MRP-8/14 in Mrp-14-/- mice significantly increased thrombus weight (Mrp-14-/- mice infused with buffer control: 8.0 ± 8.5 mg vs. Mrp-14-/- mice infused with purified MRP-8/14: 20.1 ± 9.3 mg, P = 0.032), restoring IVC thrombus weight to that observed in WT mice (18.5 ± 3.1 mg; Figure 1A). This result strongly suggests that extracellular MRP-14 action is required for venous thrombogenesis.
Figure 4. Infusion of recombinant MRP-8/14 protein rescues the thrombosis defect in Mrp-14-/- mice.
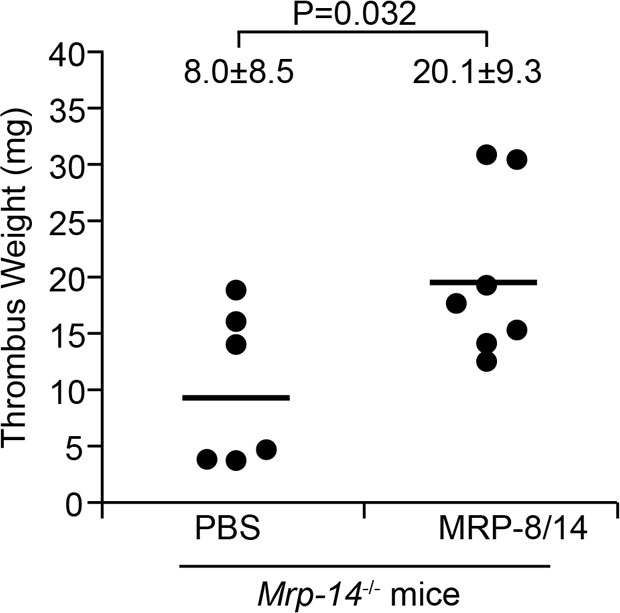
Thrombus weight (mg) of individual Mrp-14-/- mice (recipient) that received recombinant MRP-8/14 protein (0.15 μg/g body weight) prior to inferior vena cava (IVC) ligation (stasis). Thrombi were harvested 2 days after DVT induction. Each dot represents 1 mouse. Data represent mean ± SD. P value was obtained from unpaired, 2-tailed t test using Excel.
MRP-14 regulates the formation of neutrophil extracellular traps.
Having observed reduced venous thrombosis in Mrp-14-/- mice, we set out to determine the mechanism. We reported previously in our arterial thrombosis studies that platelet count, activated partial thromboplastin time, and tissue factor–induced total thrombin generation in plasma were similar in WT and Mrp-14-/-mice (11), indicating that neither platelet count nor coagulation parameters likely account for diminished DVT in Mrp-14-/- mice. However, having established that adoptive transfer of WT neutrophils corrects the defective DVT formation in Mrp-14-/- mice, we considered the possibility that MRP-14 might influence DVT by regulating the formation of neutrophil extracellular traps (NETs), which stimulate thrombus formation and coagulation and are abundant in thrombi in animal models of DVT (15) and in human venous thrombi (16). Formation of NETs occurs in a step-wise process characterized by nuclear membrane dissolution, chromatin decondensation, and cytolysis. Histone citrullination is a hallmark of chromatin decondensation in neutrophils and is considered a marker of NET formation (17). Immunofluorescence of thrombi harvested on day 2 after DVT induction showed reduced citrullinated histone H3–positive (H3Cit-positive) immunostaining in Mrp-14-/- thrombi compared with WT thrombi (Figure 5, A, B, and C), consistent with decreased NET formation in Mrp-14-/- mice. Adoptive transfer of WT neutrophils into MRP-14-/- mice restored the thrombosis defect in Mrp-14-/- mice (Figure 3A) and was associated with increased H3Cit-positive immunostaining in Mrp-14-/- thrombi (Figure 5D), consistent with increased NET formation.
Figure 5. NET formation is defective in Mrp-14-/- mice.

(A) Representative immunofluorescence (IF) images of staining for citrullinated histone 3 (His3Cit, green) and nuclei (DAPI, blue) in WT and Mrp-14-/- thrombi. (B) Representative IF images of His3Cit (green), neutrophils (7/4, red), and nuclei (DAPI, blue) in WT and Mrp-14-/- thrombi. (C) Sytox-Orange nuclear staining. Typical neutrophil extracellular trap (NET) fibers designated by arrows. (D) Transfer of WT neutrophils increases NET formation in Mrp-14-/- mice. Representative IF images of staining for His3Cit, neutrophils (7/4), and nuclei in Mrp-14-/- thrombi from mice that received Mrp-14-/- (top) or WT (bottom) neutrophils before induction of thrombus formation. Scale bars: 100 μm (A); 5 μm (B); 200 μm (D). Original magnifications for C is ×100 for left and right panels and ×370 for the middle panel.
To further investigate the role of MRP-14 in limiting the formation of NETs (i.e., NETosis), we incubated purified peritoneal-derived or blood-derived neutrophils harvested from WT and Mrp-14-/- mice with ionomycin to induce histone hypercitrullination and NETosis. The number of H3cit+ cells stimulated by ionomycin was reduced significantly in Mrp-14-/- compared with WT peritoneal-derived neutrophils (Mrp-14-/-: 8.2% ± 1.4% vs. WT: 17.6% ± 2.0%, P = 0.034, 90 minutes; Mrp-14-/-: 23.3% ± 5.8% vs. WT: 46.1% ± 3.6%, P < 0.001, 180 minutes; Figure 6, A–C) and blood-derived neutrophils (Mrp-14-/-: 14.5% ± 5.8% vs. WT: 28.8% ± 4.5%, P = 0.003; Figure 6, D and E).
Figure 6. In vitro NETosis in WT and Mrp-14-/- neutrophils.

(A) Representative images of immunofluorescence (IF) staining for citrullinated histone 3 (His3Cit, green) and nuclei (DAPI, blue) in unstimulated and ionomycin-stimulated WT or Mrp-14-/- peritoneal neutrophils. (B) Example of linear or strand-like staining of His3Cit-positive cells. (C) Quantification of His3Cit-positive cells in unstimulated (0 minutes) and ionomycin-stimulated (90- or 180-minute stimulation) peritoneal neutrophils. (D) Ionomycin-stimulated NETosis of WT or Mrp-14-/- peripheral blood neutrophils detected by IF for His3Cit (green), neutrophils (mAb 7/4, red), and nuclei (DAPI, blue). (E) Quantification of His3Cit-positive cells in unstimulated and ionomycin-stimulated (120 minutes) blood neutrophils. Each dot represents 1 mouse from 3–5 independent experiments. Data for each mouse were obtained by averaging the quantification of 4–6 images of each mouse. Images were visualized by a fluorescence microscope (Leica DM 2000, LED) and captured by a Q-Imaging digital camera (Retiga 2000R). Scale bars: 20 μm (A); 10 μm (B); 5 μm (D). Data represent mean ± SD. P values were obtained by conducting 1-way ANOVA followed by post-hoc test with the Bonferroni criterion using SPSS (version 24, IBM).
Given the observation that the infusion of recombinant MRP-14 restored the venous thrombosis defect in Mrp-14-/- mice (Figure 4), we hypothesized that MRP-14 may stimulate NETosis. To test this possibility, we incubated recombinant MRP-14 with peritoneal-derived neutrophils harvested from WT and Mrp-14-/- mice. Strikingly, MRP-14 protein alone induced the formation of NETs in both WT and Mrp-14-/- neutrophils (Figure 7A). Stimulation of WT neutrophils with MRP-14 significantly increased the percentage of H3cit+ cells (MRP-14 stimulated: 20.3% ± 4.4% vs. unstimulated: 5.9% ± 2.2%, P = 0.036). Interestingly, stimulation of Mrp-14-/- neutrophils also significantly increased the percentage of H3cit+ cells (MRP-14 stimulated: 21.9% ± 5.5% vs. unstimulated: 7.1% ± 1.6%, P = 0.013) (Figure 7B). The finding that stimulation with MRP-14 restored NET formation in Mrp-14-/- neutrophils to WT levels highlights the importance of extracellular rather than intracellular MRP-14. In addition, MRP-14 potentiated the effect of ionomycin on NET formation. Costimulation of both WT and Mrp-14-/- neutrophils with MRP-14 and ionomycin significantly increased the percentage of H3cit+ cells compared with ionomycin alone (WT neutrophils: ionomycin stimulated: 32.9% ± 6.0% vs. MRP-14 and ionomycin costimulated: 49.0% ± 9.5%, P = 0.017; Mrp-14-/- neutrophils: ionomycin stimulated: 21.6% ± 1.3% vs. MRP-14 and ionomycin costimulated: 52.3% ± 5.6%, P < 0.001) (Figure 7).
Figure 7. Effect of recombinant MRP-14 on NETosis in WT, Mrp-14-/-, and Mac-1-/- neutrophils.

(A) Representative images of NETosis in WT, Mrp-14-/-, or Mac-1-/- peritoneal neutrophils detected by immunofluorescence (IF) staining for citrullinated histone 3 (His3Cit, green) and nuclei (DAPI, blue). Scale bars: 50 μm. (B) Quantification of His3Cit-positive cells in unstimulated or ionomycin-stimulated, recombinant MRP-14–stimulated, or ionomycin and recombinant MRP-14 costimulated peritoneal neutrophils (120-minute stimulation). Each dot represents 1 mouse from 3 independent experiments. Data for each mouse were obtained by averaging the quantification of 4–6 images of each mouse. Images were captured and quantified by the EVOS FL Cell Imaging System (Invitrogen). Data represent mean ± SD. Bonferroni-corrected P values were obtained by conducting 1-way ANOVA followed by post-hoc test by Bonferroni criterion using SPSS (version 24, IBM).
Finally, Neeli and coworkers have provided evidence that the leukocyte β2 integrin Mac-1 (αMβ2, also known as CD11b/CD18 or Mac-1) may be involved in the initiation of changes in the neutrophil cytoskeleton that facilitate the breakdown of nuclear and plasma membranes for the release of NETs (18). Given the prior report that MRP-14 regulates the activation of Mac-1 (19), we hypothesized that MRP-14–stimulated NETosis is Mac-1 dependent. To test this hypothesis, we examined the effect of recombinant MRP-14 with and without ionomycin on NET formation in Mac-1–deficient (Mac-1-/-) neutrophils. NETosis in Mac-1-/- neutrophils was reduced in response to stimulation with MRP-14 alone (MRP-14 stimulated: 12.7% ± 1.8%, P = 0.147 vs. WT) and in response to MRP-14 and ionomycin costimulation (21.1% ± 4.3% His3cit+ cells, P = 0.005 vs. WT) compared with NETosis in WT neutrophils (MRP-14 stimulated 20.3% ± 4.4%, MRP and ionomycin costimulated: 49.0% ± 9.5%) (Figure 7).
Discussion
In this study, we have indicated that neutrophil-derived and platelet-derived MRP-14 regulate venous thrombogenesis. This conclusion is supported by the following data: (a) DVT is reduced in Mrp-14-/-mice in IVC ligation stasis and partial flow restriction stenosis models; (b) deep vein thrombus resolution is enhanced in Mrp-14-/- mice; (c) adoptive transfer of WT donor neutrophils into Mrp-14-/- recipient mice restores the thrombosis defect and adoptive transfer of WT donor platelets into Mrp-14-/- recipient mice partially restores the thrombosis defect; (d) infusion of purified MRP-8/14 corrected the venous thrombosis defect in Mrp-14-/- mice; (e) NET formation is impaired in thrombi and purified neutrophils from Mrp-14-/- mice; and (f) MRP-14 induces NETosis in a Mac-1–dependent manner.
The precise sequence of events that promotes DVT remained obscure previously, but recent observations in mouse models indicate that cross talk among monocytes, neutrophils, and platelets is responsible for the initiation and amplification of DVT (3). Using conditional mutants and bone marrow chimeras, the Massberg laboratory showed that intravascular activation of the extrinsic pathway of coagulation via tissue factor derived from myeloid leukocytes causes the extensive intraluminal fibrin formation characteristic of DVT. Neutrophils are indispensable for subsequent DVT propagation by binding factor XII and by supporting its activation through the release of NETs. Platelets associate with innate immune cells via glycoprotein Ibα and contribute to DVT progression by promoting leukocyte recruitment and stimulating neutrophil-dependent coagulation (20).
Since MRP-14 is abundantly expressed in both neutrophils (21) and platelets (11), our finding that both neutrophil-derived and platelet-derived MRP-14 regulate DVT provides important insights into a unifying molecular mechanism for deep venous thrombus formation. MRP-8/14 is known to be expressed by various cell types, including neutrophils (21), monocytes (21), tissue macrophages under conditions of chronic inflammation (22), mucosal epithelium (23), and involved epidermis of psoriasis (24). MRP-8/14 complexes are also present in mouse cells, and extensive biochemical characterization has confirmed that mouse MRP-14 is functionally equivalent to its human counterpart (25). Analyses of mice that lack Mrp-8 and Mrp-14 have provided important insights into the function of these proteins. Although the homozygous deletion of MRP-8 results in embryonic lethality (26), deletion of the Mrp-14 gene does not affect viability and results in the additional loss of MRP-8 protein (5, 27). Failure to produce mature MRP-8 protein in the presence of normal Mrp-8 mRNA production likely results from instability of MRP-8 in the absence of MRP-14. Thus, the Mrp-14-/- mice used in this and previous studies lack both MRP-8 and MRP-14 protein and MRP-8/14 complexes.
The established roles of intracellular and extracellular MRP-8/14 in leukocyte and platelet function provide important clues into the putative action(s) of MRP-8/14 in venous thrombosis. Experiments with leukocytes isolated from Mrp-14-/- mice have demonstrated aberrant calcium signaling and blunted calcium responses following chemokine stimulation (28). MRP-8/14 further appears to modulate calcium-coupled arachidonic acid signaling by binding to arachidonic acid in a calcium-dependent manner (29, 30) and by facilitating translocation of arachidonic acid to the cytoskeleton (31, 32). Intracellular MRP-8/14 also modulates cytoskeletal reorganization by promoting polymerization of microtubules (6).
In response to cytokines or during contact with activated endothelium, myeloid cells secrete heterodimeric MRP-8/14, which is the dominant extracellular form (33–35), through a tubulin-dependent “alternative” secretion pathway (36). Extracellular MRP-8/14 is then able to bind to receptors on target cells, including CD36 (8), RAGE (10), TLR-4 (9), special carboxylated N-glycans (37), and heparin-like glycoaminoglycans (38). Using antibody-blocking and genetic deletion experiments, we recently reported that MRP-14 binding to platelet CD36 induces platelet activation via a signaling cascade involving the MAP kinase JNK2 and src kinase Vav and is required for arterial thrombosis (11).
The adoptive transfer of WT neutrophils or the infusion of purified MRP-14 into Mrp-14-/- mice restored the reduced thrombus weight in Mrp-14-/- mice to WT levels, indicating a key role for neutrophil-derived (i.e., secreted) MRP-14 in venous thrombogenesis. The importance of neutrophil MRP-14 in DVT is particularly interesting in light of the central role of NETs in DVT. NETs are not just innocent bystanders, but rather contribute to neutrophil-driven coagulation during DVT propagation, based on antibody depletion of neutrophils and disintegration of NETs with deoxyribonuclease 1 (DNAse1) (3, 15, 39). NETs, produced to allow neutrophils to trap and disarm microbes in the extracellular environment, play an important role in innate immunity (40). Platelet-neutrophil cross talk plays an important role in this process, with platelet TLR4 detecting TLR4 ligands associated with sepsis in blood and inducing platelet binding to adherent neutrophils that leads to robust neutrophil activation and NET formation (41). The molecular basis of NET generation (known as NETosis) is a complex process requiring reactive oxygen species production (42) and neutrophil proteases (i.e., neutrophil elastase, myeloperoxidase, and peptidylarginine deiminase-4) (43). Neeli and coworkers have provided evidence that the leukocyte β2 integrin Mac-1 may be involved in the initiation of changes in the neutrophil cytoskeleton that facilitate the breakdown of nuclear and plasma membranes for the release of NETs (18). Given the prior report that MRP-14 regulates the activation of Mac-1 (19), we hypothesized that MRP-14–stimulated NETosis is Mac-1 dependent. To test this hypothesis, we examined the effect of recombinant MRP-14 with and without ionomycin on NET formation in Mac-1-/- neutrophils and showed that Mac-1-/- neutrophils were defective in NET formation in response to stimulation with MRP-14 alone and to MRP-14 and ionomycin costimulation. The neutrophil receptor responsible for Mac-1 activation by MRP-14, and in turn NETosis, is unknown and the focus of ongoing studies. As a member of the alarmin or danger-associated molecular pattern molecules family, MRP-14 is now positioned as a targetable mediator linking innate immunity with thrombosis.
Limitations.
We recognize that these two mouse models used may not reproduce all the features of human DVT, in particular because healthy animals were used with no underlying inflammatory or procoagulant condition and the thrombotic insult is surgically induced. In addition, although it has been proposed (15) that NETs may promote endogenous fibrinolysis and thrombus resolution by binding fibrin-degrading proteases, including plasminogen/plasmin, cathepsin G, and neutrophil elastase (44), we observed that thrombus resolution was enhanced in Mrp-14-/- mice compared with WT mice. Finally, we reported previously that plasma levels of MRP-8/14 are an independent predictor of future cardiovascular events (cardiovascular death, myocardial infarction, and stroke) in apparently healthy postmenopausal women (4) as well as recurrent cardiovascular events in patients presenting with acute coronary syndromes (45). Whether plasma levels of MRP-8/14 predict future VTE is unknown and the focus of ongoing nested case-control studies.
Clinical implications.
Deficiency of MRP-14 did not interfere with tail-bleeding time, platelet adhesion to and spreading on vWF or collagen, and plasma coagulation activity (i.e., aPTT and thrombin generation) (11). The identification of a new pathway of arterial and venous thrombosis involving MRP-14 that does not affect hemostatic parameters, such as bleeding time and platelet adhesion and spreading, has possible clinical implications. Thrombotic cardiovascular diseases, including myocardial infarction, stroke, and DVT, are the leading cause of death in developed countries (46). Primary drug therapies include antiplatelet and anticoagulant agents, and current drugs are associated with significant bleeding risk, which is associated with increased mortality (47–49). While new oral anticoagulant agents (NOACs, including dabigatran, rivaroxaban, apixaban, and edoxaban) have been approved for the prevention and/or treatment of DVT/PE and represent a major advance in oral anticoagulation (50–53), there are remaining challenges that need to be addressed (54). Major or clinically relevant nonmajor bleeding during treatment still occurs in up to 10.3% of patients (51). There are data specifying that extended anticoagulation is indicated for idiopathic and recurrent VTE, but some patients are too high risk for this treatment because of bleeding concerns. There is emerging experimental evidence distinguishing the molecular and cellular mechanisms of hemostasis and thrombosis (55). MRP-14 is now positioned as a targetable mediator of both arterial and venous thrombosis but not hemostasis (i.e., reduced bleeding risk).
Methods
Materials.
We used the following antibodies: anti-mouse neutrophil-specific marker (clone mAb 7/4, BIO-RAD, MCA771B); anti-mouse macrophage-specific marker Mac-3 (catalog 550292, BD Biosciences); and anti-citrullinated histone 3 (Abcam). Histopaque and Percoll were purchased from Sigma-Aldrich.
Animals.
Mrp-14-/- mice were generated in the laboratory of Nancy Hogg (Leukocyte Adhesion, London Research Institute, London, United Kingdom (5). Mac-1-/- mice were generated in the laboratory of Christie Ballantyne (Baylor College of Medicine, Houston, Texas, USA) (56). All mice had a congenic C57BL/6 background and were maintained in animal facilities at Case Western Reserve University School of Medicine. Male mice (12–15 weeks of age) were used for all DVT experiments.
Induction of stasis-induced DVT.
Stasis venous thrombosis was induced following previously published methods (12, 57). Briefly, once anesthetized, 12- to 15-week-old male mice underwent laparotomy, exteriorization of the intestines, and exposure of the infrarenal IVC. Intestines were kept moist by saline-soaked sterile gauze throughout the procedure. The IVC was then dissected away from the aorta. All visible side branches proximal to the iliac bifurcation were ligated with 7-0 polypropylene sutures. The back branches were left patent. The IVC was then ligated immediately distal to the renal veins with 6-0 polypropylene suture. Exteriorized organs were replaced, and the peritoneum was closed using 6-0 polypropylene in a continuous suture, followed by closure of the skin with a 6-0 nylon suture in a simple interrupted pattern.
Mice were sacrificed at day 2, 4, and 12, and the abdominal IVC (infrarenal to the iliac bifurcation) was harvested. The thrombus was carefully removed from the IVC, rinsed in PBS, and weighed. At the time of resection, a white-light micrograph of the thrombus was captured using a Nikon D70s camera mounted to the dissection scope. Micrographs were calibrated and used to measure thrombus length.
Induction of flow restriction–induced DVT.
Flow restriction (i.e., stenosis) venous thrombosis was induced following previously published methods (3). Thirteen- to fourteen-week-old male mice were used. After exposing the IVC by atraumatic surgery, a space holder (30-gauge, 3-mm long needle) was positioned on the outside of the vessel, and a permanent narrowing ligature (8.0 monofil polypropylene filament) was placed exactly below the left renal vein. Subsequently, the wire was removed to avoid complete vessel occlusion. Mice were sacrificed, IVC was harvested, and thrombus was excised as above.
Cell preparation for adoptive transfer.
Neutrophils were isolated from the blood of donor mice using Percol (P1644, Sigma-Aldrich. PBMCs were isolated using Histopaque (Histopaque-1077 (Sigma-Aldrich) according to the manufacturer’s protocol. Gel-filtrated mouse platelets were prepared using buffer containing prostacyclin-E1 to minimize platelet activation as described previously (11). WT or Mrp-14-/- neutrophils (1 × 106), PBMCs (1 × 106), or platelets (1 × 108) in 250-μl in volume were injected into the recipient Mrp-14-/- mice via tail vein.
In vitro induction of NETosis.
Neutrophils from 2- to 3-month-old age- and sex-matched WT, Mrp-14-/-, or Mac-1-/- mice were isolated from peripheral blood or harvested from the peritoneal cavity of mice after overnight intraperitoneal injection of 1 ml sterile 3% thioglycolate broth (58). Coverslips were coated in 24-well plates with 0.5 ml polylysine (1 mg/ml) overnight at 4°C and blocked with 5% BSA for 60 minutes at room temperature. Cells (5 × 105 cells/ml in culture medium) were allowed to adhere to the coverslips at 37°C for 60 minutes. After washing away nonadherent cells, the remaining cells were incubated with control buffer, ionomycin (4 μM, I24222, Life Technologies)), purified MRP-14 (NBP1-44500, Novus Biologicals) or ionomycin, and purified MRP-14 together for 90–180 minutes and then fixed with 4% formaldehyde for 10 minutes.
Immunofluorescence.
Fixed cells were permeabilized for 20 minutes with 0.5% Triton X-100 and then blocked with 5%–10% BSA overnight. Cells were incubated with antibody against citrullinated histone 3 (3 μg/ml, ab5103) at room temperature for 1 hour. Following the wash, biotin-conjugated secondary antibody was applied in the dark for 60 minutes at room temperature. Following another wash, fluorescein-conjugated (DTAF-conjugated) streptavidin (2 μg/ml, catalog 016-010-084, Jackson ImmunoResearch Laboratories) was added to the cells. DNA was visualized with DAPI containing mounting medium (Vector Laboratories).
Histology and immunohistochemistry.
Thrombosed veins or resected thrombi were carefully cut into 2 segments and processed accordingly for frozen sections or fixation. Thrombi were embedded in optimal cutting temperature media and flash frozen using a dry ice/isopentane slurry. Six-μm cryosections were cut for all histological analyses. Thrombi were also immersed in 4% paraformaldehyde in 0.1 M phosphate buffer, pH 7.3, embedded in paraffin and sectioned (5 μm). For immunohistochemistry, standard avidin-biotin procedures (DAKO) for mouse-specific neutrophil marker (mAb 7/4, 2.5 μg/ml) and macrophage-specific marker (Mac-3, 0.62 μg/ml) were used. For each antibody, controls included species-specific nonimmune IgG as well as omission of the primary antibody. A histologist blinded to genotype analyzed staining using a microscope equipped with a charge-coupled device camera (Zeiss AxioCam MRc5 and Axiovision 4.8) interfaced to a computer.
Analysis of complete blood count in thrombi.
Mice (2–3 months old, both male and female) were euthanized 2 days after IVC ligation. Thrombi were carefully excised and homogenized, and cells were dispersed in 1–2 ml of Tyrode’s buffer. Fifty μl of the resulting solution was applied to a CBC analyzer (HV950FS, Drew Scientific Inc.). The total number of cells was quantified and also normalized by dividing the total number by thrombus weight.
Statistics.
Data are presented as mean ± SD. Comparisons between two groups were performed by unpaired, 2-tailed Student’s t test using Excel. Comparisons among multiple groups were performed by 1-way ANOVA followed by post-hoc test using Bonferroni criterion (SPSS, version 24, IBM). P values of less than 0.05 were considered significant.
Study approval.
Animal care and procedures for this study were reviewed and approved by the Case Western Reserve University School of Medicine Institutional Animal Care and Use Committees and performed in accordance with the guidelines of the American Association for Accreditation of Laboratory Animal Care and the National Institutes of Health.
Author contributions
YW designed the research, performed experiments, analyzed data, and wrote the paper; HG and CWK performed experiments; and AS, FAJ, and DIS designed the research and wrote the paper.
Acknowledgments
This work was supported in part by National Institutes of Health grants to AS (HL052779), FAJ (HL122388), and DIS (R37 HL57506 MERIT Award and R01 HL126645) and an American Heart Association Grant-in-Aid to FAJ (13GRNT1760040).
Footnotes
Conflict of interest: Y. Wang and D.I. Simon are coinventors of technology related to MRP-14 and thrombosis that is assigned to Case Western Reserve University.
Reference information:JCI Insight. 2017;2(11):e91356. https://doi.org/10.1172/jci.insight.91356.
Contributor Information
Yunmei Wang, Email: yunmei.wang@case.edu.
Huiyun Gao, Email: huiyun.gao@case.edu.
Alvin Schmaier, Email: schmaier@case.edu.
References
- 1.Goldhaber SZ, Bounameaux H. Pulmonary embolism and deep vein thrombosis. Lancet. 2012;379(9828):1835–1846. doi: 10.1016/S0140-6736(11)61904-1. [DOI] [PubMed] [Google Scholar]
- 2.Beckman MG, Hooper WC, Critchley SE, Ortel TL. Venous thromboembolism: a public health concern. Am J Prev Med. 2010;38(4 Suppl):S495–S501. doi: 10.1016/j.amepre.2009.12.017. [DOI] [PubMed] [Google Scholar]
- 3.von Brühl ML, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. 2012;209(4):819–835. doi: 10.1084/jem.20112322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Healy AM, et al. Platelet expression profiling and clinical validation of myeloid-related protein-14 as a novel determinant of cardiovascular events. Circulation. 2006;113(19):2278–2284. doi: 10.1161/CIRCULATIONAHA.105.607333. [DOI] [PubMed] [Google Scholar]
- 5.Hobbs JA, et al. Myeloid cell function in MRP-14 (S100A9) null mice. Mol Cell Biol. 2003;23(7):2564–2576. doi: 10.1128/MCB.23.7.2564-2576.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vogl T, et al. MRP8 and MRP14 control microtubule reorganization during transendothelial migration of phagocytes. Blood. 2004;104(13):4260–4268. doi: 10.1182/blood-2004-02-0446. [DOI] [PubMed] [Google Scholar]
- 7.Lackmann M, et al. Identification of a chemotactic domain of the pro-inflammatory S100 protein CP-10. J Immunol. 1993;150(7):2981–2991. [PubMed] [Google Scholar]
- 8.Kerkhoff C, Sorg C, Tandon NN, Nacken W. Interaction of S100A8/S100A9-arachidonic acid complexes with the scavenger receptor CD36 may facilitate fatty acid uptake by endothelial cells. Biochemistry. 2001;40(1):241–248. doi: 10.1021/bi001791k. [DOI] [PubMed] [Google Scholar]
- 9.Vogl T, et al. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med. 2007;13(9):1042–1049. doi: 10.1038/nm1638. [DOI] [PubMed] [Google Scholar]
- 10.Boyd JH, Kan B, Roberts H, Wang Y, Walley KR. S100A8 and S100A9 mediate endotoxin-induced cardiomyocyte dysfunction via the receptor for advanced glycation end products. Circ Res. 2008;102(10):1239–1246. doi: 10.1161/CIRCRESAHA.107.167544. [DOI] [PubMed] [Google Scholar]
- 11.Wang Y, et al. Platelet-derived S100 family member myeloid-related protein-14 regulates thrombosis. J Clin Invest. 2014;124(5):2160–2171. doi: 10.1172/JCI70966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kessinger CW, et al. Statins improve the resolution of established murine venous thrombosis: reductions in thrombus burden and vein wall scarring. PLoS One. 2015;10(2):e0116621. doi: 10.1371/journal.pone.0116621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Croce K, et al. Myeloid-related protein-8/14 is critical for the biological response to vascular injury. Circulation. 2009;120(5):427–436. doi: 10.1161/CIRCULATIONAHA.108.814582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schnekenburger J, et al. The calcium binding protein S100A9 is essential for pancreatic leukocyte infiltration and induces disruption of cell-cell contacts. J Cell Physiol. 2008;216(2):558–567. doi: 10.1002/jcp.21433. [DOI] [PubMed] [Google Scholar]
- 15.Fuchs TA, Brill A, Wagner DD. Neutrophil extracellular trap (NET) impact on deep vein thrombosis. Arterioscler Thromb Vasc Biol. 2012;32(8):1777–1783. doi: 10.1161/ATVBAHA.111.242859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Savchenko AS, et al. Neutrophil extracellular traps form predominantly during the organizing stage of human venous thromboembolism development. J Thromb Haemost. 2014;12(6):860–870. doi: 10.1111/jth.12571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Y, et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol. 2009;184(2):205–213. doi: 10.1083/jcb.200806072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neeli I, Dwivedi N, Khan S, Radic M. Regulation of extracellular chromatin release from neutrophils. J Innate Immun. 2009;1(3):194–201. doi: 10.1159/000206974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Newton RA, Hogg N. The human S100 protein MRP-14 is a novel activator of the beta 2 integrin Mac-1 on neutrophils. J Immunol. 1998;160(3):1427–1435. [PubMed] [Google Scholar]
- 20.Brill A, et al. von Willebrand factor-mediated platelet adhesion is critical for deep vein thrombosis in mouse models. Blood. 2011;117(4):1400–1407. doi: 10.1182/blood-2010-05-287623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hogg N, Allen C, Edgeworth J. Monoclonal antibody 5.5 reacts with p8,14, a myeloid molecule associated with some vascular endothelium. Eur J Immunol. 1989;19(6):1053–1061. doi: 10.1002/eji.1830190615. [DOI] [PubMed] [Google Scholar]
- 22.Hogg N, Palmer DG, Revell PA. Mononuclear phagocytes of normal and rheumatoid synovial membrane identified by monoclonal antibodies. Immunology. 1985;56(4):673–681. [PMC free article] [PubMed] [Google Scholar]
- 23.Wilkinson MM, Busuttil A, Hayward C, Brock DJ, Dorin JR, Van Heyningen V. Expression pattern of two related cystic fibrosis-associated calcium-binding proteins in normal and abnormal tissues. J Cell Sci. 1988;91(Pt 2):221–230. doi: 10.1242/jcs.91.2.221. [DOI] [PubMed] [Google Scholar]
- 24.Brandtzaeg P, Dale I, Fagerhol MK. Distribution of a formalin-resistant myelomonocytic antigen (L1) in human tissues. II. Normal and aberrant occurrence in various epithelia. Am J Clin Pathol. 1987;87(6):700–707. doi: 10.1093/ajcp/87.6.700. [DOI] [PubMed] [Google Scholar]
- 25.Nacken W, Sopalla C, Pröpper C, Sorg C, Kerkhoff C. Biochemical characterization of the murine S100A9 (MRP14) protein suggests that it is functionally equivalent to its human counterpart despite its low degree of sequence homology. Eur J Biochem. 2000;267(2):560–565. doi: 10.1046/j.1432-1327.2000.01040.x. [DOI] [PubMed] [Google Scholar]
- 26.Baker JR, et al. Distinct roles for S100a8 in early embryo development and in the maternal deciduum. Dev Dyn. 2011;240(9):2194–2203. doi: 10.1002/dvdy.22709. [DOI] [PubMed] [Google Scholar]
- 27.Manitz MP, et al. Loss of S100A9 (MRP14) results in reduced interleukin-8-induced CD11b surface expression, a polarized microfilament system, and diminished responsiveness to chemoattractants in vitro. Mol Cell Biol. 2003;23(3):1034–1043. doi: 10.1128/MCB.23.3.1034-1043.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McNeill E, Conway SJ, Roderick HL, Bootman MD, Hogg N. Defective chemoattractant-induced calcium signalling in S100A9 null neutrophils. Cell Calcium. 2007;41(2):107–121. doi: 10.1016/j.ceca.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 29.Klempt M, Melkonyan H, Nacken W, Wiesmann D, Holtkemper U, Sorg C. The heterodimer of the Ca2+-binding proteins MRP8 and MRP14 binds to arachidonic acid. FEBS Lett. 1997;408(1):81–84. doi: 10.1016/S0014-5793(97)00394-3. [DOI] [PubMed] [Google Scholar]
- 30.Siegenthaler G, et al. A heterocomplex formed by the calcium-binding proteins MRP8 (S100A8) and MRP14 (S100A9) binds unsaturated fatty acids with high affinity. J Biol Chem. 1997;272(14):9371–9377. doi: 10.1074/jbc.272.14.9371. [DOI] [PubMed] [Google Scholar]
- 31.Kerkhoff C, Eue I, Sorg C. The regulatory role of MRP8 (S100A8) and MRP14 (S100A9) in the transendothelial migration of human leukocytes. Pathobiology. 1999;67(5-6):230–232. doi: 10.1159/000028098. [DOI] [PubMed] [Google Scholar]
- 32.Roulin K, Hagens G, Hotz R, Saurat JH, Veerkamp JH, Siegenthaler G. The fatty acid-binding heterocomplex FA-p34 formed by S100A8 and S100A9 is the major fatty acid carrier in neutrophils and translocates from the cytosol to the membrane upon stimulation. Exp Cell Res. 1999;247(2):410–421. doi: 10.1006/excr.1998.4382. [DOI] [PubMed] [Google Scholar]
- 33.Hetland G, Talgö GJ, Fagerhol MK. Chemotaxins C5a and fMLP induce release of calprotectin (leucocyte L1 protein) from polymorphonuclear cells in vitro. MP, Mol Pathol. 1998;51(3):143–148. doi: 10.1136/mp.51.3.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Voganatsi A, Panyutich A, Miyasaki KT, Murthy RK. Mechanism of extracellular release of human neutrophil calprotectin complex. J Leukoc Biol. 2001;70(1):130–134. [PubMed] [Google Scholar]
- 35.Edgeworth J, Gorman M, Bennett R, Freemont P, Hogg N. Identification of p8,14 as a highly abundant heterodimeric calcium binding protein complex of myeloid cells. J Biol Chem. 1991;266(12):7706–7713. [PubMed] [Google Scholar]
- 36.Rammes A, Roth J, Goebeler M, Klempt M, Hartmann M, Sorg C. Myeloid-related protein (MRP) 8 and MRP14, calcium-binding proteins of the S100 family, are secreted by activated monocytes via a novel, tubulin-dependent pathway. J Biol Chem. 1997;272(14):9496–9502. doi: 10.1074/jbc.272.14.9496. [DOI] [PubMed] [Google Scholar]
- 37.Srikrishna G, Panneerselvam K, Westphal V, Abraham V, Varki A, Freeze HH. Two proteins modulating transendothelial migration of leukocytes recognize novel carboxylated glycans on endothelial cells. J Immunol. 2001;166(7):4678–4688. doi: 10.4049/jimmunol.166.7.4678. [DOI] [PubMed] [Google Scholar]
- 38.Robinson MJ, Tessier P, Poulsom R, Hogg N. The S100 family heterodimer, MRP-8/14, binds with high affinity to heparin and heparan sulfate glycosaminoglycans on endothelial cells. J Biol Chem. 2002;277(5):3658–3665. doi: 10.1074/jbc.M102950200. [DOI] [PubMed] [Google Scholar]
- 39.Brill A, et al. Neutrophil extracellular traps promote deep vein thrombosis in mice. J Thromb Haemost. 2012;10(1):136–144. doi: 10.1111/j.1538-7836.2011.04544.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brinkmann V, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303(5663):1532–1535. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 41.Clark SR, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. 2007;13(4):463–469. doi: 10.1038/nm1565. [DOI] [PubMed] [Google Scholar]
- 42.Fuchs TA, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. 2007;176(2):231–241. doi: 10.1083/jcb.200606027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li P, Li M, Lindberg MR, Kennett MJ, Xiong N, Wang Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med. 2010;207(9):1853–1862. doi: 10.1084/jem.20100239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Plow EF. The major fibrinolytic proteases of human leukocytes. Biochim Biophys Acta. 1980;630(1):47–56. doi: 10.1016/0304-4165(80)90136-1. [DOI] [PubMed] [Google Scholar]
- 45.Morrow DA, et al. Myeloid-related protein 8/14 and the risk of cardiovascular death or myocardial infarction after an acute coronary syndrome in the Pravastatin or Atorvastatin Evaluation and Infection Therapy: Thrombolysis in Myocardial Infarction (PROVE IT-TIMI 22) trial. Am Heart J. 2008;155(1):49–55. doi: 10.1016/j.ahj.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lloyd-Jones D, et al. Heart disease and stroke statistics--2009 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2009;119(3):e21–181. doi: 10.1161/CIRCULATIONAHA.108.191261. [DOI] [PubMed] [Google Scholar]
- 47.Eikelboom JW, Mehta SR, Anand SS, Xie C, Fox KA, Yusuf S. Adverse impact of bleeding on prognosis in patients with acute coronary syndromes. Circulation. 2006;114(8):774–782. doi: 10.1161/CIRCULATIONAHA.106.612812. [DOI] [PubMed] [Google Scholar]
- 48.Stone GW, et al. Bivalirudin during primary PCI in acute myocardial infarction. N Engl J Med. 2008;358(21):2218–2230. doi: 10.1056/NEJMoa0708191. [DOI] [PubMed] [Google Scholar]
- 49.Mehran R, et al. Bivalirudin in patients undergoing primary angioplasty for acute myocardial infarction (HORIZONS-AMI): 1-year results of a randomised controlled trial. Lancet. 2009;374(9696):1149–1159. doi: 10.1016/S0140-6736(09)61484-7. [DOI] [PubMed] [Google Scholar]
- 50.Schulman S, et al. Dabigatran versus warfarin in the treatment of acute venous thromboembolism. N Engl J Med. 2009;361(24):2342–2352. doi: 10.1056/NEJMoa0906598. [DOI] [PubMed] [Google Scholar]
- 51.EINSTEIN–PE Investigators, et al. Oral rivaroxaban for the treatment of symptomatic pulmonary embolism. N Engl J Med. 2012;366(14):1287–1297. doi: 10.1056/NEJMoa1113572. [DOI] [PubMed] [Google Scholar]
- 52.Agnelli G, et al. Oral apixaban for the treatment of acute venous thromboembolism. N Engl J Med. 2013;369(9):799–808. doi: 10.1056/NEJMoa1302507. [DOI] [PubMed] [Google Scholar]
- 53.Hokusai-VTE Investigators, et al. Edoxaban versus warfarin for the treatment of symptomatic venous thromboembolism. N Engl J Med. 2013;369(15):1406–1415. doi: 10.1056/NEJMoa1306638. [DOI] [PubMed] [Google Scholar]
- 54.Yeh CH, Hogg K, Weitz JI. Overview of the new oral anticoagulants: opportunities and challenges. Arterioscler Thromb Vasc Biol. 2015;35(5):1056–1065. doi: 10.1161/ATVBAHA.115.303397. [DOI] [PubMed] [Google Scholar]
- 55.Sachs UJ, Nieswandt B. In vivo thrombus formation in murine models. Circ Res. 2007;100(7):979–991. doi: 10.1161/01.RES.0000261936.85776.5f. [DOI] [PubMed] [Google Scholar]
- 56.Ding ZM, et al. Relative contribution of LFA-1 and Mac-1 to neutrophil adhesion and migration. J Immunol. 1999;163(9):5029–5038. [PubMed] [Google Scholar]
- 57.Henke PK, et al. Deep vein thrombosis resolution is modulated by monocyte CXCR2-mediated activity in a mouse model. Arterioscler Thromb Vasc Biol. 2004;24(6):1130–1137. doi: 10.1161/01.ATV.0000129537.72553.73. [DOI] [PubMed] [Google Scholar]
- 58.Wang Y, et al. Leukocyte engagement of platelet glycoprotein Ibalpha via the integrin Mac-1 is critical for the biological response to vascular injury. Circulation. 2005;112(19):2993–3000. doi: 10.1161/CIRCULATIONAHA.105.571315. [DOI] [PubMed] [Google Scholar]