

Abstract
Background
Lymphotoxin alpha (LTα) is expressed in human atherosclerotic lesions and genetic variations in the LTα pathway have been linked to myocardial infarction. Activation of the P2Y2 nucleotide receptor (P2Y2R) regulates the production of LTα. in vitro. We aimed to uncover a potential pathway linking purinergic receptor to LTα-mediated inflammatory processes pivotal to the early stages of atherosclerosis in apolipoprotein E (ApoE−/−) deficient mice.
Methods and results
En face immunostaining revealed that P2Y2R and VCAM-1 are preferentially expressed in the atherosclerosis prone site of the mouse aortic sinus. Deletion of the P2Y2R gene suppresses VCAM-1 expression. Compared with ApoE−/−mice, ApoE−/−mice lacking the P2Y2R gene (ApoE−/−/P2Y2R−/−) did not develop fatty streak lesions when fed a standard chow diet for 15 weeks. Systemic and CD4+ T cell production of the pro-inflammatory cytokine lymphotoxin-alpha (LTα) were specifically inhibited in ApoE−/−/P2Y2R−/− mice. Anti-LTα preventive treatment was initiated in ApoE−/− mice with intraperitoneal administration of recombinant human tumor necrosis factor receptor 1 fusion protein (TNFR1-Fc) on 5 consecutive days before the disease onset. Remarkably, none of the TNFR1:Fc-treated ApoE−/− mice exhibited atherosclerotic lesions at any developmental stage.
Significance
ApoE−/− mice deficient in P2Y2R exhibit low endothelial cell VCAM-1 levels, decreased production of LTα and delayed onset of atherosclerosis. These data suggest that targeting this nucleotide receptor could be an effective therapeutic approach in atherosclerosis.
1. Introduction
Atherosclerosis is widely considered to be an inflammatory disease involving the recruitment of leukocytes, and the activation of pro-thrombotic pathways [1]. Targeted control of pro-inflammatory factors that have the potential to support endothelial dysfunction could have a significant impact on the limitation of vascular complications. The cardiovascular purinergic signaling system is a promising source for novel drug targets. Clinical trials have provided clear evidence that purinergic antithrombotic drugs reduce the risks of recurrent strokes and heart attacks [2–3]. These drugs are antagonists to the P2Y12 receptor that mediates platelet aggregation [4].
Leukocyte adherence to the endothelium in lesion-prone areas of the arterial wall is one of the earliest cellular responses in the formation of lesions of atherosclerosis [5]. Vascular cell adhesion molecule-1 (VCAM-1) is particularly important for firm, integrin-mediated adhesion of leukocyte to endothelial cells (EC) and subsequent trans-endothelial migration [6]. Under traumatic arterial events, nucleotide release activates a specific nucleotide receptor subtype, the P2Y2R, leading to the transmigration of blood-derived cells into the vessel wall and subsequent development of intimal hyperplasia [7]. Comparison of signal transduction pathways for wild type and different mutant P2Y2R constructs have identified structural features that enable the P2Y2R to regulate VCAM-1 expression on ECs [8].
Stimulation of P2 receptors is coupled to the release of pro-inflammatory cytokines [9–12] that are of obvious relevance to the development of atherosclerosis. In particular, activation of the P2Y2R has been shown to regulate the production lymphotoxin alpha (LTα) in macrophages [13]. LTα is a member of the TNF ligand family and is synthesized predominantly by activated T-and B-lymphocytes [14]. Genetic variations in the LTα pathway have been linked to myocardial infarction [15]. Because these findings imply a role for P2Y2R in vascular inflammation, we investigated its contribution to the early stages of development of atherosclerosis and tested whether blocking specific signal pathways linked to the activation of this nucleotide receptor delays the onset of atherosclerosis in mice.
Our data show that deletion of the P2Y2R gene modulates inflammatory processes pivotal to the development of early stage atherosclerosis, thus limiting atherosclerotic plaque formation in ApoE−/− mice. This conclusion is supported by evidence showing that loss of P2Y2R re-presses VCAM-1 expression in the lesion prone site of the aortic sinus, and selectively inhibits the production of the pro-inflammatory cytokine LTα.
Subsequently, we showed that short-term treatment with soluble recombinant human LTα antagonist inhibits fatty streak formation in ApoE-deficient mice.
This work defines a new pathway linking purinergic receptors to LTα-mediated inflammatory processes pivotal to the development of atherosclerosis.
2. Materials and methods
2.1. Animals
Animal protocols were approved by the Animal Care and Use Committee of Indiana University. C57BL/6, ApoE−/− and P2Y2R−/− mice were purchased from Jackson laboratory. P2Y2R−/−mice were bred to the ApoE−/−background to generate ApoE−/−/P2Y2R−/− mice. All animal were fed with a standard chow diet. Only males were used in experimental groups. Recombinant human TNFR1:Fc (100 μg per mouse per day, n = at least 12 mice per group) was administered intraperitoneally on 5 consecutive days beginning at 5 weeks of age. For control, PBS or 100 μg of an irrelevant human IgG was used. Following treatment, mice were maintained on standard chow diet until sacrifice at week 15.
2.2. Antibody production
A rabbit polyclonal anti-P2Y2 receptor antibodies were raised against a keyhole limpet hemocyanin-conjugated peptide (NRTVRKDLSVSSDD) corresponding to amino acids 342–355 of the mouse P2Y2R amino acid sequence (NM_01302347). Peptide synthesis, animal immunization, and sera collections were performed by Genscript (Piscataway, NJ). The antibodies were purified by affinity chromatography using an antigen peptide-conjugated column.
The validity of the newly developed P2Y2R polyclonal antibody was tested by immunohistochemistry on cross sections of aortic sinus samples from wild-type and P2Y2R−/− mice. As shown in the Online figure, both endothelial cells and smooth muscle cells in wild-type mice expressed P2Y2R. In contrast, no P2Y2R-positive staining was detectable in either cell type, thus confirming the specificity of this antibody.
2.3. Histological analysis and quantification of aortic sinus lesions
Mouse hearts (n = 12 at least) were perfused, fixed in 4% paraformaldehyde and embedded in paraffin. Sections were made and discarded until the valve cups became visible. At that point, 5-μm sections were then collected and stained with Masson’s Trichrome to identify collagen and muscle fibers. The lesions were classified into five different categories as previously described [16] to examine if there were any differences in lesion severity.
For the quantification of atherosclerotic lesions, hearts were embedded in OCT and frozen sections were stained with Oil Red O. The relative lesion area was calculated by dividing the lesion area by the total cross-sectional area. The lesion area was measured with image analysis (MetaMorph software, Universal Imaging Corp, Downingtown, Pa).
2.4. Immunohistochemical analysis of aortic sinus lesions
Immunohistochemical staining was performed by the labeled streptavidin biotin method. A rabbit polyclonal antibody against human smooth muscle α-actin was used to detect smooth muscle cells (Sigma, 1/1000 dilution). VCAM-1 immunostaining was performed on frozen sections with a rat anti-mouse VCAM-1 monoclonal antibody (BD Pharmingen; 1/200 dilution). Macrophages were identified by immunostaining with a rat anti-mouse Mac-3 monoclonal antibody (M3/84, 550292, BD PharMingen, San Diego, CA; 1/100 dilution).
2.5. Lymphocyte preparation and cytokine analysis
Purified CD4+ cells were obtained from single cell suspensions of splenocytes by negative selection using the EasySep™ Mouse CD4+ T Cell Isolation Kit (StemCell Technologies). CD4+ T cells (1 × 106/ml) were cultured in 48-well culture plates in 0.5 ml of RPMI 1640 medium containing 10% fetal calf serum, 5 mmol/L-glutamine, 50 IU penicillin G, and 50 μg streptomycin and were stimulated with 5 μg/ml plate- bound anti-CD3 mAb (BD Biosciences, San Diego, CA) for 1–3 days. Concentrations of LTα in CD4+ T cell supernatant or mouse blood plasma were determined with ELISA using a kit (#MBS266789, BioSource).
2.6. Real-time polymerase chain reaction
Total RNA was extracted from aortic sinus samples using RNeasy mini kit (Qiagen). High quality RNA extracted was used for synthesis of single-strand cDNA with Superscript II (Invitrogen). Real time PCR was conducted with 45 ng of cDNA using an ABI Prism 7900HT PCR machine (Applied Biosystems). The sequences of PCR primers for VCAM-1 were 5′-CATGGAATTCGAACCCAAACA-30 (sense) and 5′-GGCTGACCAAGACGGTTGTATC-3′ (anti-sense) as described elsewhere (15). The following primer sequences were used for cytokines mRNA amplification: TNF-α-5′-ATGAGCACAGAAAGCATGATCCGCGAC-3′ (sense), 5′-TCACAGAGCAATGACTCCAAAGTAGACCTG-3′ (antisense); TNF-β-5′-CCCATGGCATCCTGAAAC-3′ (sense), 5′-GGAGGCCTGGAA TCCAAT-3′ (antisense).
The PCR primer sequences for GAPDH were 5′-TGACG TGCCGCCTGGAGAAAC-3′ (sense) and 5′-CCGGCATCGAAGGT GGAAG AGT-3′ (anti-sense).
2.7. En face immunofluorescent analysis of VCAM-1 expression
Aortic tissue segments were opened longitudinally and incubated with PBS-T containing 10% donkey serum. Tissue segments were then rinsed 3 times with PBS-T and incubated overnight at 4 °C with anti-mouse VCAM-1 polyclonal antibodies (Santa Cruz Biotechnology Inc; 1:100) diluted in PBS-T. Anti-donkey secondary antibody labeled with Alexa Fluor fluorophore molecules (Molecular Probes Inc.) was then applied for 45 min at room temperature. The segments were mounted on slides, covered with Vectorshield mounting medium (Vector Laboratories Inc) and observed with a FV1000 confocal system (Olympus). At least 4 animals were used. Five pictures of each field were captured at different focal lengths and VCAM-1 positive cells per 1000 endothelial cells were counted.
2.8. Promoter reporter assay
Murine VCAM-1 promoter sequence has been documented [17] and is available on NCBI (accession number U42327). Genomic DNA was obtained from C57BL/6 mice. The VCAM-1 promoter was amplified by PCR as previously described [18] and cloned into the pGL3-Basic vector (Promega) containing Firefly luciferase reporter gene. The resultant construct was co-transfected with the pRL-TK Vector (Promega) containing Renilla luciferase reporter gene into the cultured ECs. A Nucleofector electroporator and a basic Nucleofector kit (Amaxa Inc., Gaithersburg, MD) were used for transfection of primary ECs according to the instructions of the manufacturer. Transfected ECs were first incubated with or without the addition of UTP. Transfected cells were then lysed, and the Firefly-to-Renilla luciferase activity ratios in the lysates were measured with dual-luciferase assay kit (Promega) for evaluation of VCAM-1 gene promoter activity. Five independent experiments in duplicates were performed.
2.9. Cell culture
Mouse aortic ECs were isolated and cultured as described previously [19]. Briefly, aortas cut into circular rings were opened longitudinally and placed luminal side down onto collagen gels. After 10 days, aortic segments were removed, and collagen gels were digested with 0.3% collagenase H. ECs were grown in complete medium and used at passages 2 to 4.
2.10. Statistical analysis
All results are expressed as mean ± SEM. Differences between the values of P < 0.05 were considered significant with 2-tailed Student t test.
3. Results
3.1. P2Y2 receptor is preferentially expressed in atherosclerosis susceptible region of the aorta
We confirmed previous studies that show that VCAM-1 is preferentially expressed in endothelial cells at lesion-prone sites [20]. As demonstrated by en face immunostaining, VCAM-1 was abundantly expressed in sinus aortic ECs of wild-type mice (Fig. 1A). In contrast, VCAM-1 was not detected in the descending thoracic aorta (Fig. 1A). Flow cytometry substantiated these results by demonstrating that the number of cells expressing surface VCAM-1 was higher in aortic sinus ECs as compared with ECs from the descending thoracic aorta (Fig. 1B). As P2Y2R expressed in the vascular endothelium is known to regulate VCAM-1 expression [8,21], we examined the spatial distribution of P2Y2R in the mouse aorta using a newly developed antibody. En face immunostaining showed that P2Y2R was abundantly expressed in ECs at the curvature of the aortic arch as compared to a more modest expression found in the region of the descending thoracic aorta (Fig. 2A–B). These results indicate P2Y2R is highly expressed in the VCAM-1 enriched atherosclerosis prone-region of the aorta.
Fig. 1.

Differential expression of VCAM-1 in regions of the mouse aorta. A, representative en face immunostaining for VCAM-1 expression in the aorta of wild-type mice. Endothelial cell nuclei and VCAM-1 are in blue and green, respectively. Scale bar, 20 μm. P < 0.05. n = 15 visual fields. B, Flow cytometry analysis of EC surface expression of VCAM-1 in the aortic sinus and descending thoracic aorta of wild-type mice. Graphs show that the number of cells expressing VCAM-1 on the cell surface was greater in the aortic sinus as compared to the descending aorta. Endothelial cells incubated with only a second antibody were used as controls.
Fig. 2.
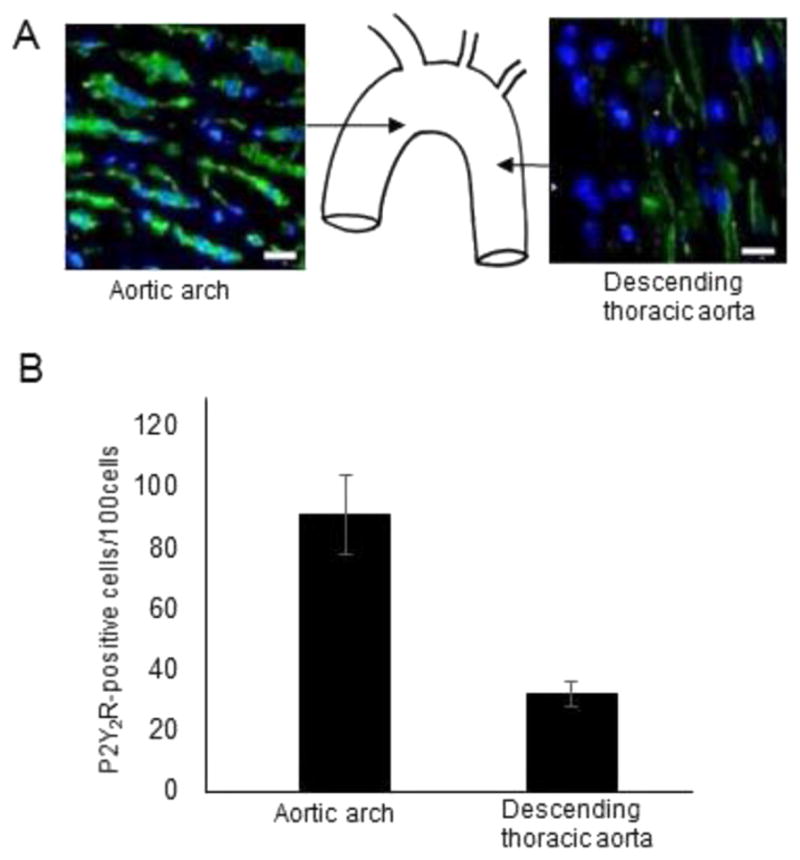
High P2Y2R expression in the inner curvature of the aortic arch in wild-type mice. A–B, representative en face images and quantitative histograms of P2Y2R-positive cells in different areas of the aortic sinus and descending thoracic aorta represented in the schematic. Endothelial cell nuclei and P2Y2R are in blue and green, respectively. Scale bar, 20 μm. P < 0.05. n = 15 visual fields from 4 different mice.
3.2. P2Y2 receptor gene deletion suppresses basal expression of VCAM-1 in murine endothelium
To determine if P2Y2R signaling is responsible for the high levels of VCAM-1 expression in the lesion-prone aortic arch, we examined the effects of P2Y2R gene deletion on VCAM-1 expression. Real time-PCR revealed a striking decrease in VCAM-1 mRNA levels in the aortic sinus of P2Y2R−/− mice as compared to wild-type mice (Fig. 3A). Remarkably, ECs in the aortic sinus of P2Y2R−/− exhibited no detectable VCAM-1 immuno-staining (Fig. 3B). The complete lack of VCAM1 expression suggests P2Y2R plays a dominant role in VCAM1 expression in the endothelial cells of the aortic sinus.
Fig. 3.

Effect of P2Y2R gene deletion on VCAM-1 in different regions of the mouse aorta. A, Real-time PCR analysis of VCAM-1 mRNA levels in the aortic sinus of wild-type (n = 4) and P2Y2R−/− (n = 3) mice. P < 0.05. B, representative en face immunostaining for VCAM-1 expression in the arch and descending thoracic aorta in P2Y2R−/− mice. No VCAM-1-positive cell was detected in either part of the aorta. Endothelial cell nuclei are in shown blue. Scale bar, 20 μm. P < 0.05. n = 15 visual fields from 4 different mice.
3.3. Nucleotide-dependent transcriptional regulation of the VCAM-1 gene is mediated by P2Y2 receptor
We previously reported that P2Y2R activation increased expression of VCAM-1 [8,21]. To explore the mechanism of nucleotide-induced upregulation of VCAM-1, we examined the effect of U UTPγS documented. A plasmid containing a luciferase reporter gene driven by a murine VCAM-1 promoter segment [17] was used to examine the effect of the P2Y2R agonist UTPγS on VCAM-1 promoter activity. Aortic sinus endothelial cells were transiently transfected with the pGL3- VCAM-1 luciferase reporter gene. As shown in Fig. 4, UTPγS induced a dose-dependent stimulation of reporter gene activity, with maximal induction occurring at a concentration of 10−3 mol/L (Fig. 4). In contrast, UTPγS has no such effect on VCAM-1 promoter activity in P2Y2R−/− ECs (Fig. 4). These data indicate that P2Y2R regulates transcription of VCAM-1 in ECs.
Fig. 4.

VCAM-1 promoter reporter assay. Luciferase activity in endothelial cells transfected with a pGL3 vector containing murine VCAM-1 promoter and stimulated with UTP-γ-S. T Error bars represent standard error of mean from triplicates. P < 0.05.
3.4. P2Y2 receptor deficiency prevents fatty streak formation in ApoE−/− mice
Because endothelial cell surface expression of VCAM-1 is one of the initial steps in the pathogenesis of atherosclerosis, we asked whether P2Y2R may contribute to atherosclerosis disease onset. To do this we examined the developmental of early atherosclerosis in ApoE−/− and ApoE−/−/P2Y2R−/− mice fed a standard chow diet for 15 weeks. There was no statistical differences in body weight or plasma lipid profiles between the two genotypes (Table 1). A gross morphological evaluation revealed the presence of characteristic fatty streak lesions in ApoE−/−mice, as previously defined [16] (Fig. 5A; Table 2). Fatty streak lesions were observed in all ApoE−/− mice examined (n = 12) and consisted of a mixture of macrophages and a few smooth muscle cells with deposition of connective tissue matrix (Fig. 5A). We observed strong.
Table 1.
Body weight and plasma lipid analysis in chow-fed ApoE−/− and ApoE−/−/P2Y2R−/− mice.
| Week 15 (n = 12) | ApoE−/− | ApoE−/−/P2Y2R−/− |
|---|---|---|
| Body weight (g) | 28.5 ± 0.6 | 31.2 ± 0.5 |
| Total Cholesterol (mg/dl) | 409 ± 32 | 388 ± 25 |
| Triglycerides (mg/dl) | 58 ± 6 | 63 ± 8 |
Fig. 5.

Analysis of atherosclerotic lesions in the aortic sinus of male ApoE−/− and ApoE−/−/P2Y2R−/− littermate mice fed standard chow diet for 15 weeks. A, Aortic sinus cross sections stained with Masson’s trichrome for gross morphological analysis and classification of lesions as shown in Table 2. B–C, Cross sections were stained with Oil Red O and the relative lesion area was calculated by dividing the lesion area by the total cross-sectional area. D, Representative images of immunohistological staining of atherosclerotic lesions in the aortic sinus. Adjacent sections were stained with, Mac-3 antibody, VCAM-1 and smooth muscle α-actin antibodies, respectively. Each scale bar represents 100 μm.
Table 2.
Classification of atherosclerotic lesions in chow-fed ApoE−/− and ApoE−/−/P2Y2R−/− mice.
| Standard diet (n = 12) (n = 12) | ApoE−/− | ApoE−/−/P2Y2R−/− | |
|---|---|---|---|
| No lesion | – | 9 | No foam cell |
| Category 1 formation | – | 3 | Early foam cell |
| Category 2 | 12 | – | Regular fatty streaks |
| Category 3 | – | – | Mild plaque |
| Category 4 | – | – | Moderate plaque |
| Category 5 | – | – | Severe plaque |
Sinus aortic lesions were classified into the following categories as previously described [16]. (1) Early foam cell formation: up to 10 foam cell per section are present in the intima; (2) regular fatty streaks: >10 cells present in the intima; (3) mild plaque: extension of foam cells into the media: (4) moderate plaque: foam cells in the media, fibrosis, cholesterol clefts, mineralization and/or necrosis of the media; (5) severe plaque: similar to category 4 but more extensive and deeper into the media.
VCAM-1 immunoreactivity in the intima and underlying media layer (Fig. 5A–D). Cross sections of aortic sinus specimens stained with Oil-red-O showed a mean lesion area of 0.06 mm2 ± 0.005 in these ApoE−/− mice (Fig. 5B, p < 0.05). By comparison, only 25% of the ApoE−/−/P2Y2R−/− mice (3/12 animals) exhibited any type of atherosclerotic lesion in the aortic sinus (Fig. 5A; Table 2). Moreover the lesions that formed were more immature and characterized by having less than 10 foam cells/cross section (Table 2). As expected, normal C57BL/6 mice did not develop any intimal lesions (Fig. 5A). Further examination of sections collected at every 100 μm over a 1-mm segment from the beginning of the aortic root showed no evidence of lesion formation in the descending aortic arch, thus ruling out the possibility that P2Y2R deficiency results in atherosclerosis onset at an alternative site at the aortic root. These findings demonstrate that loss of P2Y2R attenuates atherosclerosis development in ApoE−/− mice and prevents the formation of fatty streaks.
3.5. Inhibition of systemic production of LTα in ApoE−/− mice lacking the P2Y2 receptor gene
In addition to promoting VCAM-1 expression, P2Y2R activation induces expression and secretion of the inflammatory cytokine LTα in mouse peritoneal macrophages [13]. We thus asked whether P2Y2R deficiency affects systemic cytokine levels in ApoE−/− mice. ELISA was used to measure LTα and TNF-α plasma levels of individual standard chow-fed mice at 4 weeks of age (baseline) and 15 weeks. The mean plasma cytokine levels measured in the same mice at both time points are summarized in Table 3. The mean plasma levels of TNF-α in ApoE−/− and ApoE−/−/P2Y2R−/− groups were similar at baseline and similarly increased at 15 weeks (n = 8 mice/genotype, P < 0.05). ApoE−/− mice exhibited low LTα plasma concentrations at baseline, which increased significantly after 15 weeks whereas LTα was not detectable either at baseline or after 15 weeks in ApoE−/−/P2Y2R−/−mice. Further examination of plasma levels of inflammatory cytokines using a Multiplex assay showed the effect of P2Y2R deletion to be selective to LTα, as no significant difference in the levels of 13 other cytokines (including interleukin (IL)-2, IL-3, IL-4, IL-6, TNF-α and IFN-γ) was observed between ApoE−/− and ApoE−/−/P2Y2R−/− mice (Data not shown).
Table 3.
Plasma levels of LTα and TNF-α in chow-fed ApoE−/− and ApoE−/−/P2Y2R−/− mice.
| pg/ml
|
||||
|---|---|---|---|---|
| TNF-α
|
Ltα
|
|||
| ApoE−/− | ApoE−/−/P2Y2R−/− | ApoE−/− | ApoE−/−/P2Y2R−/− | |
| 4 weeks | 6.1 ± 0.7 (n = 12) | 6.5 ± 0.5 (n = 10) | 3.7 ± 0.8 (n = 12) | n.d |
| 15 weeks | 9.3 ± 1.5 (n = 11) | 8.9 ± 0.9 (n = 10) | 7.9 ± 1.8 (n = 12) | n.d |
P < 0.01.
3.6. P2Y2 receptor deficiency impairs T cell-mediated LTα production in ApoE−/− mice
As LTα is synthesized predominantly by T-cells [14], the ability of P2Y2R to regulate LTα and TNF-α production was investigated in CD4+ T cells activated with anti-CD3 mAb. Cytokine levels in the culture supernatants were measured after 1, 2, and 3 days using an ELISA. We found that ApoE−/− CD4+ T cells released high levels of LTα and the maximal levels were observed at 3 days post stimulation (Fig. 6A). By comparison, LTα production was notably inhibited in CD4+T cells of ApoE−/−/P2Y2R−/− mice (Fig. 6A). In contrast, although TNF-α production in ApoE−/−CD4+ T cells occurred as early as 12 h after stimulation and reached its highest level after 48 h (Fig. 6), the timing and amount of secreted TNF-α were similar between CD4+ T cells in ApoE−/− and ApoE−/−/P2Y2R−/− mice. CD4+ T cell TNF-α mRNA expression after 5, 10, or 24 h of activation also showed no difference between the two genotypes whereas LTα mRNA levels in ApoE−/−/P2Y2R−/− CD4+ T activated cells remained very low even after 24 h as compared to those in CD4+ T cells from ApoE−/− mice (Fig. 6B, p < 0.05).
Fig. 6.

Time course of LTα, and TNF-α production in activated CD4+ T cells. A, CD4+ T cells were stimulated with anti-CD3 mAb, and the culture supernatants were collected after 1, 2, and 3 days for cytokine measurement. Data shown are mean ± SEM of 4 experiments, P < 0.05. B, CD4+ T cells were stimulated with anti-CD3 mAb and collected at the indicated time periods. Total RNA was extracted and subjected to Real-time PCR for the detection of LTα, and TNF-α mRNAs. Cytokine mRNA levels were quantified by densitometric analysis.
3.7. Preventive treatment with TNFR1:Fc protects mice against fatty streak lesions
As demonstrated above (Table 2 and Fig. 6), loss of P2Y2R did not affect systemic TNF-α levels or TNF-α produced by activated CD4+ T cells, suggesting that LTα but not TNF-α mediates the inflammatory processes associated with P2Y2R activation. We next asked whether neutralization of LTα before the time of disease onset protects against atherosclerosis.
Recombinant TNFR1:Fc, constructed by linkage of the cDNA encoding this protein with a DNA fragment encoding the Fc region of human IgG1, binds TNF-α and LT-α with high affinity and acts as antagonist of TNF-α and LT-α both in vitro and in vivo assays [22]. Preliminary experiments were conducted to assess the effects of increasing doses of TNFR1:Fc protein on systemic LT-α levels. We found a TNFR1:Fc dose of 100 μg per mouse to be highly effective in clearing LT-α in the blood (Data not shown). Based on these results, ApoE−/− mice were injected with 100 μg of TNFR1:Fc or isotype-matched human irrelevant IgG control by IP once a day for 5 consecutive days at 5 weeks of age. Mortality, body weight, serum concentrations of total cholesterol, HDL and LDL cholesterol and triglycerides did not differ on the day of sacrifice (15 weeks of age) between the two groups. (Table 4). Similarly, examination of blood parameters showed no statistical differences in blood leukocyte, lymphocyte, platelet, or erythrocyte counts between treatments (Table 4). Remarkably, none of the TNFR1:Fc-treated mice exhibited any type of atherosclerotic lesion (n = 8) (Fig. 7). No significant difference in the atherosclerotic lesions was found between the animals injected with human IgG or PBS (Data not shown).
Table 4.
Lipids and hematological parameters in ApoE−/− mice treated with TNFR1:Fc or human IgG.
| Parameter | TNFR1:Fc-treatment (n = 15) ± SD | IgG-treatment (n = 10) ± SD | P value |
|---|---|---|---|
| Leukocytes (/nl) | 3.08 ± 0.59 | 3.63 ± 0.39 | n.s. |
| Erythrocytes (/pl) | 7.72 ± 1.09 | 7.11 ± 0.84 | n.s. |
| Triglycerides (mg/dl) | 101.67 ± 38.87 | 78.50 ± 24.04 | n.s. |
| Total cholesterol (mg/dl) | 344.44 ± 54.94 | 365.33 ± 36.20 | n.s. |
| Body weight (g) | 28.80 ± 2.40 | 29.30 ± 2.10 | n.s. |
Fig. 7.

Effect of TNFR1:Fc on the development of atherosclerosis in ApoE−/− mice. TNFR1:Fc or human IgG1 was administered (100 μg/mouse for 5 consecutive days) to 5 week-old ApoE−/− mice fed a standard chow diet. Mice were sacrificed at 15 weeks of age. A, Aortic sinus cross sections were stained with Masson’s trichrome for gross morphological evaluation. B–C, Cross sections were stained with Oil Red O and the relative lesion area was then quantified. P < 0.05 vs IgG-injected controls.
4. Discussion
In the present study, we report that deletion of the P2Y2R gene modulates inflammatory processes pivotal to the early stages of development of atherosclerosis, thus limiting atherosclerotic plaque formation in ApoE mice. Mechanistically, inhibition of the atherosclerotic lesion may be caused by the repression of endothelial cell activation as monitored by VCAM-1 expression. The absence of P2Y2R results in the selective loss of the cytokine LTα. The importance of attenuated LTα in this process was further shown by the inhibition of fatty streak formation that was observed following short-term treatment with the LTα/TNF antagonist. Of significance, this effect was achieved when anti-LTα therapy was initiated before any obvious sign of intimal accumulation of foam cells.
Nucleotide release occurs in response to both physiological and pathological stimuli [23–24]. We previously reported that nucleotide-induced activation of P2Y2Rs contributes to monocyte recruitment in vivo [7]. We further provided mechanistic insights into these observations by showing that binding of extracellular nucleotides to the P2Y2R stimulates endothelial VCAM-1 expression and promotes the adherence of monocyte to ECs [20]. VCAM-1 is particularly important for firm, integrin-mediated adhesion of leukocytes to endothelial cells and their subsequent trans-endothelial migration [6] an important early step in the development of atherosclerosis. P2Y2R activation was found to stimulate transcription of VCAM through activation of its promoter (Fig. 4). The P2Y2R agonist UTPγS stimulated the activity of VCAM-1 promoter in a P2Y2R dependent manner as UTP-γ-S failed to activate the VCAM-1 promoter in ECs from P2Y2R−/− mice. Interestingly, we observed that P2Y2R is differentially expressed in the mouse aorta, being most highly expressed in the aortic sinus where VCAM-1 is also preferentially expressed. These findings provide mechanistic insights that help explain why the aortic sinus is particularly susceptible to the development of atherosclerotic plaque. Because of the importance VCAM-1 in the early stages of atherosclerosis [6], we postulated that deletion of the P2Y2R gene could delay fatty streak formation. When maintained on a regular chow diet, within 15 weeks ApoE−/− mice developed fatty streak lesions mainly comprised of macrophages and a few SMCs (Fig. 5, Table 2). In contrast, deletion of P2Y2R suppressed basal expression of VCAM-1 in the aortic sinus and attenuated both lesion development and maturation (Fig. 5, Table 2). In addition to decreased VCAM-1 expression, we also found that P2Y2R deficiency in ApoE−/− mice, led to a striking decrease in the plasma levels of the pro-inflammatory cytokine LTα. The effect of P2Y2R deletion was selective to LTα, as no significant difference in the levels of other examined cytokines was observed between ApoE−/− and ApoE−/−/P2Y2R−/−mice. Unlike TNFα, which is produced by a large variety of cells, including non-lymphoid cells, production of LTα is restricted to lymphocytes and, to a lesser extent, NK cells and B cells [14,25]. We showed that the levels of LTα released by activated ApoE−/−CD4+ T cells were very significant and appear to be much higher than those of TNF-α. ApoE−/−/P2Y2R−/− CD4+ T cells produced very low levels of LTα compared to ApoE−/− CD4+ T cells, whereas TNF-α levels and its time course of release were similar in both groups of T cells. The selective attenuation of LTα but not TNFα signal in T lymphocytes from P2Y2R-deficient mice suggests that P2Y2R may be involved in T-cell receptor (TCR)-dependent T cell activation. There is evidence that P2 receptor-mediated signaling is involved in TCR-dependent T cell activation. Murine T cell activation by stimulation of TCR results in both CD25 expression and interleukin (IL)-2 production. However, the P2Y6 antagonist MRS2578 significantly blocked the increases of both CD25 expression and IL-2 production, and P2X7 antagonists A438079 and oxidized ATP inhibited IL-2 production rather than CD25 expression, suggesting the involvement of P2Y6 and P2X7 receptors in different processes of T cell activation [26].
Our results strongly suggest that decrease in LTα production is likely contributing to the attenuated atherosclerosis observed in ApoE−/−/P2Y2R−/− mice. In further support of this, LTα/TNF antagonism was found to attenuate atherosclerosis development in ApoE−/− mice (Fig. 7). Recombinant TNFR1:Fc, a chimeric protein comprised of the ligand binding portion of TNFR1 fused to the Fc region of human IgG1, binds TNF and LTα with high affinity and acts as an antagonist of TNF and LTα both in vitro and in vivo [22]. TNFR1:Fc is a potent inhibitor of arthritis, administration of this cytokine antagonist over brief periods causes sustained amelioration of disease incidence and severity [22]. Notably, etanercept, a dual TNF-α and LT-α inhibitor, has proven to be very efficient against rheumatoid arthritis [27]. As we found that soluble TNFR1:Fc administration efficiently blocked fatty streak formation in ApoE−/− mice, which are predisposed to atherosclerosis, this presents the exciting possibility that TNFR1:Fc may be an effective preventative therapy for patients at high risk of developing atherosclerosis. Furthermore, preliminary observations (not shown) indicate that TNFR1:Fc treatment caused a significant decrease in both membrane and plasma levels of VCAM-1 suggesting that TNFR1:Fc exerts its action at least in part through downregulation of VCAM-1, thus blocking monocyte/macrophage recruitment. These data suggest that P2Y2R, regulate VCAM-1 expression both directly via regulating its promoter activity and indirectly through regulating LTα expression or secretion. Further support for a role in LTα in atherosclerosis is provided by previous studies which showed that a single nucleotide polymorphism (SNP) in the galectin gene that is required for LTα secretion is associated with susceptibility to myocardial infarction [15]. However, the relevance of these SNPs to the pathogenesis of myocardial infarction is controversial [28]. Our data reveal a new pathway whereby P2Y2R activation regulates LTα secretion, suggesting that mechanisms other than SNPs in the galectin-2 gene control LTα secretion in vivo.
The role of TNF in atherosclerosis is still unclear. Previous studies have shown that deletion of the TNFα gene has no effect on the development of atherosclerosis [29]. However, subsequent studies concluded that inhibition of TNα reduce atherosclerotic lesions in ApoE−/− mice [30]. LTα and TNFα genes are clustered within 12 kb of genomic DNA inside of the major histocompatibility complex [31]. Indeed, impaired TNF production by LTα total knockout mice was previously reported [32]. Unlike the conventional LTα−/− mice, LTαΔ/Δ animals generated using Cre-LoxP technology are capable of producing normal levels of systemic TNFα upon lipopolysaccharide challenge [33]. Therefore, is likely that the close proximity between the two genes accounts for the discrepancies in previous studies regarding the roles of LTα and TNF in atherosclerosis [28–29]. However, our data clearly indicate that LTα but not TNF-α mediates the inflammatory processes associated with P2Y2R activation.
In conclusion, this study supports the hypothesis that nucleotide release during endothelial injury activates the P2Y2R, which triggers production of the pro-inflammatory cytokine LTα and upregulates expression of VCAM-1, thus modulating inflammatory processes in the early development of atherosclerosis. Anti-LTα treatment prevents atherosclerosis by overriding detrimental inflammatory processes evoked by P2Y2R activation. Our findings suggest that targeting this nucleotide receptor could be an effective therapeutic approach in atherosclerosis.
Acknowledgments
Funding sources
This work was supported by a grant from the National Institutes of Health (1R01HL112883) to Cheikh I. Seye.
Footnotes
Funding sources: This work was supported by a grant from the National Institute of Health (1R01HL112883) to Cheikh I. Seye.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.vph.2016.06.001.
References
- 1.Jain M, Ridker P. Anti-inflammatory effects of statins: clinical evidence and basic mechanisms. Nat Rev Drug Discov. 2005;4:977–987. doi: 10.1038/nrd1901. [DOI] [PubMed] [Google Scholar]
- 2.Kam PC, Nethery CM. The thienopyridine derivatives (platelet adenosine diphosphate receptor antagonists), pharmacology and clinical developments. Anaesthesia. 2003;58:28–35. doi: 10.1046/j.1365-2044.2003.02960.x. [DOI] [PubMed] [Google Scholar]
- 3.Kunapuli SP, Ding Z, Dorsam RT, Kim S, Murugappan S, Quinton TM. ADP receptors-targets for developing antithrombotic agents. Curr Pharm Des. 2003;9:2303–2316. doi: 10.2174/1381612033453947. [DOI] [PubMed] [Google Scholar]
- 4.Gachet C. ADP receptors of platelets and their inhibition. Thromb Haemost. 2001;86:222–232. [PubMed] [Google Scholar]
- 5.Gimbrone M. Vascular endothelium: an integrator of pathophysiologic stimuli in atherosclerosis. Am J Cardiol. 1995;75:67–70. doi: 10.1016/0002-9149(95)80016-l. [DOI] [PubMed] [Google Scholar]
- 6.Cybulsky MI, Liyama K, Li H, Zhu S, Chen M, Liyama M, et al. A major role for VCAM-1, but not ICAM-1, in early atherosclerosis. J Clin Invest. 2001;107(10):1255–1262. doi: 10.1172/JCI11871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seye CI, Kong Q, Erb L, Garrad RC, Krugh B, Wang M, et al. Functional P2Y2 nucleotide receptors mediate uridine 5′-triphosphate-induced intimal hyperplasia in collared rabbit carotid arteries. Circulation. 2002;106:2720–2726. doi: 10.1161/01.cir.0000038111.00518.35. [DOI] [PubMed] [Google Scholar]
- 8.Seye CI, Yu N, Gonzalez FA, Erb L, Weisman GA. The P2Y2 nucleotide receptor mediates vascular cell adhesion molecule-1 expression through interaction with VEGF receptor-2 (KDR/Flk-1) J Biol Chem. 2004;279:35679–35686. doi: 10.1074/jbc.M401799200. [DOI] [PubMed] [Google Scholar]
- 9.Perregaux D, Gabel CA. Interleukin-1β maturation and release in response to ATP and nigericin. Evidence that potassium depletion mediated by these agents is a necessary and common feature of their activity. J Biol Chem. 1994;269:15195–15203. [PubMed] [Google Scholar]
- 10.Perregaux D, Gabel CA. Post-translational processing of murine IL-1: evidence that ATP- induced release of IL-1α and IL-1β occurs via a similar mechanism. J Immunol. 1998;160:2469–2477. [PubMed] [Google Scholar]
- 11.Ferrari D, Chiozzi P, Falzoni S, Dal ZM, Melchiorri L, Baricordi OR, et al. Extracellular ATP triggers IL-1β release by activating the purinergic P2Z receptor of human macrophages. J Immunol. 1997;159:1451–1458. [PubMed] [Google Scholar]
- 12.Warny M, Aboudola S, Robson SC, Sevigny J, Communi D, Soltoff SP, et al. P2Y6 nucleotide receptor mediates monocyte IL-8 production in response to UDP or lipopolysaccharide. J Biol Chem. 2001;276:26051–26056. doi: 10.1074/jbc.M102568200. [DOI] [PubMed] [Google Scholar]
- 13.Seye CI, Agca Y, Agca C, Derbigny W. P2Y2 receptor-mediated lymphotoxin-α (LTA) secretion regulates intercellular cell adhesion molecule (ICAM)-1 expression in vascular smooth muscle cells. J Biol Chem. 2012;287:10535–10543. doi: 10.1074/jbc.M111.313189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ware CF, VanArsdale TL, Crowe PD, Browning J. The ligands and receptors of the lymphotoxin system. Curr Top Microbiol Immunol. 1995;198:175–218. doi: 10.1007/978-3-642-79414-8_11. [DOI] [PubMed] [Google Scholar]
- 15.Ozaki K, Inoue K, Sato H, Iida A, Ohnishi1 Y, Sekine A, et al. Functional variation in LGALS2 confers risk of myocardial infarction and regulates lymphotoxin-α secretion in vitro. Nature. 2004;429:72–75. doi: 10.1038/nature02502. [DOI] [PubMed] [Google Scholar]
- 16.Van Vlijmen BJ, et al. Diet-induced hyperlipoproteinemia and atherosclerosis in apolipoprotein E3-Leiden transgenic mice. J Clin Invest. 1994;93:1403–1410. doi: 10.1172/JCI117117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Korenaga R, Ando J, Kosaki K, Isshiki M, Takada Y, Kamiya A. Negative transcriptional regulation of the VCAM-1 gene by fluid shear stress in murine endothelial cells. Am J Phys. 1997;273:C1506–C1515. doi: 10.1152/ajpcell.1997.273.5.c1506. [DOI] [PubMed] [Google Scholar]
- 18.Hosking BM, Mary Wang S-C, Downes M, Koopman P, George EO, Muscat GE. The VCAM-1 Gene that encodes the vascular cell adhesion molecule is a target of the Sry-related high mobility group box Gene, Sox18. J Biol Chem. 2004;279:5314–5322. doi: 10.1074/jbc.M308512200. [DOI] [PubMed] [Google Scholar]
- 19.Kreisel D, Krupnick AS, Szeto WY, Popma SH, Sankaran D, Krasinskas AM, et al. A simple method for culturing mouse vascular endothelium. J Immunol Methods. 2001;254:31–45. doi: 10.1016/s0022-1759(01)00371-4. [DOI] [PubMed] [Google Scholar]
- 20.Suo J, Ferrara DE, Sorescu D, Guldberg RE, Taylor WR, Giddens DP. Hemodynamic shear stresses in mouse aortas: implications for atherogenesis. Arterioscler Thromb Vasc Biol. 2007;27:346–351. doi: 10.1161/01.ATV.0000253492.45717.46. [DOI] [PubMed] [Google Scholar]
- 21.Seye CI, Yu N, Jain R, Kong Q, Minor T, Newton J, et al. The P2Y2 nucleotide receptor mediates UTP-induced vascular cell adhesion molecule-1 expression in coronary artery endothelial cells. J Biol Chem. 2003;278:24960–24965. doi: 10.1074/jbc.M301439200. [DOI] [PubMed] [Google Scholar]
- 22.Wooley P, Dutcher J, Widmir M, Gillis S. Influence of a recombinant human soluble tumor necrosis factor receptor FC fusion protein on type I collagen-induced arthritis in mice. J Immunol. 1993;151:6602–6607. [PubMed] [Google Scholar]
- 23.Ralevic V, Burnstock G. Roles of P2-receptors in the cardiovascular system. Circulation. 1993;84:1–14. doi: 10.1161/01.cir.84.1.1. [DOI] [PubMed] [Google Scholar]
- 24.Di Virgilio F, Solini A. P2 receptors: new potential players in atherosclerosis. Br J Pharmacol. 2002;135:831–842. doi: 10.1038/sj.bjp.0704524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ohshima Y, Yang LP, Avice MN, Kurimoto M, Nakajima T, Sergerie M, et al. Naïve human CD4+ T cells are a major source of lymphotoxin alpha. J Immunol. 1999;162:3790–3794. [PubMed] [Google Scholar]
- 26.Tsukimoto M, Tokunaga A, Harada H, Kojima S. Blockade of murine T cell activation by antagonists of P2Y6 and P2X7 receptors. Biochem Biophys Res Commun. 2009;384(4):512–518. doi: 10.1016/j.bbrc.2009.05.011. (10) [DOI] [PubMed] [Google Scholar]
- 27.Mikuls TR, Moreland LW. TNF blockade in the treatment of rheumatoid arthritis: infliximab versus etanercept. Expert Opin Pharmacother. 2001;2:75–84. doi: 10.1517/14656566.2.1.75. [DOI] [PubMed] [Google Scholar]
- 28.Li W, Xu J, Wang X, Chen J, Zhang C, Sun Kand Hui R. Lack of association between lymphotoxin-α, galectin-2 polymorphisms and coronary artery disease: a meta-analysis. Atherosclerosis. 2010;2010(208):433–436. doi: 10.1016/j.atherosclerosis.2009.08.014. [DOI] [PubMed] [Google Scholar]
- 29.Schreyer SA, Vickand CM, LeBoeuf RC. Loss of lymphotoxin-α but not tumor necrosis factor-α reduces atherosclerosis in mice. J Biol Chem. 2002;(277):12364–1236. doi: 10.1074/jbc.M111727200. [DOI] [PubMed] [Google Scholar]
- 30.Brånén L, Hovgaard L, Nitulescu M, Bengtsson E, Nilsson J, Jovinge S. Inhibition of tumor necrosis factor-alpha reduces atherosclerosis in apolipoprotein E knockout mice. Arterioscler Thromb Vasc Biol. 2004;24(11):2137–2212. doi: 10.1161/01.ATV.0000143933.20616.1b. [DOI] [PubMed] [Google Scholar]
- 31.Muller U, Jongeneel CV, Nedospasov SA, Fisher Lindahl K, Steinmetz M. Tumor necrosis factor and lymphotoxin genes map close to H-2D in the mouse major histocompatibility complex. Nature. 1987;325:265–267. doi: 10.1038/325265a0. [DOI] [PubMed] [Google Scholar]
- 32.Alexopoulou L, Pasparakis M, Kollias G. Complementation of lymphotoxin alpha knockout mice with tumor necrosis factor-expressing transgenes rectifies defective splenic structure and function. J Exp Med. 1998;188:745–754. doi: 10.1084/jem.188.4.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liepinsh DJ, Grivennikov SI, Klarmann KD, Lagarkova MA, Drutskaya MS, Lockett SJ, et al. Novel lymphotoxin alpha (LTα) knockout mice with unperturbed tumor necrosis factor expression: reassessing LTα biological functions. Mol Cell Biol. 2006;26:4214–4225. doi: 10.1128/MCB.01751-05. [DOI] [PMC free article] [PubMed] [Google Scholar]