

Abstract
Preparation of a broad range of enantioenriched β-fluoro amines (α,β-disubstituted) is described in which the nitrogen and fluorine atoms are attached to sp3-hybridized carbons. The key finding is a chiral bifunctional Brønsted acid/base catalyst that can deliver β-amino-α-fluoro nitroalkanes with high enantio- and diastereoselection. A denitration step renders the nitro group ‘traceless’ and delivers secondary, tertiary, or vinyl alkyl fluorides embedded within a vicinal fluoro amine functional group. A concise synthesis of each possible stereoisomer of a β-fluoro lanicemine illustrates the potential ease with which fluorinated small molecules relevant to neuroscience drug development can be prepared in a stereochemically-comprehensive manner.
Graphical abstract
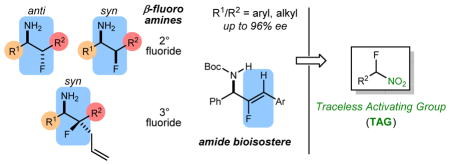
The introduction of the fluorine atom into small molecules has received increased attention in recent years,1 in recognition of its ability to impart specific and dramatic pharmacological effects in bioactive pharmaceutical and agrochemical small molecules. Replacement of a single hydrogen with fluorine in a small molecule can increase metabolic stability, improve lipophilicity, and improve bioavailability.2 Such a substitution, however, remains a formidable challenge, especially when the fluorine-bearing carbon is stereogenic.3 β-Fluoro amines are a specific class of fluorinated compounds4 that display remarkable CNS-penetrant properties. Additionally, the β-fluoro amine Sofosbuvir is an RNA polymerase inhibitor that is in part responsible for the recent and unprecedented high cure rates of the hepatitis C virus.5 β-Fluoro amines exhibit decreased amine basicity and enhanced binding interactions.6 From a synthetic standpoint, enantioselective methods to prepare chiral fluorocarbons have focused on C-F bond formation using both electrophilic7 and nucleophilic8,9 fluorine reagents.1 Herein we report an approach to saturated β-fluoro amines by carbon-carbon bond formation,10,11 reliant on a traceless nitroalkane activation strategy. A broad range of stereoenriched β-fluoro amines can be accessed, but of particular note is the convergency with which fluorinated derivatives of 1-phenethyl amines can be prepared,12 since these are common substructures of neurologically-active small molecules. β-Fluoro amines in which both carbons are chiral have few direct preparative solutions.
A convergent approach to the β-fluoro amine substructure involves formation of the central carbon-carbon bond, but requires both relative and absolute stereocontrol. Among possible fluoroalkyl nucleophiles activated by a traceless activating group (TAG,13 Scheme 1), esters, gem-diesters and nitroalkanes can be fluorinated, but only substrates bearing an α-fluoro carbonyl have been utilized broadly in enantioselective synthesis.14 Lu reported enantioselective reactions using α-fluorinated nitroalkanes, but an additional activating group (ester or aryl) at the fluoromethyl carbon was required in the additions to nitro-olefins for a reaction to occur.15 This highlights fluorine’s limited effect on nitroalkane acidity relative to its higher halogen counterparts (Scheme 1).16,17 Moreover, fluorine can actually deacidify carbon acids (Scheme 1): fluoronitromethane is less acidic than bromonitromethane, and fluoro dinitromethane is less acidic than dinitromethane.18
Scheme 1.
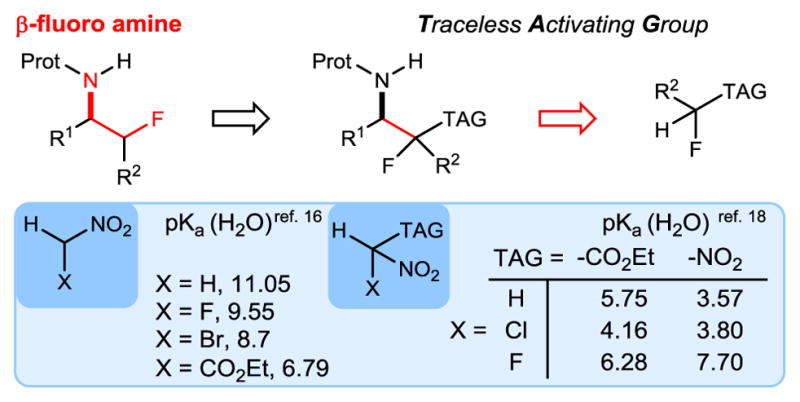
Considerations for use of a Traceless Activating Group Strategy for Enantioselective β-Fluoro Amine Synthesis
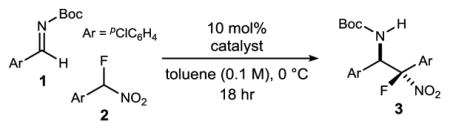
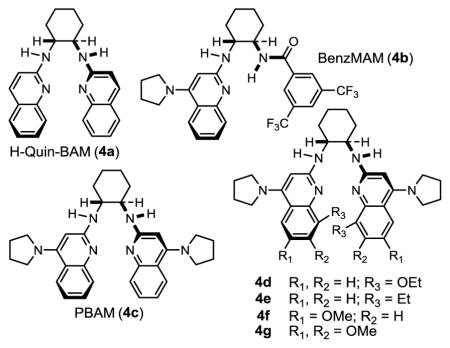
α-Fluoro nitroalkanes were prepared from their hydrocarbon parents by a deprotonation (KOH, aq CH3CN) and fluorination (Selectfluor) treatment as outlined by Guo.19 α-Fluoro aryl nitromethane 2 was used to first benchmark reactivity in a reaction with aldimine 1 (Table 1). Brønsted base catalyst H,Quin-BAM (4a)20 provided a low level of conversion, and equally low levels of diastereoselection (2.1:1 dr) and enantioselection (25% ee) (Table 1, entry 1). Increasingly Brønsted basic catalysts were examined, leading to good levels of conversion, but with little improvement to selectivity (Table 1, entries 2–3).21 Examination of the reactivity of catalyst 4c was extended to its acid salt (Table 1, entry 4), for which a corresponding increase in enantioselection, but not diastereoselection, was observed. Diastereoselection could be improved at the sacrifice of enantioselection by adding an additional electron-donating substituent to the catalyst (Table 1, entry 5), but its acid salt was unexpectedly less selective. The behavior was also observed with its 8-alkyl counterpart (Table 1, entry 7). It was not until a further increase in Brønsted basicity of the acid salts was sought that 6,7(MeO)2PBAM·HNTf2 (4g·HNTf2) emerged as the key to formation of 3 with high yield (97%), diastereoselection (4.8:1), and enantioselection (91/86% ee) (Table 1, c.f. entries 8–9). Use of the free base provided inferior results (Table 1, entry 10), in line with similar comparisons outlined above, but in stark contrast to earlier work with aryl nitromethanes that showed equal selectivity when comparing free base to its salt.22,23 A temperature decrease led to improved selectivity (Table 1, entry 11). Catalyst loading could be decreased at some expense to selectivity (Table 1, entries 12–13). Assignment of absolute configuration was made using X-ray analysis of 3.
Table 1.
Diastereo- and Enantioselective Synthesis of β-Amino-α-Fluoro Nitroalkanes: Catalyst Discovery and Development
| |||||
|---|---|---|---|---|---|
| entry | catalystbase | catalyst acida | yieldb | d.r.c | ee (%)c |
| 1 | HQuinBAM (4a) | – | <20 | 2.1:1 | 25/24 |
| 2 | BenzMAM (4b) | – | 93 | 4.4:1 | 25/36 |
| 3 | PBAM (4c) | – | 96 | 1.4:1 | 41/71 |
| 4 | 4c | HNTf2 | 71 | 1.9:1 | 83/81 |
| 5 | 8(EtO)PBAM (4d) | – | 97 | 2.8:1 | 68/68 |
| 6 | 4d | HNTf2 | 80 | 1.5:1 | 40/41 |
| 7 | 8EtPBAM (4e) | HNTf2 | 42 | 1.6:1 | 70/71 |
| 8 | 6(MeO)PBAM (4f) | HNTf2 | 96 | 2.3:1 | 86/84 |
| 9 | 6,7(MeO)2PBAM (4g) | HNTf2 | 97 | 4.8:1 | 91/86 |
| 10 | 4g | – | 84 | 1.4:1 | 63/77 |
| 11 | 4g | HNTf2 | 71d | 5.8:1 | 94/87 |
| 12 | 4g | HNTf2 | 73e | 4.1:1 | 88/77 |
| 13 | 4g | HNTf2 | 62f | 3.3:1 | 86/71 |
Catalyst prepared as the 1:1 base:acid salt. Reactions are 0.1 M toluene unless otherwise noted.
Isolated yield.
Diastereomeric ratios determined by 1H NMR, and enantiomeric excess (ee) determined by HPLC using a chiral stationary phase. The major diastereomer/enantiomer of 3 was determined by X-ray crystallographic analysis.
Reaction temp was −20 °C and ran for 40 h.
5 mol % catalyst employed.
2 mol % catalyst employed.
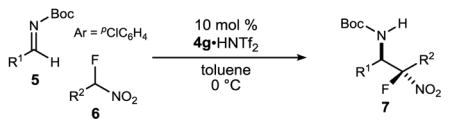
An investigation of the level of generality inherent to the reaction and catalyst reagent is summarized in Table 2. The overall objective was to target four general classes of β-fluoro amines, defined by the type of substituent – aryl or alkyl – at the aminomethyl (R1) and fluoromethyl (R2) carbons (R1,R2≠H). For example, the combination of aryl aldimine and aryl fluoronitromethane leads to a permutation with an aryl substituent at the aminomethyl and fluoromethyl carbons (3,7a–f). In this context, an electronically diverse group of aryl aldimines performed well (Table 2) en route to masked-fluoro stilbene amine compounds (vide infra). A highlight is the 3-pyridyl imine, resulting in relatively high diastereoselection at 6.9:1 dr and providing a heterocycle-containing product in high ee (Table 2, entry 6). A heterocycle is also allowable on the nitroalkane component, even when the pyridine nitrogen is in close proximity to the nitro group (Table 2, entry 7). This case was anticipated to be the most challenging from a catalyst selectivity standpoint, as it could serve as a frustrating hydrogen bond acceptor in the transition state. This feature may have contributed to the relatively low diastereoselection at 2.5:1 dr, but both diastereomers were formed with high enantioselection (95/94% ee), and in 81% yield (Table 2, entry 7).
Table 2.
Examination of Various Fluoro Nitroalkanes and Boc-imines in the Enantioselective aza-Henry Reactiona
| ||||||
|---|---|---|---|---|---|---|
| entry | product | product | temp | yieldb | drc | ee (%)c |
| 1 |
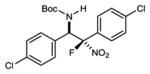
|
3 | 0 °C | 97 | 4.8:1 | 91 |
| 2 |
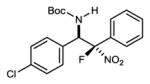
|
7a | 0 °C | 93 | 5.2:1 | 95 |
| 3 |
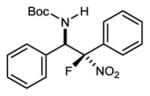
|
7b | 0 °C | 87 | 4.5:1 | 95 |
| 4 |
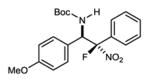
|
7c | 0 °C | 86 | 5.6:1 | 96 |
| 5 |
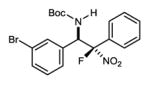
|
7d | 0 °C | 92 | 4.8:1 | 92 |
| 6 |
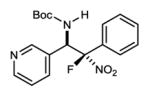
|
7e | 0 °C | 86 | 6.9:1 | 95 |
| 7 |
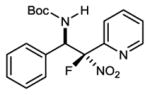
|
7f | 0 °C | 81 | 2.5:1 | 95 |
| 8 |
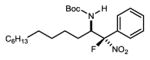
|
7g | 0 °C | 21e | 6.1:1 | 84 |
| 9 | −20 °C | 70d,e | 9.0:1 | 93 | ||
| 10 |
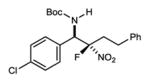
|
7h | 24 °C | 85 | 5.0:1 | 93 |
| 11 |
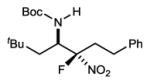
|
7i | 24 °C | 21e | >10:1 | 84 |
| 12 |
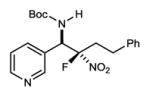
|
7j | 24 °C | 84 | 4.4:1 | 96 |
Catalyst prepared as the 1:1 acid salt. Reactions were 0.1 M in dry toluene for 18 h, unless otherwise noted. Relative and absolute configuration assigned by analogy to 3 (X-ray).
Isolated yield.
Diastereomeric ratios determined by 1H NMR, and enantiomeric excess (ee, major enantiomer shown) determined by HPLC using a chiral stationary phase.
48 h reaction time.
Yield over 2-steps from α-amido sulfone (see Supporting Information).
N-Boc aldimines could provide an alkyl substituent at the aminomethyl carbon, but are challenging electrophiles due in part to their sensitivity to tautomerization to unreactive N-Boc enamides. Under the standard conditions, α-fluoro phenyl nitromethane engaged the imine to produce the product in moderate yield (53%, two steps from the α-amido sulfone) at 0 °C and serviceable stereoselection (6.1:1 dr, 84% ee) (Table 2, entry 8). Gratifyingly, improved stereocontrol and yield were obtained at −20 °C (9:1 dr, 93% ee, 70% yield over 2 steps: Table 2, entry 9) as a result of decreased imine tautomerization.24 Alkyl fluoronitroalkanes can be easily prepared (2 steps from various alkyl bromides)19 and are also suitable here for the first time in enantioselective catalysis, in direct contrast to previously reported attempts15 (Table 2, entries 10–12). Notably, the alkyl fluoronitroalkanes react only at warmer temperatures (24 °C), allowing for an even more simplified reaction setup. Regardless, high levels of stereocontrol can be readily obtained (7h; 5:1 dr, 93% ee, 85% yield) (Table 2, entry 10).25
The coupling of two aliphatic partners (Bocimine and fluoronitroalkane) is the most challenging pair due to the lower reactivity of each (21% yield, 2 steps), yet very useful levels of stereocontrol can be achieved (7i; >10:1 dr, 84% ee) (Table 2, entry 11). In a final example, an alkyl fluoronitroalkane can engage a heteroaromatic pyridyl Bocimine at room temperature to afford the adduct (7j) in good yield (84%) (Table 2, entry 12: 4.4:1 dr, 96% ee). Preliminary work to assess reactivity of α-fluoro nitroalkanes relative to their nonfluorinated counterparts replicated the trend suggested by relative pKa (Scheme 1).26 This aligns with the hypothesis that a more Brønsted basic catalyst is necessary for activation, and may explain the lack of prior success.15
The addition reactions in Table 2 are the basis for a collective preparation of the four stereoisomers of β-fluoro amines, a need that is particularly pressing in therapeutic development where a small molecule’s conformation and protonation state affect its pharmacologic properties.27 We targeted fluorinated lanicemine (8), a stereoisomeric series of compounds unknown in the literature. Lanicemine (AZD-6765) is a potent, low-trapping NMDA receptor antagonist (Scheme 2).28 Preparation of both fluorine diastereomers would be considered important in exploratory studies for several reasons, including the conformational effects that result from the chiral fluoromethyl group.29 Using the (R,R) and (S,S) antipodes of 4g·HNTf2 separately, each enantiomer of the addition product 7f was obtained with high enantioselection (95–96% ee, Scheme 2). Conversions of β-fluoro-β-nitro amines to β-fluoro amines are unprecedented,30 but were readily performed under reductive conditions using free radical-mediated denitration.31 It is this step that establishes the nitro functionality as a TAG.13 Boc-deprotection produced the desired β-fluoro-lanicemine derivatives 8 in good yield overall.32
Scheme 2.
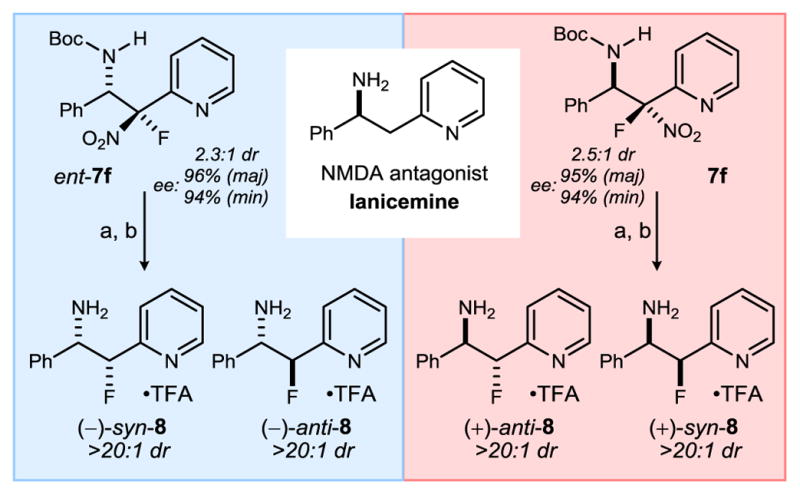
Lanicemine (AZD6765) and Preparation of All Four Stereoisomers of ‘β-Fluoro-Lanicemine’ (8).
Reaction conditions: a) Bu3SnH (4 equiv.), AIBN (0.4 equiv), benzene, 80 °C, 180 min (dr ≈ 2:1 anti:syn, 74%); b) TFA, CH2Cl2, 180 min (98%). In each case, diastereomers 8 were purified by reverse phase preparatory HPLC as their TFA salt adducts (see SI). Enantiomeric excess (ee) for stereoisomers 8 follow from ee of 7f/ent-7f.
The potential versatility of the nitro group as a TAG was probed further (Scheme 3). Reductive allylation of 7a formed the tertiary fluoro-β-amine 9 in >10:1 dr and 53% yield.33 Under basic conditions, conversion of 10 to vinyl fluoride 11 was accomplished by regio- and stereoselective elimination of the nitro group.34 This transformation establishes a new enantioselective approach to an amide isostere of phenyl glycine in this case.13j,35,36,37
Scheme 3.
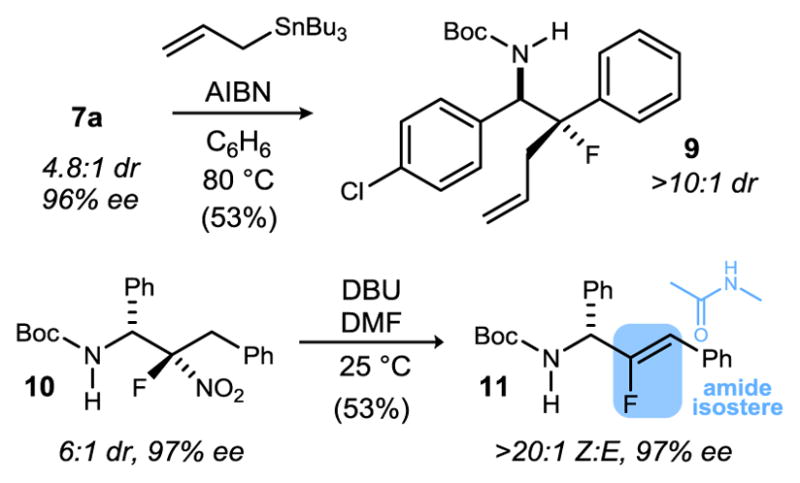
Transformations of Enantioenriched α-Amino Fluoronitroalkanes: Denitrative Functionalizations
In summary, a convenient stereocontrolled synthesis of β-fluoro amines has been developed, particularly those involving sp3-hybridized secondary and tertiary fluoromethyls. The approach required development of the catalyzed addition of α-fluoro nitroalkanes to imines, one that allowed each permutation of aryl and alkyl substituent to be used for each reactant. High diastereoselection and enantioselection is possible when using a more Brønsted basic ligand (4g) within the Brønsted acid catalyst (4g·HNTf2), which counteracted the acid-weakening effect of fluorine on nitroalkane acidity. A selection of denitrative functionalizations provided entry to several fluorinated pharmacophores.
Supplementary Material
Acknowledgments
BAV was supported in part by a Lamar Field Research Award. We are grateful to the National Institute of General Medical Sciences (NIH GM 084333) for financial support, Dr. Maren Pink (Indiana University Molecular Structure Center) for X-ray analysis, and Jade Bing for data collection.
Footnotes
The authors declare no competing financial interests.
Procedures, complete experimental details, and analytical data for all new compounds are provided. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Reviews: Champagne PA, Desroches J, Hamel JD, Vandamme M, Paquin JF. Chem Rev. 2015;115:9073. doi: 10.1021/cr500706a.Yang XY, Wu T, Phipps RJ, Toste FD. Chem Rev. 2015;115:826. doi: 10.1021/cr500277b.
- 2.Reviews: Purser S, Moore PR, Swallow S, Gouverneur V. Chem Soc Rev. 2008;37:320. doi: 10.1039/b610213c.Wang J, Sánchez-Roselló M, Aceña JL, del Pozo C, Sorochinsky AE, Fustero S, Soloshonok VA, Liu H. Chem Rev. 2014;114:2432. doi: 10.1021/cr4002879.Hagmann WK. J Med Chem. 2008;51:4359. doi: 10.1021/jm800219f.Gillis. 2015:6312.
- 3.Hu XG, Thomas DS, Griffith R, Hunter L. Angew Chem Int Ed. 2014;53:6176. doi: 10.1002/anie.201403071. [DOI] [PubMed] [Google Scholar]
- 4.Percy JM. Sci Synth. 2006;34:379. [Google Scholar]
- 5.Clark JL, Hollecker L, Mason JC, Stuyver LJ, Tharnish PM, Lostia S, McBrayer TR, Schinazi RF, Watanabe KA, Otto MJ, Furman PA, Stec WJ, Patterson SE, Pankiewiez KW. J Med Chem. 2005;48:5504. doi: 10.1021/jm0502788. [DOI] [PubMed] [Google Scholar]
- 6.Morgenthaler M, Schweizer E, Hoffmann-Roder A, Benini F, Martin RE, Jaeschke G, Wagner B, Fischer H, Bendels S, Zimmerli D, Schneider J, Diederich F, Kansy M, Mueller K. ChemMedChem. 2007;2:1100. doi: 10.1002/cmdc.200700059. [DOI] [PubMed] [Google Scholar]
- 7.(a) Phipps RJ, Hiramatsu K, Toste FD. J Am Chem Soc. 2012;134:8376. doi: 10.1021/ja303959p. [DOI] [PubMed] [Google Scholar]; (b) Wu J, Wang YM, Drljevic A, Rauniyar V, Phipps RJ, Toste FD. Proc Natl Acad Sci U S A. 2013;110:13729. doi: 10.1073/pnas.1304346110. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Fadeyi OO, Lindsley CW. Org Lett. 2009;11:943. doi: 10.1021/ol802930q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) O’Reilly MC, Lindsley CW. Tetrahedron Lett. 2013;54:3627. doi: 10.1016/j.tetlet.2013.04.116. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Rauniyar V, Lackner AD, Hamilton GL, Toste FD. Science. 2011;334:1681. doi: 10.1126/science.1213918. [DOI] [PubMed] [Google Scholar]
- 8.Stereospecific ring opening by fluoride: Alvernhe G, Kozlowska-Gramsz E, Lacombe-Bar S, Laurent A. Tetrahedron Lett. 1978:5203.Wade TN. J Org Chem. 1980;45:5328.Alvernhe GM, Ennakoua CM, Lacombe SM, Laurent AJ. J Org Chem. 1981;46:4938.Duthion B, Pardo DG, Cossy J. Org Lett. 2010;12:4620. doi: 10.1021/ol1019579.Huang HT, Lacy TC, Błachut B, Ortiz GX, Wang Q. Org Lett. 2013;15:1818. doi: 10.1021/ol4003866.Kalow JA, Schmitt DE, Doyle AG. J Org Chem. 2012;77:4177. doi: 10.1021/jo300433a.
- 9.Enantioselective meso-aziridine ring opening by fluoride (up to 84% ee): Kalow JA, Doyle AG. Tetrahedron. 2013;69:5702.
- 10.Enantioselective addition of fluoromethyl acids: Prakash GKS, Wang F, Stewart T, Mathew T, Olah GA. Proc Natl Acad Sci U S A. 2009;106:4090. doi: 10.1073/pnas.0900179106.Appayee C, Brenner-Moyer SE. Org Lett. 2010;12:3356. doi: 10.1021/ol101167z.
- 11.Diastereoselective addition of fluoromethyl acids: Garcia Ruano JL, Parra A, Alonso I, Fustero S, del Pozo C, Arroyo Y, Sanz-Tejedor A. Chem - Eur J. 2011;17:6142. doi: 10.1002/chem.201100455.
- 12.Cresswell AJ, Davies SG, Lee JA, Roberts PM, Russell AJ, Thomson JE, Tyte MJ. Org Lett. 2010;12:2936. doi: 10.1021/ol100862s. [DOI] [PubMed] [Google Scholar]
- 13.Recent leading references for TAG-based strategies: Carboxylic acid: Chu L, Ohta C, Zuo Z, MacMillan DWC. J Am Chem Soc. 2014;136:10886. doi: 10.1021/ja505964r.Chernyak N, Dudnik AS, Huang C, Gevorgyan V. J Am Chem Soc. 2010;132:8270. doi: 10.1021/ja1033167.Luo J, Preciado S, Larrosa I. J Am Chem Soc. 2014;136:4109. doi: 10.1021/ja500457s.Neely JM, Rovis T. J Am Chem Soc. 2014;136:2735. doi: 10.1021/ja412444d.Cyano: Li J, Zhao H, Jiang X, Wang X, Hu H, Yu L, Zhang Y. Angew Chem. 2015;127:6404. doi: 10.1002/anie.201500961.Pyridyl: Huang C, Chernyak N, Dudnik AS, Gevorgyan V. Adv Synth Catal. 2011;353:1285. doi: 10.1002/adsc.201000975.N-Oxide: Sharma R, Kumar R, Kumar I, Sharma U. Eur J Org Chem. 2015:7519.Nitro: Shen B, Johnston JN. Org Lett. 2008;10:4397. doi: 10.1021/ol801797h.Sulfone: Mizuta S, Shibata N, Goto Y, Furukawa T, Nakamura S, Toru T. J Am Chem Soc. 2007;129:6394. doi: 10.1021/ja071509y.Jacobsen CB, Nielsen M, Worgull D, Zweifel T, Fisker E, Jørgensen KA. J Am Chem Soc. 2011;133:7398. doi: 10.1021/ja110624k.Prakash GKS, Gurung L, Jog PV, Tanaka S, Thomas TE, Ganesh N, Haiges R, Mathew T, Olah GA. Chem Eur J. 2013;19:3579. doi: 10.1002/chem.201204621.Huang W, Ni C, Zhao Y, Hu J. New J Chem. 2013;37:1684.
- 14.Leading references: Jiang Z, Pan Y, Zhao Y, Ma T, Lee R, Yang Y, Huang KW, Wong MW, Tan CH. Angew Chem Int Ed. 2009;48:3627. doi: 10.1002/anie.200900964.Hatano M, Ishihara K. Synthesis. 2010;2010:3785.Yoon SJ, Kang YK, Kim DY. Synlett. 2011;2011:420.Han X, Kwiatkowski J, Xue F, Huang KW, Lu Y. Angew Chem Int Ed. 2009;48:7604. doi: 10.1002/anie.200903635.Pan Y, Zhao Y, Ma T, Yang Y, Liu H, Jiang Z, Tan CH. Chem Eur J. 2010;16:779. doi: 10.1002/chem.200902830.Trost BM, Saget T, Lerchen A, Hung CI. Angew Chem. 2016;128:791. doi: 10.1002/anie.201509719.
- 15.(a) Kwiatkowski J, Lu YX. Chem Commun. 2014;50:9313. doi: 10.1039/c4cc03513e. [DOI] [PubMed] [Google Scholar]; (b) Kwiatkowski J, Lu YX. Org Biomol Chem. 2015;13:2350. doi: 10.1039/c4ob02486a. [DOI] [PubMed] [Google Scholar]
- 16.Bordwell FG, Bartmess JE. J Org Chem. 1978;43:3101. [Google Scholar]
- 17.For base-catalyzed additions of bromonitromethane: Review: Soengas RG, Acúrcio RC, Silva AMS. Eur J Org Chem. 2014:6339.Selected recent examples: Russo A, Meninno S, Tedesco C, Lattanzi A. Eur J Org Chem. 2011:5096.Manzano R, Andres JM, Alvarez R, Muruzabal MD, de Lera AR, Pedrosa R. Chem - Eur J. 2011;17:5931. doi: 10.1002/chem.201100241.Dobish MC, Villalta F, Waterman MR, Lepesheva GI, Johnston JN. Org Lett. 2012;14:6322. doi: 10.1021/ol303092v.Leighty MW, Shen B, Johnston JN. J Am Chem Soc. 2012;134:15233. doi: 10.1021/ja306225u.Yu H, Wang Q, Wang Y, Song H, Zhou Z, Tang C. Chem - Asian J. 2013;8:2859. doi: 10.1002/asia.201300778.Das U, Tsai YL, Lin W. Org Biomol Chem. 2013;11:44. doi: 10.1039/c2ob26943k.Makley DM, Johnston JN. Org Lett. 2014;16:3146. doi: 10.1021/ol501297a.Schwieter KE, Johnston JN. Chem Sci. 2015;6:2590. doi: 10.1039/c5sc00064e.
- 18.(a) Adolph HG, Kamlet MJ. J Am Chem Soc. 1966;88:4761. [Google Scholar]; (b) Slovetskii VI, Okhobystina LV, Feinzil’berg AA, Ivanov AI, Birynkova LJ, Novikov SS. Izvest Akad Nauk SSSR, Otdel khim Nauk. 1965:2063. [Google Scholar]
- 19.(a) Huan F, Hu HW, Huang YG, Chen QY, Guo Y. Chin J Chem. 2012;30:798. [Google Scholar]; (b) Hu HW, Huang YG, Guo Y. J Fluorine Chem. 2012;133:108. [Google Scholar]
- 20.Nugent BM, Yoder RA, Johnston JN. J Am Chem Soc. 2004;126:3418. doi: 10.1021/ja031906i. [DOI] [PubMed] [Google Scholar]
- 21.Differences in reactivity in this reaction when using aryl nitromethane, nitroalkane, and their α-fluoro derivatives was also observed when preparing the racemic standards. For example, α-fluoro phenyl nitromethane converted with either DMAP or DBU promoter, while (3-fluoro-3-nitropropyl)benzene failed to convert using DMAP, 60 °C, 16 hours.
- 22.Davis TA, Johnston JN. Chem Sci. 2011;2:1076. doi: 10.1039/C1SC00061F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vara BA, Mayasundari A, Tellis JC, Danneman MW, Arredondo V, Davis TA, Min J, Finch K, Guy RK, Johnston JN. J Org Chem. 2014;79:6913. doi: 10.1021/jo501003r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schwieter KE, Johnston JN. ACS Catalysis. 2015;5:6559. doi: 10.1021/acscatal.5b01901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Competition experiments designed to assess relative reactivity of aliphatic and α-fluoro nitroalkanes were carried out and support previous claims of the decreased acidity of α-fluoro nitroalkanes. The results are summarized in the Supporting Information.
- 26.(a) Adolph HG, Kamlet MJ. J Am Chem Soc. 1966;88:4761. [Google Scholar]; (b) Hine J, Mahone LG, Liotta CL. J Am Chem Soc. 1967;89:5911. [Google Scholar]; (c) Lorand JP, Urban J, Overs J, Ahmed QA. J Org Chem. 1969;34:4176. [Google Scholar]
- 27.Analogous to the rationale for preparation of vic-fluorohydrin stereoisomers of indinavir: Myers AG, Barbay JK, Zhong B. J Am Chem Soc. 2001;123:7207. doi: 10.1021/ja010113y.
- 28.Sanacora G, Smith MA, Pathak S, Su HL, Boeijinga PH, McCarthy DJ, Quirk MC. Mol Psychiatry. 2014;19:978. doi: 10.1038/mp.2013.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.O’hagan D. Chem Soc Rev. 2008;37:308. doi: 10.1039/b711844a. [DOI] [PubMed] [Google Scholar]
- 30.α-Bromo nitroalkanes are selectively debrominated to nitroalkanes using free radical-mediated stannane reduction: Dong L-t, Lu R-j, Du Q-s, Zhang J-m, Liu S-p, Xuan Y-n, Yan M. Tetrahedron. 2009;65:4124.See also: Ono N, Kaji A. Synthesis. 1986;1986:693.
- 31.For α-nitroester denitration, see refs. 13h, 15.
- 32.Comparative analysis (1H, 19F NMR) for free base and TFA salt of β-fluorostilbene amine is provided in the SI. A complete study, including activity, of stereoisomers 8 will be reported in due course.
- 33.Reductive destannation of 7a favors the anti-fluoro amine (2:1 dr), assigned by chemical correlation (see SI). Assignment of relative configuration for 9 is tentative, based on comparison of relative 19F shifts and analogy, see SI and ref. 8b.
- 34.Takeuchi Y, Nagata K, Koizumi T. J Org Chem. 1989;54:5453. [Google Scholar]
- 35.(a) Couve-Bonnaire S, Cahard D, Pannecoucke X. Org Biomol Chem. 2007;5:1151. doi: 10.1039/b701559c. [DOI] [PubMed] [Google Scholar]; (b) Drouin M, Hamel JD, Paquin JF. Synlett. 2016;27:821. [Google Scholar]; (c) Nihei T, Nishi Y, Ikeda N, Yokotani S, Ishihara T, Arimitsu S, Konno T. Synthesis. 2016;48:865. [Google Scholar]
- 36.Fluoroalkene stereoisomers can exhibit mechanistic differences when engaging a receptor: Silverman RB, Bichler KA, Leon AJ. J Am Chem Soc. 1996;118:1253.
- 37.For a recent approach to the synthesis of terminal vinyl fluorides, see: Koh MJ, Nguyen TT, Zhang H, Schrock RR, Hoveyda AH. Nature. 2016;531:459. doi: 10.1038/nature17396.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.