

Abstract
Glucagon-like peptide-1 (GLP-1) promotes insulin secretion from pancreatic β-cells in a glucose dependent manner. Several pathways mediate this action by rapid, kinase phosphorylation-dependent, but gene expression-independent mechanisms. Since GLP-1-induced insulin secretion requires glucose metabolism, we aimed to address the hypothesis that GLP-1 receptor (GLP-1R) signalling can modulate glucose uptake and utilization in β-cells. We have assessed various metabolic parameters after short and long exposure of clonal BRIN-BD11 β-cells and rodent islets to the GLP-1R agonist Exendin-4 (50 nM). Here we report for the first time that prolonged stimulation of the GLP-1R for 18 hours promotes metabolic reprogramming of β-cells. This is evidenced by up-regulation of glycolytic enzyme expression, increased rates of glucose uptake and consumption, as well as augmented ATP content, insulin secretion and glycolytic flux after removal of Exendin-4. In our model, depletion of Hypoxia-Inducible Factor 1 alpha (HIF-1α) impaired the effects of Exendin-4 on glucose metabolism, while pharmacological inhibition of Phosphoinositide 3-kinase (PI3K) or mTOR completely abolished such effects. Considering the central role of glucose catabolism for stimulus-secretion coupling in β-cells, our findings suggest that chronic GLP-1 actions on insulin secretion include elevated β-cell glucose metabolism. Moreover, our data reveal novel aspects of GLP-1 stimulated insulin secretion involving de novo gene expression.
Introduction
Insulin release is a complex and highly controlled process1. It is dependent on stimulus-secretion coupling, whereby glucose catabolism within pancreatic β-cells generates the primary signal for secretion. ATP generated through glycolysis and mitochondrial respiration provides the key signal for closure of ATP-sensitive K+ channels, subsequently causing membrane depolarization and activation of voltage-dependent Ca2+ channels (VDCCs), Ca2+ influx and exocytosis of insulin vesicles2. Therefore, the normal response to carbohydrate containing meals involves a rise in blood glucose concentration and increased β-cell glucose metabolism, subsequently promoting insulin secretion. However the latter responses are impaired in type 2 diabetes (T2D), where β-cell dysfunction plays a major role3. Thus, any treatment capable of augmenting glucose metabolism in β-cells may result in clinical benefit from improvement in insulin secretion and overall glucose homeostasis.
Glucagon-like peptide-1 (GLP-1) physiologically induces glucose-dependent insulin secretion from β-cells and GLP-1 analogues ameliorate hyperglycaemia in T2D patients4–6. GLP-1 exerts its actions by binding to a G-protein coupled receptor (GLP-1R) expressed on the surface of many cells including β-cells, which, upon stimulation, leads to rapid activation of adenylyl cyclase thus increasing cAMP levels7. cAMP directly activates protein kinase A (PKA) and cAMP-regulated guanine nucleotide exchange factor 2 (Epac2), that act in concert to generate downstream signals resulting in increased insulin secretion8. Mechanisms include ATP sensitive K+ channel closure, facilitation of VDCCs opening, inhibiting membrane repolarization via Kv channels and Ca2+-induced Ca2+ release from cytoplasmic storage sites9–14.
All known mechanisms of GLP-1-induced insulin secretion depend on glucose metabolism. Thus, it is attractive to hypothesize that GLP-1 signalling could enhance flux through the glycolytic pathway to generate metabolic stimulus-secretion factors. However, it has previously been reported that acute exposure to GLP-1 does not affect energy metabolism in β-cells15. On the other hand, prolonged (16 h) stimulation with GLP-1 was shown to promote Hypoxia-Inducible Factor 1 (HIF-1) activity via induction of the mammalian Target of Rapamycin (mTOR)16. HIF-1 is a heterodimeric transcriptional factor composed of two subunits, HIF-1α and HIF-1β17. It induces metabolic reprogramming in response to hypoxia and growth factor signalling18, 19, partly by promoting transcriptional activation of glycolytic genes20. This piece of evidence suggests that GLP-1 may induce late metabolic changes, not yet elucidated, downstream to HIF-1α activation.
Here we show that chronic stimulation of the GLP-1R increases glycolysis and ATP production in β-cells through transcriptional activation and expression of glycolytic genes. Pharmacological inhibition of the PI3K/mTOR pathway abolished such effects, suggesting that the metabolic actions of GLP-1 depend on mTOR activity. In addition, we observed that HIF-1α protein levels accumulate downstream of mTOR in response to GLP-1R signalling, whereas we also demonstrated that depletion of HIF-1α impaired effects on glycolysis and transcriptional regulation of glycolytic genes. We propose that chronic exposure to GLP-1 signalling promotes mTOR-dependent metabolic reprograming via activation of the HIF-1 transcriptional program. Such metabolic reprograming persists after removal of receptor stimulation, this was evidenced by increased levels of insulin secretion and glucose utilization following withdrawal of Exendin-4.
Results
Prolonged GLP-1R Signalling Stimulates Glycolysis via Up-Regulation of Glycolytic Enzymes
GLP-1 requires the presence of glucose for stimulation of insulin secretion14; thus we investigated if GLP-1 could modulate glucose metabolism in β-cells. Rat insulin-secreting BRIN-BD11 cells or isolated murine islets were stimulated for 18 hours with 50 nM of the GLP-1 analogue Exendin-4 and a high (20 mM) glucose concentration. Media was changed and cells cultured for additional 24 hours in the absence of Exendin-4. The rationale for this approach was to investigate if GLP-1 signalling could cause metabolic reprograming that would persist after removal of receptor stimulation (Fig. 1A). Glucose consumption, lactate production, total ATP content and insulin secretion were determined after 24 hours of incubation in the absence of Exendin-4 (Fig. 1B–G). All parameters were significantly enhanced in cells pre-conditioned with Exendin-4 relative to control cells treated with high glucose alone. Exendin-4 may affect β-cell number by stimulating cellular proliferation21, 22. However, in our experimental model with BRIN-BD11 cells, 18 h exposure to 50 nM Ex-4 did not induce significant changes in cell numbers (Fig. S1). All primary outcomes presented herein were normalized by total DNA content, eliminating cell number effects from the interpretation of our data.
Figure 1.
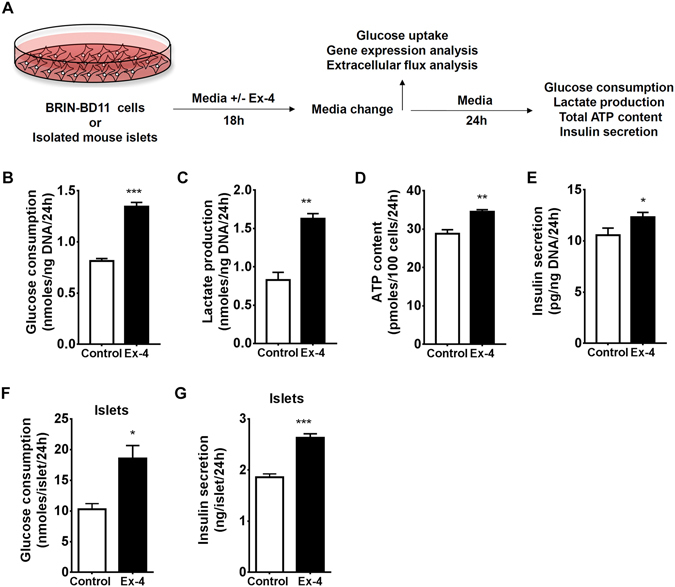
Chronic GLP-1R signalling stimulates β-cell glycolytic metabolism and function. (A) Schematic diagram outlining experimental procedures. (B) Glucose consumption, (C) lactate production, (D) ATP content, and (E) insulin secretion were assessed in BRIN-BD11 cells. (F) Glucose consumption and (G) insulin secretion were assessed in murine islets. Data represent mean ± SEM, n ≥ 3; *P < 0.05; **P < 0.01; ***P < 0.001.
Next, we assessed glycolytic flux by measuring extracellular acidification rates (ECAR) using the XFe96 flux analyzer. Exendin-4 pre-conditioned cells responded to both glucose and oligomycin injections by increasing their ECAR levels substantially higher than control cells (Fig. 2A). Basal glycolysis and glycolytic capacity can be calculated by subtraction of ECAR values obtained after glucose and oligomycin injections from basal ECAR levels respectively. ECAR measurements indicated that basal glycolysis (Fig. 2B) and glycolytic capacity (Fig. 2C) were both increased by Exendin-4 pre-conditioning. In order to confirm that the observed glycolytic effects require prolonged exposure to GLP-1R activation, we performed flux analysis after acute injection of Exendin-4 (Fig. 2D). Calculation of basal glycolysis and glycolytic capacity rates revealed an absence of any significant change after acute injection of Exendin-4 (Fig. 2E and F). To validate these findings, we assessed glucose uptake rates through flow cytometric measurement of a fluorescently labelled glucose analogue (2-NBDG). This demonstrated that cells pre-treated with Exendin-4 for 18 h responded by an approximate 25% increase in the rate of 2-NBDG uptake, whereas exposure to Exendin-4 for only 30 minutes did not elicit any significant change (Fig. 2G). Interestingly, cellular complexity and size, as estimated by side scatter (SSC) and forward scatter (FSC) respectively, both were increased as a result of 18 h Exendin-4 pre-treatment, but not after 30 minutes of exposure (Fig. 2H and I). Altogether, this dataset suggests that prolonged, but not acute, activation of GLP-1R induces metabolic reprogramming leading to increased glycolysis and cellular hypertrophy in BRIN-BD11 cells. To confirm that the observed effects were mediated through direct GLP-1R activation, or indeed, that they are not secondary autocrine effects of insulin, we performed 2-NBDG uptake experiments in the presence of the GLP1R antagonist Exendin (9–39) and the potassium channel opener, diazoxide. The effects of Exendin-4 were abolished in the presence of Exendin (9–39), but remained intact when insulin secretion was abrogated by diazoxide (Fig. 2J). This confirms that the observed metabolic reprogramming was dependent on chronic activation of GLP-1R signalling and independent of its acute stimulation of insulin secretion, which may result in autocrine insulin signalling.
Figure 2.
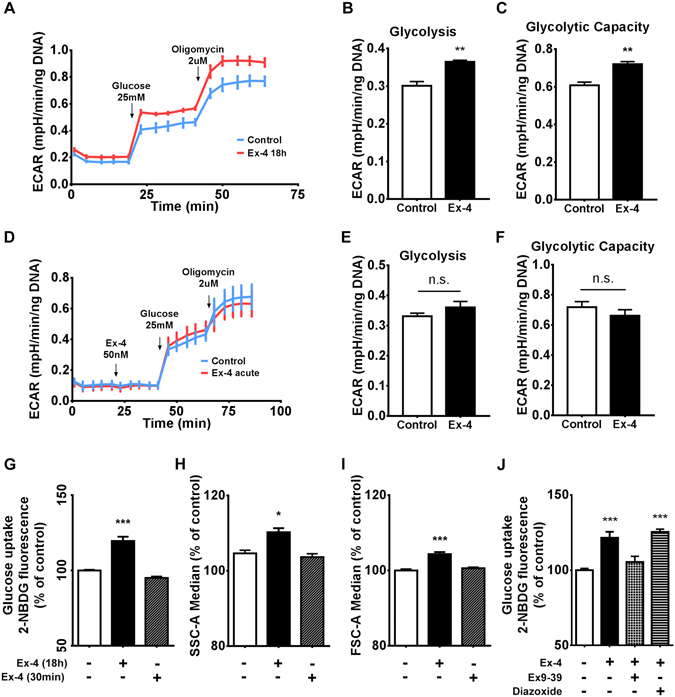
Prolonged, but not acute, Exendin-4 stimulation promotes metabolic reprogramming. Following 18 hours of incubation in the presence or absence of 50 nM Exendin-4, BRIN-BD11 cells were subjected to extracellular flux analysis. (A) ECAR was determined after sequential injection of 25 mM glucose and 2 µM oligomycin. (B and C) Glycolytic rate and capacity were calculated as detailed in Methods. (D) ECAR profiles were generated using control untreated cells to evaluate the effect of acute injection of 50 nM Exendin-4 compared to cells injected with control media, followed by 25 mM glucose and 2 µM oligomycin. (E and F) Glycolytic rate and capacity were calculated. (G) Uptake of 2-NBDG was determined by flow cytometry analysis after preconditioning in the presence or absence of 50 nM Exendin-4 for either 18 h or 30 min. (H and I) The same treatments were evaluated in respect to SSC-A and FSC-A to estimate cellular complexity and size respectively. (J) 2-NBDG uptake was determined after co-incubation for 18 h with 1 µM Exendin (9–39) or 250 µM diazoxide. Data represent mean ± SEM, n ≥ 3; n.s. = non-significant *P < 0.05; **P < 0.01; ***P < 0.001.
Flux through the glycolytic pathway is tightly controlled through regulation of glycolytic enzyme activity by allosteric modulation, post-translational modifications as well as transcriptional control. The time required for each of these regulatory mechanisms to occur is typically in milliseconds, seconds, and hours, respectively23. The extended time required for the effects observed here to take place suggests that they include regulation of gene expression. Hence, we assessed mRNA and protein expression profiles of genes involved in the glycolytic pathway. All genes analysed give rise to enzymes that catalyse rate-controlling steps in glycolysis, and therefore changes in their expression levels have the potential to affect the glycolytic flux. Four of the six genes investigated by quantitative RT-PCR were significantly up-regulated by Exendin-4 pre-conditioning, including Lactate dehydrogenase A (Ldha), Aldolase A (Aldoa), Phosphofructokinase (Pfkp) and Glucose-6-phosphate isomerase (Gpi) (Fig. 3A). Furthermore, we used a commercial glycolysis antibody panel to detect Pfkp, Pyruvate kinase M1 and M2 (Pkm1, Pkm2), Hexokinase 1, Hexokinase 2 and Pyruvate dehydrogenase α1 subunit (Pdh) by immunoblot analysis. A time-dependent increase in protein levels of the analysed glycolytic proteins was observed in response to Exendin-4 stimulation (Fig. 3B–H). Glucose transporter 2 (Glut2) and Glucokinase, which respectively mediate two early rate-controlling steps in the glycolytic pathway, glucose uptake and carbon 6 phosphorylation respectively were also investigated. Immunoblot analysis, however, indicated no significant changes in the protein levels of Glut2 and Glucokinase in response to Exendin-4 treatment (Fig. S2).
Figure 3.
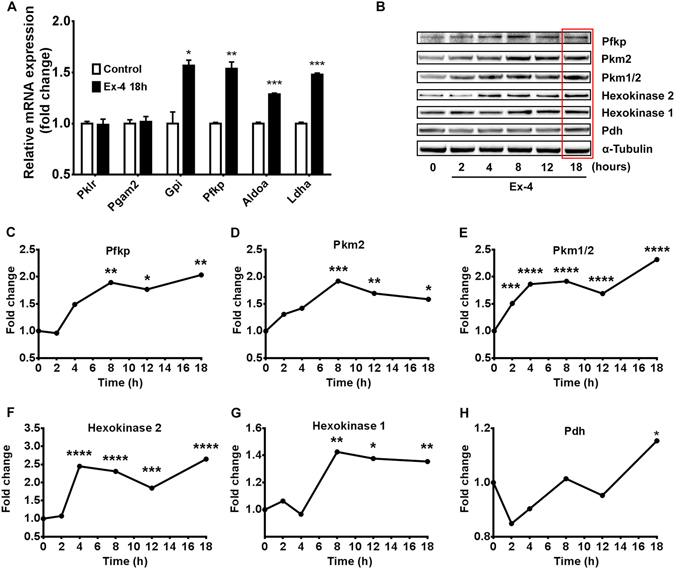
18 h of GLP-1R stimulation induces mRNA and protein expression of glycolytic enzymes. (A) mRNA expression of the glycolytic enzymes Pyruvate Kinase (Pklr), Lactate dehydrogenase A (Ldha), Aldolase A (Aldoa), Phosphofructokinase (Pfkp), Glucose-6-phosphate isomerase (Gpi) and Phosphoglycerate mutase 2 (Pgam2) was assessed by qRT-PCR in BRIN-BD11 cells exposed (or not) to 50 nM Exendin-4 for 18 hours. (B) Protein levels of glycolytic enzymes were analysed by immunoblot following 0, 2, 4, 8, 12 and 18 hours of exposure to 50 nM Exendin-4. Expression levels of (C) Pfkp, (D and E) Pyruvate Kinase M1 and M2 (Pkm1, Pkm2), (F) Hexokinase 1, (G) Hexokinase 2 and (H) Pyruvate dehydrogenase α1 subunit (Pdh) were quantified by band densitometry analysis. Data represent mean ± SEM, n ≥ 3; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Maximal Mitochondrial Respiration and Reserve Capacity are induced after Prolonged GLP-1R Activation
Extracellular flux analysis can be used to assess mitochondrial bioenergetics through measurements of oxygen consumption rates (OCR) following the injection of specific mitochondrial inhibitors24. We measured OCR in cells either pre-conditioned, or not, with Exendin-4 after injection of glucose, oligomycin, FCCP and a combination of rotenone and antimycin A (Fig. 4A). This strategy allowed us to estimate several mitochondrial parameters in response to GLP-1R stimulation. Maximal mitochondrial respiration and reserve capacity were strikingly enhanced by Exendin-4 preconditioning (Fig. 4B and C, respectively), whereas mitochondrial ATP turnover was slightly increased (Fig. 3D). On the other hand, basal respiration (Fig. 4E), proton leak (Fig. 4F) and non-mitochondrial OCR (Fig. 4G) were unaffected. Interestingly, maximal respiration and reserve capacity are both estimated from OCR measurements after injection of FCCP, an uncoupling agent that elevates mitochondria to their maximum electron transport capacity. Under these conditions, the production of glycolytic derived pyruvate becomes rate limiting for the Krebs cycle and, consequently, to mitochondrial electron transport. The observed effects in maximal respiration and reserve capacity most likely occur due to increased glycolysis and consequent augmented flux of pyruvate into the Krebs cycle.
Figure 4.
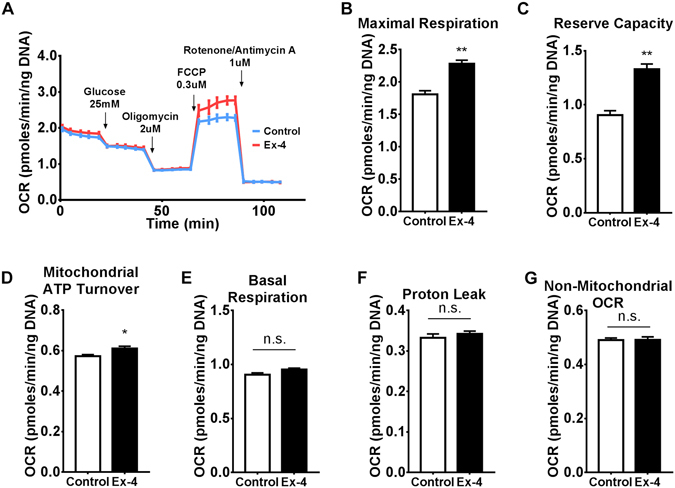
Maximal respiration and reserve capacity are increased after 18 h exposure to Exendin-4. (A) Following 18 h of incubation in the presence or absence of 50 nM Exendin-4, BRIN-BD11 cells were subjected to extracellular flux analysis. OCR was determined sequentially after injection of 25 mM glucose followed by a set of mitochondrial inhibitors (2 µM oligomycin, 0.3 µM FCCP and 1 µM each of rotenone and antimycin A) to generate a mitochondrial stress profile. (B–G) Mitochondrial parameters were calculated from the mitochondrial stress profile as described in Methods. (B) Maximal respiration, (C) reserve capacity, (D) mitochondrial ATP turnover, (E) basal respiration, (F) proton leak, (G) non-mitochondrial OCR. Data represent mean ± SEM, n = 3; n.s. = non-significant; *P < 0.05; **P < 0.01.
PI3K/mTOR Axis Mediates GLP-1R Signalling Actions on Glucose Metabolism
GLP-1 can regulate cell size, function and proliferation in β-cells via PI3K/mTOR pathway25, 26. Moreover, mTOR controls nutrient sensing and energy metabolism in various cell types27. Co-incubation of Exendin-4 with either LY294002 or Torin-1, two inhibitors of PI3K and mTOR respectively, completely abrogated the effect of Exendin-4 on glucose consumption (Fig. 5A) and uptake (Fig. 5B). The ECAR profile (Fig. 5C) revealed that mTOR inhibition slightly impaired responsiveness to glucose, but entirely abrogated cellular ability to increase ECAR to compensate for oligomycin-induced blockade of mitochondrial ATP production. Most importantly, Torin 1 abolished the observed increase in ECAR in response to Exendin-4. Estimation of basal glycolysis (Fig. 5D) and glycolytic capacity (Fig. 5E) indicated that inhibition of mTOR blunted such metabolic adaptations induced by GLP-1R activation.
Figure 5.
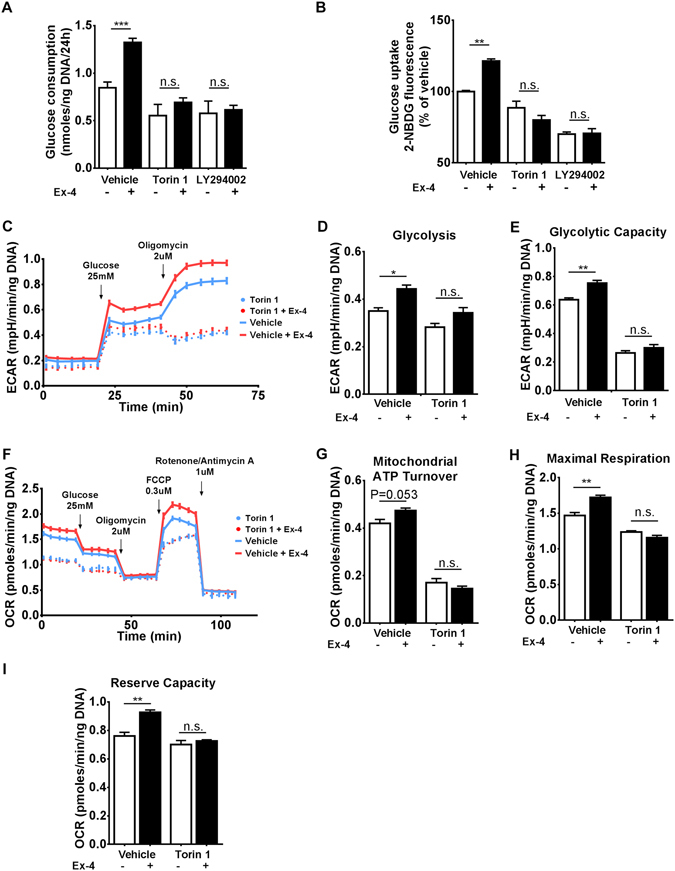
Inhibition of the PI3K/mTOR axis abolishes the effects of GLP-1R signalling on cellular metabolism. (A) BRIN-BD11 cells were pre-incubated with either vehicle, 1 µM Torin 1, or 50 µM LY294002 for 30 min, followed by addition (or not) of 50 nM Exendin-4 for 18 hours, as indicated. Then, media was changed to RPMI containing 20 mM of glucose for additional 24 hours in the absence of Exendin-4 and glucose consumption was determined. (B–I) Cells were treated similarly to (A), but instead of the additional 24 hours incubation time, 2-NBDG uptake or extracellular flux analysis were performed immediately following the 18 hours incubation period and parameters calculated as described in Methods. Data represent mean ± SEM, n ≥ 3; n.s. = non-significant; *P < 0.05; **P < 0.01; ***P < 0.001.
Next, we assessed the role of mTOR in mediating Exendin-4 effects on mitochondrial bioenergetics (Fig. 5F). mTOR inhibition not only impaired mitochondrial ATP turnover, but abolished the positive effect of Exendin-4 on this parameter (Fig. 5G). In addition, the effects of Exendin-4 on maximal mitochondrial respiration (Fig. 5H) and reserve capacity (Fig. 5I) were completely blocked by inhibition of mTOR.
Torin-1 is a potent inhibitor of both mTOR complexes 1 and 2, namely mTORC1 and mTORC2. In order to investigate specifically the role of mTORC1, we determined Exendin-4 effects on glucose consumption and uptake under inhibition of mTORC1 by Rapamycin, a compound that does not inhibit mTORC2 at the concentration tested (100 nM) (Fig. S2A and B, respectively). The results of these experiments demonstrate that in spite of the role of mTORC1 in the control of energy metabolism, as evidenced by a significant reduction of overall glucose consumption and uptake following Rapamycin treatment; Exendin-4 could still exhibit stimulatory activity in the same setting, suggesting that either mTORC2 alone or both mTOR complexes must be blocked in order to completely abrogate GLP-1R signalling action on glycolysis. Our data confirm the importance of mTOR activity in the regulation of β-cell energy metabolism and provide evidence that chronic GLP-1R signalling effects are mediated by the PI3K/mTOR pathway.
HIF-1α Accumulates Downstream of mTOR and is required for GLP-1-Induced Metabolic Reprograming
HIF-1 targets expression of glycolytic enzymes28; furthermore, GLP-1 has been shown to promote HIF-1α translation via induction of mTOR in mouse islets16. We hypothesized that HIF-1α mediates Exendin-4 effects on glycolysis in our model. Immunoblot analysis confirmed that Exendin-4 treatment induced HIF-1α protein levels similar to those achieved with Forskolin (Fig. 6A). Gene expression analysis revealed no changes in HIF-1α mRNA, suggesting that GLP-1R signalling induces HIF-1α protein by a post-transcriptional mechanism (Fig. 6B), consistent with previous observations16. Next, we assessed how HIF-1α levels are modulated by the activity of PI3K/mTOR axis in this setting. Inhibition of either PI3K or mTOR abolished accumulation of HIF-1α triggered by Exendin-4, suggesting that such event occurs downstream of mTOR (Fig. 6C). In addition, the two inhibitors caused HIF-1α protein to drop further below basal levels, indicating that mTOR pathway controls HIF-1α additionally in unstimulated conditions. Moreover, phosphorylation of mTOR at S2448 was enhanced by Exendin-4, confirming that GLP-1R signalling increases the activation state of mTOR, as previously demonstrated16, 29, 30. Knockdown of HIF-1α expression by siRNA transfection resulted in an approximate reduction in HIF-1α protein levels by 60% (Fig. 6D) and mRNA levels by 80% (Fig. 6E). We then sought to determine how HIF-1α knockdown influences the effects of Exendin-4 on glycolytic gene expression. Induction of Pdh and Pfkp by Exendin-4 was significantly blunted by HIF-1α knockdown (Fig. 6D). Protein levels of Pkm1, Pkm2, Hexokinase 1 and Hexokinase 2 were not affected (Fig. S4). Furthermore, depletion of HIF-1α caused a reduction in mRNA expression of glycolytic genes investigated, although only Pgam2 was statistically significant (Fig. 6F). This was accompanied by a reduction in glucose consumption and uptake in response to Exendin-4 (Fig. 6G and H, respectively), and also blocked the effects of Exendin-4 on cellular complexity and size (Fig. 6I and J, respectively). These findings indicate that HIF-1α was necessary for the observed effects of GLP-1R signalling on glucose metabolism.
Figure 6.
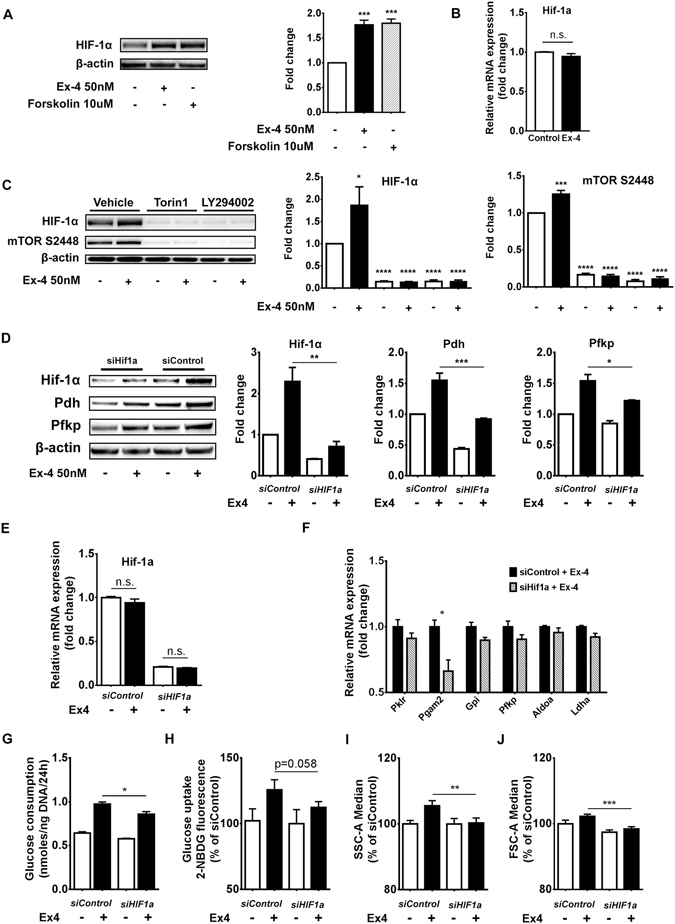
Increase in glucose metabolism induced by GLP-1R signalling is mediated by HIF-1α. (A) BRIN-BD11 cells were treated as indicated for 18 hours and Hif1α protein levels were determined by immunoblot analysis. (B) After treatment with Exendin-4 for 18 hours HIF-1α mRNA expression was evaluated by qRT-PCR. (C) BRIN-BD11 cells were pre-incubated with either vehicle, 1 µM Torin 1 or 50 µM LY294002 for 30 min as indicated, followed by addition (or not) of 50 nM Exendin-4 for 18 hours. Immunoblot shows Hif1α protein expression and phosphorylation levels of mTOR at serine 2448. (D–J) BRIN-BD11 cells were transfected with either HIF-1α specific siRNAs (siHIF1a) or non-targeting siRNAs (siControl) for 24 h, and subsequently exposed to 50 nM Exendin-4 (or not) for 18 h. (D) Exendin-4’s ability to promote protein levels of Pdh and Pfkp was significantly blunted in cells transfected with siHIF1a as compared to siControl transfected cells. (E) At the mRNA level, siHIF1a transfection caused a reduction of approximately 80% in HIF-1α expression. (F) mRNA expression of glycolytic enzymes was determined by qRT-PCR analysis. (G–J) Determination of glucose consumption, 2-NBDG uptake, SSC-A and FSC-A following HIF-1α knockdown. Data are mean ± SEM, n ≥ 3; n.s. = non-significant; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.001.
Discussion
Treatment of T2D patients with GLP-1 analogues results in various beneficial effects, including improvement in β-cell function and glucose homeostasis7. Yet, molecular mechanisms underlining GLP-1 mediated stimulation of insulin secretion are still not fully characterized. GLP-1-induced insulin secretion is believed to rely on the direct effects of cAMP signalling on ATP sensitive K+ channels and Ca2+ handling14. Such mechanisms occur very rapidly, within 5–15 min upon receptor binding, indicating that the actions are not mediated by altered gene expression, but likely by phosphorylation of protein components of the secretory machinery13. GLP-1 is known to acutely induce glucose competence in β-cells, both in isolated islets and in human subjects31, 32. Data presented herein support the notion that prolonged exposure to GLP-1R stimulation can also induce glucose competence by a secondary mechanism involving up-regulation of glycolytic enzymes, and consequently glycolytic flux, arguably the most important stimulus-secretion coupling factor generating pathway in β-cells. Other lines of evidence also suggest that GLP-1 may have additional effects other than the well-known acute insulin secretion stimulation. For instance, GLP-1 analogues promoted persistent improvement of β-cell function four weeks after treatment interruption, both in rodents and humans21, 33. Although this concept has been recently challenged with the publication of clinical trials showing that Liraglutide34 and Exendin-435 enhancement of β-cell function was lost after treatment cessation. However, it may be that the effect cannot be sustained for several weeks after treatment interruption.
Here we provide evidence that prolonged GLP-1R activation induces a robust metabolic adaptation in β-cells. In our model, cells primed with Exendin-4 presented higher glycolytic rates when returned to culture media in the absence of GLP-1R stimulation. This is concluded from evidence provided by glucose consumption, 2-NBDG uptake and lactate production measurements as well as ECAR and OCR profiles (Figs 1 and 2). BRIN-BD11 cells were used throughout this study for their well characterised responses to multiple stimuli, cell culture based convenience and their established intact response to GLP-1R activation both in terms of cAMP and resulting stimulated insulin secretion36, 37. We also validated findings such as Exendin-4 effects on glucose consumption and insulin secretion in primary isolated mouse islets. We believe that the increase in glucose metabolism occurs via up-regulation of glycolytic enzymes, since the observed metabolic effects were accompanied by significant upregulation in mRNA and protein expression of critical glycolytic genes (Fig. 3). In addition, our data also showed that acute Exendin-4 stimulation does not elicit change in metabolic flux (Fig. 2). Corroborating this hypothesis, HIF-1α, a transcriptional factor known to target glycolytic genes, accumulates under GLP-1R stimulation. Furthermore, HIF-1α depletion impacted glucose consumption and uptake, as well as glycolytic gene expression in the same setting (Fig. 6). The dataset presented here also indicate that the PI3K/mTOR pathway plays a crucial role in mediating GLP-1R signalling effects on energy metabolism. This is evident from experiments describing the effects of specific PI3K and mTOR inhibitors, which completely abrogated the metabolic actions of Exendin-4 (Fig. 5). Accordingly, HIF-1α accumulation was also abrogated upon PI3K/mTOR inhibition, further demonstrating its importance for the observed metabolic adaptations (Fig. 6). We and others have demonstrated that GLP-1R signalling elevated ATP levels38, 39, strengthening the hypothesis that the incretin hormone may promote β-cell function through regulation of energy metabolism, especially considering the central role that ATP generation and an increase in ATP/ADP ratio play during GSIS. Indeed, in our model, prolonged GLP-1R stimulation promoted a sustained increase in insulin secretion after Exendin-4 removal; this was observed both in BRIN-BD11 cells and murine islets (Fig. 1). We suspect that metabolic/bioenergetic reprograming, observed in this study, could also mediate short-term maintenance of β-cell function upon withdrawal of GLP-1 analogues, as documented in humans and rodents by others21, 33. Clearly though, such sustained effect will be completely lost after a few weeks of treatment interruption, as recent clinical evidence suggests34, 35.
Previously, it has been reported that acute exposure to GLP-1 did not significantly affect the metabolome40, nor bioenergetics of rodent β-cells15. Gheni et al. demonstrated that exposure to GLP-1 for 30 min did not induce changes to glycolytic and TCA cycle intermediates in MIN6-K8 cells40. A short-term (90 min) stimulation with GLP-1 did not cause changes in glucose and lipid oxidation in rodent islets15, although, the same study reported a significant increase in glucose utilization, which perhaps has not been given sufficient attention. Accordingly, our data also showed that acute GLP-1R stimulation did not cause changes to glucose metabolism. Hence, we speculate that the increase in glucose utilization observed by Peyot et al.15 reflects a different mechanism, reported recently, in which GLP-1 induces rapid post-translational activation of glucokinase41, 42. Data presented herein support a novel mechanism dependent on metabolic adaptations observed after a longer time period, which is consistent with previously identified delayed phase of gene expression changes mediated via HIF-1α16.
Interestingly, a recent study by the Mandrup laboratory has shown that exposure to high glucose (25 mM) elicits a biphasic response in INS-1E cells43. In that study, the glucose-sensing transcription factor carbohydrate response element binding protein (ChREBP) has been shown to initiate a regulatory program driving an early transcriptional activation of metabolic genes, which is followed by a second wave of transcriptional regulation wherein cell-cycle genes are activated and β-cell identity genes are repressed. In our model, β-cells were exposed to Exendin-4 and high glucose concomitantly. Therefore, the metabolic adaptations observed here probably occurred in the background of the recently identified glucose-induced transcriptional program. This suggests that ChREBP and downstream associated transcription factors might also play a role in the observed metabolic shift. This hypothesis is also supported by our HIF-1α knockdown experiments, in which GLP-1R signalling-induced gene expression and metabolic adaptations were only partially perturbed, pointing to a possible involvement of additional transcriptional factors. Future studies aiming at identifying global changes in gene regulation in response to prolonged exposure to GLP-1R stimulation can determine with greater detail the remaining players in the herein identified metabolic reprograming process.
Our study describes a novel action of GLP-1 signalling in β-cells, which stimulates metabolic/bioenergetic reprograming resulting in a high glycolytic/high ATP generating phenotype, as detailed in (Fig. 7). Such metabolic reprograming likely accounts for a major part of the mechanism(s) underlying GLP-1 stimulated insulin secretion. Our study provides evidence for an important mechanism by which GLP-1 analogues improve glucose homeostasis in T2D patients, unveiling new aspects that could be useful for developing new therapeutic strategies.
Figure 7.
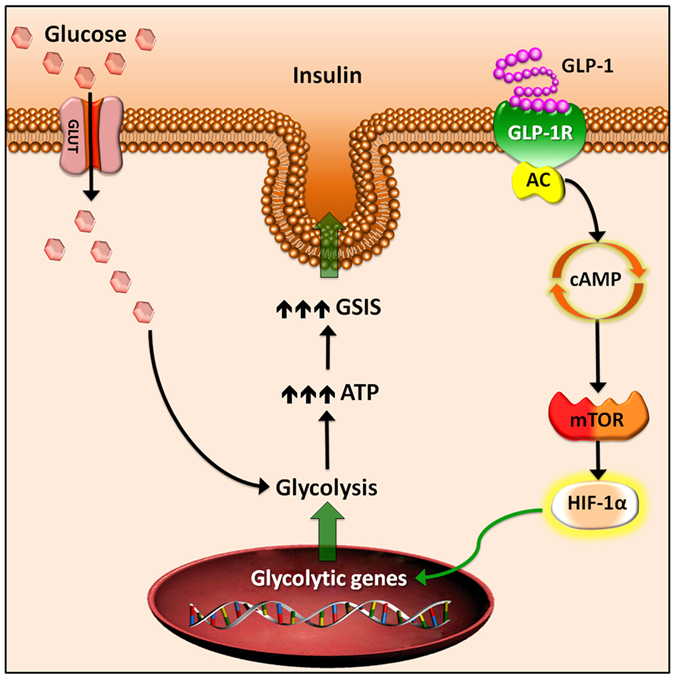
GLP-1R signalling promotes β-cell glucose metabolism via mTOR-dependent HIF-1α activation. Upon GLP-1 binding, the G-protein coupled GLP-1R receptor activates adenylyl cyclase (AC), increasing intracellular levels of cAMP. Signalling mediated by cAMP promotes the mTOR pathway in β-cells, which, in turn, induces translational activation of HIF-1α. In the nucleus, HIF-1α drives transcriptional up-regulation of glycolytic genes. Increased pool of glycolytic enzymes in the cytosol allows for an elevated glycolytic flux and glycolytic capacity, resulting in faster generation of ATP and enhanced glucose stimulated insulin secretion (GSIS).
Methods
Animals
Male C57BL/6J mice at 5–7 weeks of age were used for the purpose of pancreatic islets isolation. All animals were cared according to the guidelines for use and care of laboratory animals44. The study was approved by Curtin Ethics Committee for the use of animals in research (AEC-2014-27).
Reagents
All chemicals were purchased from Sigma-Aldrich, unless indicated otherwise. Rabbit polyclonal antibody against HIF-1α was obtained from (Novus Biologicals, Littleton, CO, USA, #NB100–449), while phosphorylated mTOR S2448, β-actin, α-Tubulin, as well as Glycolysis Antibody Sampler Kit to detect Pfkp, Pkm2, Pkm 1/2, Hexokinase 1, Hexokinase 2 and Pdh were all obtained from (Cell Signaling Technology, Beverly, MA, USA, #2971, #4970, #2148, #8337 respectively). Secondary antibodies consisted of horseradish peroxidase conjugated goat anti-rabbit IgG (Agilent’s Dako, Glostrup, Denmark). Torin 1, LY294002 and Forskolin were purchased from (ApexBio, Houston, TX, USA). ON-TARGETplus Rat Hif1a siRNA – SMARTpool (siHif1a) and Non-targeting Pool siRNA (siControl) were purchased from (Dharmacon, Lafayette, CO, USA - L-091718-02 and D-001810-10-05, respectively).
Cell culture
Clonal insulin-secreting BRIN-BD11 cells were maintained in RPMI 1640 medium containing 11.1 mM glucose, supplemented with 10% foetal bovine serum (FBS), 100 U/ml penicillin and 0.1 mg/ml streptomycin, pH 7.4, as described previously45. Cells were maintained in T75 sterile tissue culture flasks at 37 °C in a humidified atmosphere of 5% CO2 and 95% air.
Islet isolation
Islets were isolated by bile duct perfusion followed by collagenase P digestion as described previously46. After purification, islets were cultured overnight at 37 °C in a humidified atmosphere of 95% air and 5% CO2 in RPMI 1640 medium supplemented with 10% FBS, 100 units/ml penicillin and 0.1 mg/ml streptomycin. Islets were subsequently hand-picked and placed into individual micro centrifuge tubes for glucose consumption and insulin secretion assays.
Glucose consumption, lactate production, insulin secretion and total ATP content assays
After pre-conditioning in the presence or absence of 50 nM Exendin-4 for 18 hours, BRIN-BD11 cells or isolated mouse islets were cultured in RPMI containing 20 mM of glucose in the absence of Exendin-4 for additional 24 hours. Tissue culture media supernatants from the final 24 hours of culture were collected and assayed by Amplex Red Glucose/Glucose Oxidase Assay Kit (Life Technologies, Gaithersburg, MD, USA), Lactate Assay kit (Sigma-Aldrich) and Rat Ultrasensitive Insulin ELISA (Mercodia, Uppsala, Sweden) to measure total glucose, lactate and insulin respectively. Cells were lysed with RIPA buffer (Astral Scientific, Sydney, Australia) and total DNA quantified using Quanti-iT PicoGreen (Life Technologies, Gaithersburg, MD, USA). Results were reported as nmole of glucose and lactate, or pg of insulin. Results were normalized by total DNA or number of islets. For total ATP content measurements, cells were lysed with CellTiter-Glo® Reagent (Promega Corporation, Madison, WI, USA) and total ATP determined. All measurements were performed according to manufacturer’s instructions.
Glucose uptake assay
Glucose uptake was monitored with the fluorescent deoxyglucose analogue 2-(N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino)-2-deoxyglucose (2-NBDG) (Thermo Fisher Scientific, San Jose, CA, USA)47. After pre-conditioning, cells were washed and incubated in glucose-free DMEM medium containing 20 μM 2-NBDG for 30 min at 37 °C. Unlabelled controls were generated by incubating cells as described above, however in the absence of 2-NBDG. Cells were recovered by trypsinization and resuspended in 100 μL of PBS containing 1 μg/mL propidium iodide (PI). Ten thousand cells were evaluated by FACS LSR Fortessa flow cytometer (BD Biosciences, Heidelberg, Germany) and data were analysed in the FlowLogic FCS analysis software (Inivai Technologies, Melbourne, Australia). Median 2-NBDG, side scatter (SSC) and forward scatter (FSC) fluorescence were obtained after gating for PI negative cells.
Extracellular flux analysis
Bioenergetic parameters were determined using the XFe96 Extracellular Flux Analyzer (Seahorse Bioscience, North Billerica, MA). Cells were seeded into specialized 96-well plates at a density of 104 cells/well and allowed to adhere overnight. After pre-conditioning in the presence or absence of 50 nM Exendin-4 for additional 18 hours, culture medium was changed to serum-free DMEM containing 1 mM sodium pyruvate, without glucose and sodium bicarbonate. Plates were then incubated for one hour at 37 °C in a CO2-free atmosphere. Basal oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were determined. Next, OCR and ECAR profiles in response to injection of glucose (25 mM), oligomycin (2 µM), carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone (FCCP) (0.3 µM), and a combination of antimycin A (1 µM) and rotenone (1 µM) were evaluated. OCR and ECAR were measured using five 2 min cycles of mix and measurement following each injection. Normalization was performed by assessing total DNA using Quanti-iT PicoGreen (Life Technologies, Gaithersburg, MD, USA). Data analyses and calculations were performed as previously described48.
Quantitative RT-PCR
RNA was extracted from cell lysates using RNeasy Mini Kit (Qiagen, Chatsworth, CA, USA) and was reverse-transcribed using QuantiTect Reverse Transcription Kit (Qiagen, Chatsworth, CA, USA). Gene expression levels were quantified by qPCR using QuantiFast SYBR Green PCR Kit (Qiagen, Chatsworth, CA, USA) and reactions were performed on a ViiaTM 7 real time PCR system (Life Technologies, Gaithersburg, MD, USA). Gene-specific amplification was achieved using RT2 qPCR Primer Assays (Qiagen, Chatsworth, CA, USA). Catalogue numbers for each primer assays are presented in (Supplementary Table 1). Relative gene expression was determined by normalizing the expression of each target gene to β-actin.
Immunoblot analysis
HIF-1α protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions49. In order to avoid further HIF-1α degradation during whole cell lysates preparation, cells were lysed directly with NuPAGE® LDS Sample Buffer (1X) (Thermo Fisher Scientific, San Jose, CA, USA) in the presence of protease and phosphatase inhibitors cocktail (Cell Signaling Technology, Danvers, MA, USA), similarly to ref. 50. Protein extracts were separated by electrophoresis on 4–12% NuPAGE Bis-Tris Mini Gel and then transferred onto nitrocellulose membranes using iBlot transfer stacks (Life Technologies, Gaithersburg, MD, USA). Membranes were probed with various primary antibodies overnight at 4 °C. All washes and secondary antibody incubations were performed using the SNAP i.d. quick immunoblot vacuum system (Millipore, Billerica, MA, USA). Bands were developed using Clarity Western ECL substrate (Bio-Rad Laboratories, Hercules, CA, USA). Visualization and quantitative densitometry analysis were performed with Molecular Imager® Gel Doc™ XR System v5.2.1 (Bio-Rad Laboratories, Hercules, CA, USA).
Statistical analysis
Variables are reported as mean ± S.E.M. Differences across groups were tested using ANOVA with a Tukey post hoc test when more than two experimental groups were analysed and using Student’s t-test when only two experimental groups were compared. Statistical significance was set at P < 0.05 (two-tailed). All analyses were performed using GraphPad Prism software v. 6.0.
Electronic supplementary material
Acknowledgements
We thank the School of Biomedical Sciences and CHIRI for financial support and excellent research facilities, respectively. This study was partially supported by the Brazilian National Council for Scientific and Technological Development (CNPq) (grant number 402626/2012-5).
Author Contributions
R.C. and P.N. designed research; R.C., Y.C., J.R., V.C., K.K., L.E., and R.S. performed research; R.C., Y.C., J.R., V.C., and P.N. analyzed data; R.C., P.I.H.B., J.G., C.M., and P.N. wrote the paper. R.C. and Y.C. had equal contribution to this study. R.C. and P.N. are the guarantors of this study and take full responsibility for the work as a whole, including the study design, access to data, and the decision to submit and publish the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Rodrigo Carlessi and Younan Chen contributed equally to this work.
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-02838-2
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Rodrigo Carlessi, Email: rodrigo.carlessi@curtin.edu.au.
Philip Newsholme, Email: philip.newsholme@curtin.edu.au.
References
- 1.Henquin JC. Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes. 2000;49:1751–1760. doi: 10.2337/diabetes.49.11.1751. [DOI] [PubMed] [Google Scholar]
- 2.Ashcroft FM, et al. Stimulus-Secretion Coupling in Pancreatic Beta-Cells. J Cell Biochem. 1994;55:54–65. doi: 10.1002/jcb.240550007. [DOI] [PubMed] [Google Scholar]
- 3.Granhall C, Rosengren AH, Renstrom E, Luthman H. Separately inherited defects in insulin exocytosis and beta-cell glucose metabolism contribute to type 2 diabetes. Diabetes. 2006;55:3494–3500. doi: 10.2337/db06-0796. [DOI] [PubMed] [Google Scholar]
- 4.Drucker DJ, Nauck MA. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet. 2006;368:1696–1705. doi: 10.1016/S0140-6736(06)69705-5. [DOI] [PubMed] [Google Scholar]
- 5.DeFronzo RA, et al. Effects of exenatide (exendin-4) on glycemic control and weight over 30 weeks in metformin-treated patients with type 2 diabetes. Diabetes care. 2005;28:1092–1100. doi: 10.2337/diacare.28.5.1092. [DOI] [PubMed] [Google Scholar]
- 6.Vilsboll T, et al. Liraglutide, a long-acting human glucagon-like peptide-1 analog, given as monotherapy significantly improves glycemic control and lowers body weight without risk of hypoglycemia in patients with type 2 diabetes. Diabetes care. 2007;30:1608–1610. doi: 10.2337/dc06-2593. [DOI] [PubMed] [Google Scholar]
- 7.Baggio LL, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology. 2007;132:2131–2157. doi: 10.1053/j.gastro.2007.03.054. [DOI] [PubMed] [Google Scholar]
- 8.Seino S, Shibasaki T. PKA-dependent and PKA-independent pathways for cAMP-regulated exocytosis. Physiological reviews. 2005;85:1303–1342. doi: 10.1152/physrev.00001.2005. [DOI] [PubMed] [Google Scholar]
- 9.Light PE, Fox JEM, Riedel MJ, Wheeler MB. Glucagon-like peptide-1 inhibits pancreatic ATP-sensitive potassium channels via a protein kinase A- and ADP-dependent mechanism. Molecular endocrinology. 2002;16:2135–2144. doi: 10.1210/me.2002-0084. [DOI] [PubMed] [Google Scholar]
- 10.Kang GX, Leech CA, Chepurny OG, Coetzee WA, Holz GG. Role of the cAMP sensor Epac as a determinant of K-ATP channel ATP sensitivity in human pancreatic beta-cells and rat INS-1 cells. J Physiol-London. 2008;586:1307–1319. doi: 10.1113/jphysiol.2007.143818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.MacDonald PE, Wheeler MB. Voltage-dependent K+ channels in pancreatic beta cells: Role, regulation and potential as therapeutic targets. Diabetologia. 2003;46:1046–1062. doi: 10.1007/s00125-003-1159-8. [DOI] [PubMed] [Google Scholar]
- 12.Kang GX, et al. A cAMP and Ca2+ coincidence detector in support of Ca2+ induced Ca2+ release in mouse pancreatic beta cells. J Physiol-London. 2005;566:173–188. doi: 10.1113/jphysiol.2005.087510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu, B. et al. Synaptotagmin-7 phosphorylation mediates GLP-1-dependent potentiation of insulin secretion from beta-cells. Proceedings of the National Academy of Sciences of the United States of America, doi:10.1073/pnas.1513004112 (2015). [DOI] [PMC free article] [PubMed]
- 14.Meloni AR, DeYoung MB, Lowe C, Parkes DG. GLP-1 receptor activated insulin secretion from pancreatic ss-cells: mechanism and glucose dependence. Diabetes Obesity & Metabolism. 2013;15:15–27. doi: 10.1111/j.1463-1326.2012.01663.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peyot ML, et al. Glucagon-like peptide-1 induced signaling and insulin secretion do not drive fuel and energy metabolism in primary rodent pancreatic beta-cells. PloS one. 2009;4:e6221. doi: 10.1371/journal.pone.0006221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Van de Velde S, Hogan MF, Montminy M. mTOR links incretin signaling to HIF induction in pancreatic beta cells. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:16876–16882. doi: 10.1073/pnas.1114228108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Semenza GL. HIF-1: mediator of physiological and pathophysiological responses to hypoxia. Journal of applied physiology. 2000;88:1474–1480. doi: 10.1152/jappl.2000.88.4.1474. [DOI] [PubMed] [Google Scholar]
- 18.Semenza GL. HIF-1: upstream and downstream of cancer metabolism. Curr Opin Genet Dev. 2010;20:51–56. doi: 10.1016/j.gde.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Faubert B, et al. Loss of the tumor suppressor LKB1 promotes metabolic reprogramming of cancer cells via HIF-1 alpha. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:2554–2559. doi: 10.1073/pnas.1312570111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Semenza GL, Roth PH, Fang HM, Wang GL. Transcriptional Regulation of Genes Encoding Glycolytic-Enzymes by Hypoxia-Inducible Factor-1. Journal of Biological Chemistry. 1994;269:23757–23763. [PubMed] [Google Scholar]
- 21.Xu G, Stoffers DA, Habener JF, Bonner-Weir S. Exendin-4 stimulates both beta-cell replication and neogenesis, resulting in increased beta-cell mass and improved glucose tolerance in diabetic rats. Diabetes. 1999;48:2270–2276. doi: 10.2337/diabetes.48.12.2270. [DOI] [PubMed] [Google Scholar]
- 22.Tourrel C, Bailbe D, Meile MJ, Kergoat M, Portha B. Glucagon-like peptide-1 and exendin-4 stimulate beta-cell neogenesis in streptozotocin-treated newborn rats resulting in persistently improved glucose homeostasis at adult age. Diabetes. 2001;50:1562–1570. doi: 10.2337/diabetes.50.7.1562. [DOI] [PubMed] [Google Scholar]
- 23.Berg, J. M., Tymoczko, J. L. & Stryer, L. Biochemistry. 5th edn, (Freeman, W.H. 2002).
- 24.Ferrick DA, Neilson A, Beeson C. Advances in measuring cellular bioenergetics using extracellular flux. Drug Discov Today. 2008;13:268–274. doi: 10.1016/j.drudis.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 25.Nie J, et al. SAD-A kinase controls islet beta-cell size and function as a mediator of mTORC1 signaling. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:13857–13862. doi: 10.1073/pnas.1307698110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Buteau J, Foisy S, Joly E, Prentki M. Glucagon-like peptide 1 induces pancreatic beta-cell proliferation via transactivation of the epidermal growth factor receptor. Diabetes. 2003;52:124–132. doi: 10.2337/diabetes.52.1.124. [DOI] [PubMed] [Google Scholar]
- 27.Yuan HX, Xiong Y, Guan KL. Nutrient sensing, metabolism, and cell growth control. Mol Cell. 2013;49:379–387. doi: 10.1016/j.molcel.2013.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Semenza GL. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. The Journal of clinical investigation. 2013;123:3664–3671. doi: 10.1172/JCI67230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xie J, et al. Exendin-4 stimulates islet cell replication via the IGF1 receptor activation of mTORC1/S6K1. Journal of molecular endocrinology. 2014;53:105–115. doi: 10.1530/JME-13-0200. [DOI] [PubMed] [Google Scholar]
- 30.Miao XY, et al. The human glucagon-like peptide-1 analogue liraglutide regulates pancreatic beta-cell proliferation and apoptosis via an AMPK/mTOR/P70S6K signaling pathway. Peptides. 2013;39:71–79. doi: 10.1016/j.peptides.2012.10.006. [DOI] [PubMed] [Google Scholar]
- 31.Holz GGt, Kuhtreiber WM, Habener JF. Pancreatic beta-cells are rendered glucose-competent by the insulinotropic hormone glucagon-like peptide-1(7-37) Nature. 1993;361:362–365. doi: 10.1038/361362a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kjems LL, Holst JJ, Volund A, Madsbad S. The influence of GLP-1 on glucose-stimulated insulin secretion: effects on beta-cell sensitivity in type 2 and nondiabetic subjects. Diabetes. 2003;52:380–386. doi: 10.2337/diabetes.52.2.380. [DOI] [PubMed] [Google Scholar]
- 33.Bunck MC, et al. Effects of Exenatide on Measures of beta-Cell Function After 3 Years in Metformin-Treated Patients With Type 2 Diabetes. Diabetes care. 2011;34:2041–2047. doi: 10.2337/dc11-0291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Retnakaran R, Kramer CK, Choi H, Swaminathan B, Zinman B. Liraglutide and the preservation of pancreatic beta-cell function in early type 2 diabetes: the LIBRA trial. Diabetes care. 2014;37:3270–3278. doi: 10.2337/dc14-0893. [DOI] [PubMed] [Google Scholar]
- 35.Gudipaty L, et al. Effect of exenatide, sitagliptin, or glimepiride on beta-cell secretory capacity in early type 2 diabetes. Diabetes care. 2014;37:2451–2458. doi: 10.2337/dc14-0398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McClenaghan NH, et al. Characterization of a novel glucose-responsive insulin-secreting cell line, BRIN-BD11, produced by electrofusion. Diabetes. 1996;45:1132–1140. doi: 10.2337/diab.45.8.1132. [DOI] [PubMed] [Google Scholar]
- 37.Green BD, et al. Lys(9) for Glu(9) substitution in glucagon-like peptide-1(7-36)amide confers dipeptidylpeptidase IV resistance with cellular and metabolic actions similar to those of established antagonists glucagon-like peptide-1(9–36)amide and exendin (9–39) Metabolism-Clinical and Experimental. 2004;53:252–259. doi: 10.1016/j.metabol.2003.09.015. [DOI] [PubMed] [Google Scholar]
- 38.Hodson DJ, et al. Incretin-modulated beta cell energetics in intact islets of Langerhans. Molecular endocrinology. 2014;28:860–871. doi: 10.1210/me.2014-1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tsuboi T, et al. Glucagon-like peptide-1 mobilizes intracellular Ca2+ and stimulates mitochondrial ATP synthesis in pancreatic MIN6 beta-cells. Biochemical Journal. 2003;369:287–299. doi: 10.1042/bj20021288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gheni G, et al. Glutamate acts as a key signal linking glucose metabolism to incretin/cAMP action to amplify insulin secretion. Cell Rep. 2014;9:661–673. doi: 10.1016/j.celrep.2014.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Park JH, et al. Glucagon-Like Peptide-1 Enhances Glucokinase Activity in Pancreatic beta-Cells through the Association of Epac2 with Rim2 and Rab3A. Endocrinology. 2012;153:574–582. doi: 10.1210/en.2011-0259. [DOI] [PubMed] [Google Scholar]
- 42.Ding SY, Nkobena A, Kraft CA, Markwardt ML, Rizzo MA. Glucagon-like peptide 1 stimulates post-translational activation of glucokinase in pancreatic beta cells. The Journal of biological chemistry. 2011;286:16768–16774. doi: 10.1074/jbc.M110.192799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schmidt SF, et al. Integrative Genomics Outlines a Biphasic Glucose Response and a ChREBP-RORgamma Axis Regulating Proliferation in beta Cells. Cell Rep. 2016;16:2359–2372. doi: 10.1016/j.celrep.2016.07.063. [DOI] [PubMed] [Google Scholar]
- 44.National Research Council (U.S.). Committee for the Update of the Guide for the Care and Use of Laboratory Animals, Institute for Laboratory Animal Research (U.S.) & National Academies Press (U.S.). xxv, 220 p. (National Academies Press, Washington, D.C. 2011).
- 45.Dixon G, Nolan J, McClenaghan N, Flatt PR, Newsholme P. A comparative study of amino acid consumption by rat islet cells and the clonal beta-cell line BRIN-BD11 - the functional significance of L-alanine. Journal of Endocrinology. 2003;179:447–454. doi: 10.1677/joe.0.1790447. [DOI] [PubMed] [Google Scholar]
- 46.Szot, G. L., Koudria, P. & Bluestone, J. A. Murine pancreatic islet isolation. Journal of visualized experiments: JoVE255, doi:10.3791/255 (2007). [DOI] [PMC free article] [PubMed]
- 47.Zou CH, Wang YJ, Shen ZF. 2-NBDG as a fluorescent indicator for direct glucose uptake measurement. J Biochem Bioph Meth. 2005;64:207–215. doi: 10.1016/j.jbbm.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 48.Keane KN, Calton EK, Cruzat VF, Soares MJ, Newsholme P. The Impact of Cryopreservation on Human Peripheral Blood Leukocyte Bioenergetics. Clinical science. 2015 doi: 10.1042/CS20140725. [DOI] [PubMed] [Google Scholar]
- 49.Salceda S, Caro J. Hypoxia-inducible factor 1 alpha (HIF-1 alpha) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions - Its stabilization by hypoxia depends on redox-induced changes. Journal of Biological Chemistry. 1997;272:22642–22647. doi: 10.1074/jbc.272.36.22642. [DOI] [PubMed] [Google Scholar]
- 50.Schepens B, Tinton SA, Bruynooghe Y, Beyaert R, Cornelis S. The polypyrimidine tract-binding protein stimulates HIF-1 alpha IRES-mediated translation during hypoxia. Nucleic acids research. 2005;33:6884–6894. doi: 10.1093/nar/gki1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.