

Abstract
Histone modifications, including lysine methylation, are epigenetic marks that influence many biological pathways. Accordingly, many methyltransferases have critical roles in various biological processes, and their dysregulation is often associated with cancer. However, the biological functions and regulation of many methyltransferases are unclear. Here, we report that a human homolog of the methyltransferase SET (SU(var), enhancer of zeste, and trithorax) domain containing 3 (SETD3) is cell cycle-regulated; SETD3 protein levels peaked in S phase and were lowest in M phase. We found that the β-isoform of the tumor suppressor F-box and WD repeat domain containing 7 (FBXW7β) specifically mediates SETD3 degradation. Aligning the SETD3 sequence with those of well known FBXW7 substrates, we identified six potential non-canonical Cdc4 phosphodegrons (CPDs), and one of them, CPD1, is primarily phosphorylated by the kinase glycogen synthase kinase 3 (GSK3β), which is required for FBXW7β-mediated recognition and degradation. Moreover, depletion or inhibition of GSK3β or FBXW7β resulted in elevated SETD3 levels. Mutations of the phosphorylated residues in CPD1 of SETD3 abolished the interaction between FBXW7β and SETD3 and prevented SETD3 degradation. Our data further indicated that SETD3 levels positively correlated with cell proliferation of liver cancer cells and liver tumorigenesis in a xenograft mouse model, and that overexpression of FBXW7β counteracts the SETD3's tumorigenic role. We also show that SETD3 levels correlate with cancer malignancy, indicated by SETD3 levels that the 54 liver tumors are 2-fold higher than those in the relevant adjacent tissues. Collectively, these data elucidated that a GSK3β-FBXW7β-dependent mechanism controls SETD3 protein levels during the cell cycle and attenuates its oncogenic role in liver tumorigenesis.
Keywords: cell cycle, cell proliferation, liver cancer, protein degradation, protein phosphorylation, protein stability, methyltransferase
Introduction
Genetic information in eukaryotes is packaged in a highly conserved DNA-protein complex, chromatin, which supports and controls crucial functions of the genome. Dynamic alterations in chromatin occur during multiple genetic processes, such as transcription, DNA replication, DNA repair, and cell division, which are modulated, in part, by post-translational modifications of the DNA-associated histone proteins (1, 2). Histone modifications, including lysine methylation, provide a platform for the recruitment of a diverse array of epigenetic modifiers that influence these fundamental biological processes (3). In addition to the crucial role in transcription, the function of histone modifications in the cell cycle is of general interest.
The cell cycle ensures proper cell division. Cell division is tightly controlled by cyclin/cyclin-dependent kinase complexes, which regulate a strict order of temporal events (4). The cyclin/cyclin-dependent kinases undergo periodic proteolysis to maintain a normal cell cycle progression. Two important types of E3 enzymes are responsible for destroying the cell cycle regulators: the anaphase-promoting complex (APC)4 or cyclosome and the Skp, Cullin, F-box contaning complex (SCF). The APC or cyclosome is essential for metaphase-to-anaphase transition and for the transition of the cells from M phase to G1. Accordingly, the SCF complex is believed to predominantly regulate G1-S progression, but it is active throughout most phases of the cell cycle. The SCF complex consists of three invariable components (Cul1, Rbx1, and Skp1) and one of the variable F-box proteins, the substrate-targeting subunit that defines the specificity of each SCF ligase (5, 6). For example, the WD-40 repeat-containing F-box protein FBXW7 (also known as Fbw7, CDC4, Ago, or Sel-10) mediates the degradation of a network of proteins, such as cyclin E1, c-Myc, c-Jun, Notch, and KLF2 (5–8). Several known FBXW7 substrates also require a priming phosphorylation by the GSK3β kinase to generate a CDC4 phosphodegron (CPD) site for FBXW7 recognition (9, 10). The spatio-temporal regulation of these proteins ensures the orderly execution of cell division, cell growth, and differentiation.
In addition to the well-known cell cycle regulators, the contribution from histone modifications to cell cycle progression is evident. One such histone modification is methylation of histone H4 at Lys-20 (H4K20). Mono-methylation of H4K20 (H4K20me1) is solely catalyzed by the methyltransferase Set8 (also known as PR-Set7 or SETD8) (11–14). H4K20me1 is cell cycle-regulated, with the lowest levels in G1/S and a peak in M phase, indicating its critical roles in mitotic entry and genomic stability (15, 16). Interestingly, a line of studies found that Set8 itself also fluctuated, and its degradation was mediated by different E3 complexes during different cell cycle stages (17–25). Therefore, the temporal degradation of Set8 is critical for maintaining chromatin compaction, controlling DNA replication licensing and responding to DNA damage response, which indicates a complicated regulatory network of Set8.
As described above, histone lysine methylation is predominantly carried out by a group of proteins called methyltransferases that contain a SET domain named after SU(var), enhancer of zeste, and trithorax, the first three proteins shown to harbor this domain in Drosophila (26). Many methyltransferases have been found to have physiological significance. A large body of evidence has indicated that their dysregulation is involved in the development and progression of various diseases, including cancer (3). Although over 50 putative SET domain-containing proteins have been identified, the physiological and cellular functions of many of these proteins are still unknown. Of note, a subfamily of methyltransferases containing a Rubis-subs-bind domain is of particular interest since the characterization of SETD6 (26). SETD6 has been reported to function in the NF-κB signaling pathway, stem cell renewal, oxidative stress response, and Wnt signaling cascade via methylating different histones or non-histone substrates, indicating its biological significance (27–30). Recently, Kim et al. (31) identified another member protein of this family, called SETD3, in zebrafish. They showed that SETD3 might methylate histone H3 at Lys-36 (H3K36) and play roles in cell death and cell cycle regulation. Additionally, SETD3 was reported to be the enzyme responsible for H3K4 and H3K36, and it functioned in mouse muscle differentiation (32). Moreover, SETD3 may be involved in a chromosome translocation in B-cell lymphomas (33). Very recently, it has been reported that human SETD3 (hSETD3) can methylate the transcriptional factor FoxM1 and regulates VEGF expression under normoxic and hypoxic conditions (34). However, other biological functions of hSETD3 and how hSETD3 is regulated have not yet been determined.
In this study, we showed that hSETD3 is cell cycle-regulated. The SETD3 levels displayed a dynamic cell cycle profile; they peaked in S phase, declined during late S and G2 phase, and were the lowest in M phase. We further found that SETD3 levels were regulated in a GSK3β- and SCFFBXW7β-dependent manner. GSK3β-mediated phosphorylation and FBXW7β-mediated ubiquitination of SETD3 were required for its proteolysis. Interestingly, SETD3 levels were increased in human liver cancer cells. Overexpression of SETD3 in liver cancer cells promoted cell proliferation and tumorigenesis in liver cancer cells. Moreover, we revealed that SETD3 protein levels were correlated with high malignancy and poor prognosis in liver tumors. Thus, we established a link between the GSK3β-SCFFBXW7β axis and a potential oncogenic protein SETD3 both in the cell cycle and in tumorigenesis.
Results
hSETD3 protein levels fluctuate in the cell cycle
To explore the function of hSETD3, we first amplified its full-length gene from a human brain cDNA library by PCR and cloned it into the pcDNA3.1 vector. The nucleotide sequences of the Setd3 gene were confirmed by sequencing. Then the rabbit polyclonal antibody against SETD3 was generated, and the specificity of the purified α-SETD3 antibody was examined. A sharp band was detected in cells transfected with mock RNA but not in cells transfected with shRNA targeting SETD3 (supplemental Fig. S1A). The capacity of the α-SETD3 antibody to recognize endogenous SETD3 in cells was significantly reduced by preincubation with recombinant His-tagged hSETD3 but not with an equal amount of BSA (supplemental Fig. S1B). Moreover, using immunofluorescence staining analysis, we showed that hSETD3 predominantly localized in the cytoplasm, and a small portion of hSETD3 formed puncta in the nucleus. The signal intensities were dramatically diminished when SETD3 was knocked down in cells using shRNA, validating the specificity of the observed signals (supplemental Fig. S1C).
Previous studies of SETD3 in zebrafish suggested it might play a role in cell cycle regulation (31). Therefore, HeLa S3 cells were arrested at the G1/S boundary (G1/S), during prometaphase (M), or during S phase, and then the SETD3 protein levels were examined. We observed that the SETD3 levels were low in M phase, increased at the G1/S boundary, and peaked in S phase (Fig. 1A). To elucidate how SETD3 levels fluctuate in the cell cycle, we arrested the cells at the G1/S boundary, and then we released them into fresh media. The cells were collected at the indicated time points, and the cell cycle profile was analyzed by Western blotting and fluorescence-activating cell sorting (FACS) analysis. We observed that the levels of SETD3 gradually increased from the G1/S boundary, peaked in S phase (5 h after release), and then declined in the G2 phase (Fig. 1, B and C). The SETD3 levels gradually increased after thymidine-nocodazole (T/N) release, with the lowest levels in M phase (Fig. 1, D and E). To confirm that the SETD3 levels indeed peaked in S phase, we treated cells with hydroxyurea (HU) and then released them into fresh media. The SETD3 levels were reduced when the cells entered G2 phase (1 h after release), and the SETD3 protein reduction was slightly delayed compared with that of cyclin E1, suggesting that SETD3 may reach a maximum in S phase (Fig. 1, F and G). Together, these results indicate that SETD3 fluctuates during the cell cycle.
Figure 1.
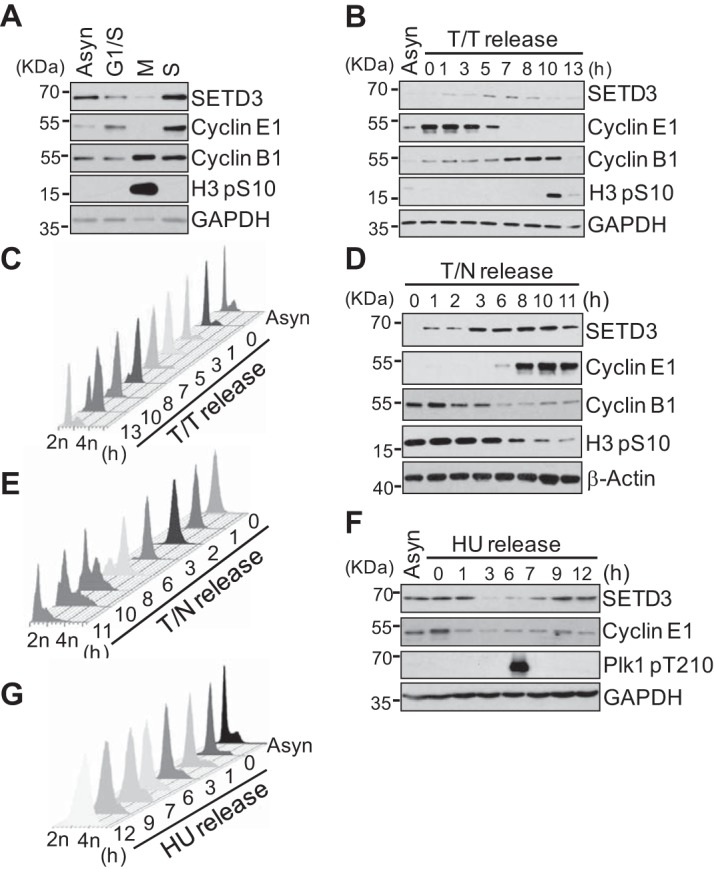
Protein levels of SETD3 fluctuate throughout the cell cycle. A, asynchronized (Asyn) or synchronized HeLa S3 cells at the G1/S boundary, late prometaphase (M), or S phase (S) were subjected to Western blotting using the indicated antibodies. B and C, HeLa S3 cells were synchronized at the G1/S boundary by a double thymidine arrest, released into fresh media, and harvested at the indicated times (T/T release). Protein levels were analyzed by Western blotting (B), and the cell cycle profile was assayed by FACS with propidium iodine (PI) staining (C). D and E, HeLa S3 cells were synchronized in late prometaphase by a thymidine-nocodazole arrest, released into fresh media, and harvested at the indicated times (T/N release). Protein levels were analyzed by Western blotting (D), and the cell cycle profile was assayed by FACS analysis with PI staining (E). F and G, HeLa S3 cells were synchronized in S phase by HU arrest, released into fresh media, and harvested at the indicated times (HU release). Protein levels were analyzed by Western blotting (F), and the cell cycle profile was assayed by FACS analysis with PI staining (G). The cyclin E1, cyclin B1, H3 phospho-S10, and Plk1 phospho-T210 antibodies were used for the cell cycle index. GAPDH and β-actin served as a loading control.
SCFFBXW7β ligase complex is required for SETD3 degradation
Next, we investigated whether alteration of the SETD3 protein level is mediated by proteasome-dependent proteolysis. HeLa S3 cells were arrested in M phase by nocodazole for 16 h and then released into fresh media containing DMSO or the proteasome inhibitor MG132 for 5 h before harvesting. SETD3 was stabilized with MG132 treatment, indicating that SETD3 is degraded by the proteasome (Fig. 2A). SETD3 protein is relatively unstable, as cells treated with the protein synthesis inhibitor cycloheximide (CHX) displayed SETD3 decay (supplemental Fig. S2A). In addition, exogenous GFP-tagged SETD3 in 293T cells was polyubiquitinated, as it appeared as a smear following immunoprecipitation (IP) using α-GFP antibodies and immunoblotting with an α-HA antibody recognizing ubiquitin (Fig. 2B). These results demonstrated that SETD3 is indeed degraded by the ubiquitin-conjugated proteasome pathway.
Figure 2.

SETD3 is degraded in a SCFFBXW7β- and proteasome-dependent manner. A, proteasome-dependent degradation of SETD3. Cells were synchronized in M phase and then treated with DMSO or 20 μm MG132 for 5 h before harvesting. Protein levels were analyzed by Western blotting. Asynchronous cells served as a control. B, in vivo ubiquitination assays were performed using 293T cells co-transfected with SETD3-GFP and an empty vector or HA-ubiquitin with or without MG132 treatment. SETD3 was immunoprecipitated with a GFP antibody, and the ubiquitinated levels were detected by immunoblots with α-HA. Cell lysates (Input) served as a control. C, several E3 ligases were co-transfected with FLAG-SETD3 and GFP in 293T cells, and protein levels of SETD3 were examined. GFP levels served as a control. The expressions of the indicated E3 ligases were detected by immunoblotting. Asterisks represent the corresponding proteins of expressed E3 ligases. D, three isoforms of FBXW7 were individually co-transfected with SETD3-GFP and the GFP vector. Relative protein levels of SETD3 were examined. E, knockdown of FBXW7 increased endogenous SETD3 protein levels. HeLa cells were transfected with control siRNA or three validated siRNAs targeting FBXW7β. Endogenous SETD3 levels were analyzed by Western blotting with GAPDH as a loading control. F, cells transfected with siControl or siFBXW7β RNA were treated with CHX. SETD3 stability was monitored at the indicated time points. The levels of cyclin E1 were used as a control (left panel). Two independent experiments were quantified by densitometry of Western blot analysis using ImageJ and presented as mean ± S.D. (right panel). G, SETD3 interacts with subunits of the SCFFBXW7β complex examined by reciprocal co-IP experiments. Co-IP assays were performed using SETD3 or FBXW7 antibody, and the subunits of the SCFFBXW7β complex, including Cul1, Rbx1, FBXW7, and Skp1 (left panel), or SETD3 (right panel) were analyzed by Western blotting. Ten percent of the total cell lysates (Input) and nonspecific IgG IP served as a control. Asterisk represents the light chain of IgG. H, co-IP assay was performed using M2 FLAG resin in 293T cells expressing SETD3-GFP and FLAG-FBXW7β. The interaction between SETD3 and FBXW7β was analyzed by Western blotting. Five percent of total cell lysates (5% input) were loaded as a control. I, F-box domain of FBXW7β is required for SETD3 degradation. Empty vector, wild-type, or F-box deletion mutant of FBXW7β was co-transfected with SETD3-GFP and the GFP vector, and the protein levels of SETD3 were examined. J, pathological mutants of FBXW7β construct derived from somatic mutations showed reduced ability to destruct SETD3. Cells expressing SETD3-GFP were co-transfected with empty vector, WT, or FBXW7β-GST mutants. Exogenous SETD3 levels in the indicated cells were analyzed by Western blotting probed with α-GFP and α-SETD3 antibodies. K, 293T cells were transfected with an empty vector, SETD3-GFP, HA-ubiquitin, or FLAG-FBXW7β as indicated, followed by IP with α-GFP. The ubiquitinated SETD3 was detected by Western blotting with α-HA.
To explore the mechanism of SETD3 degradation, we first attempted to identify the E3 ligase responsible for SETD3 fluctuation in the cell cycle. Given that the SETD3 protein levels declined after S phase but before G2/M, we speculated that an SCF ligase complex may mediate SETD3 degradation. To test this hypothesis, we co-transfected several F-box proteins or the APC activators (Cdc20 and Cdh1) with FLAG-SETD3 into 293T cells, and the relative SETD3 protein levels were examined by Western blotting with an α-SETD3 antibody. To eliminate the effect of transfection variation of the SETD3 plasmid, equal amounts of a GFP construct were co-transfected into every sample. As shown in Fig. 2C, overexpression of FBXW7β, but not other indicated F-box proteins, decreased the level of SETD3, indicating that FBXW7β may mediate SETD3 degradation.
The FBXW7 gene encodes three isoforms, α-, β-, and γ-, and the α- and γ-isoforms have been extensively shown to be adaptor proteins that recognize different substrates (6). However, only the β-isoform of FBXW7 efficiently decreased SETD3, despite the low levels of the β-isoform (Fig. 2D). In contrast, knockdown of FBXW7β increased endogenous SETD3 levels (Fig. 2E). Degradation of SETD3 is dependent on FBXW7β, as knockdown of FBXW7β compromised SETD3 protein turnover in the presence of CHX (Fig. 2F). Given that no specific antibody recognizing FBXW7β was commercially available, knockdown efficacy was examined by comparing FBXW7β mRNA levels in siControl samples versus siRNAs targeting FBXW7β samples using real-time quantitative PCR analyses (supplemental Fig. S2, B and C). Furthermore, we found that endogenous SETD3 interacts with subunits of the SCF complex, including Cul1, Rbx1, Skp1, and FBXW7, and endogenous FBXW7 can also bind SETD3 (Fig. 2G). In addition, GFP-SETD3 co-precipitated with FLAG-FBXW7β in cells (Fig. 2H). Thus, these data indicated that the SCFFBXW7β complex directly ubiquitinates SETD3 and mediates SETD3 degradation.
F-box proteins, including FBXW7β, contain at least one F-box domain, and the F-box domain is believed to mediate interactions with SCF substrates. To determine whether the F-box domain of FBXW7β is important for SETD3 degradation, we generated an FBXW7β mutant lacking an F-box domain (named FBXW7βΔF-box). Overexpression of this mutant abolished the reduction in SETD3 levels, demonstrating the importance of the F-box domain in SETD3 degradation (Fig. 2I). From the Catalog of Somatic Mutations in Cancer database, a total of 187 missense FBXW7β mutations were found in various cancer tumors, and 141 mutations occurred in the WD-40 domain (amino acids 287–627). Interestingly, several pathological mutations in this region, including R385C, R425C, R385H, and R399Q, were the top frequent mutations in the identified list (data not shown). Of note, these Arg residues have also been shown to be critical for substrate binding (35). Thus, the R385C, R399Q, and R425C mutants of FBXW7β were transfected into cells, and their impact on SETD3 protein levels was analyzed. Compared with wild-type FBXW7β, these mutants modestly reduced exogenous SETD3 levels, suggesting that the WD-40 domain of FBXW7β may be needed for SETD3 binding (Fig. 2J).
Next, we determined whether the SCFFBXW7β complex was required for SETD3 degradation. We found that overexpression of FBXW7β in cells facilitated poly-ubiquitination of SETD3 (Fig. 2K). Moreover, FBXW7β-mediated polyubiquitin chains on SETD3 are predominantly mediated through Lys-48-conjugated ubiquitin because ubiquitin containing Lys-48 as the only remaining Lys residue (K48O) was conjugated efficiently, but ubiquitin containing Lys-63 only (K63O) or all Lys-to-Arg (K0) mutations was not (supplemental Fig. S2D). Thus, we concluded that SETD3 is ubiquitinated in an SCFFBXW7β-dependent manner.
SETD3 is degraded in a GSK3β-dependent manner
As shown in previous studies, GSK3β is the priming kinase that phosphorylates most, if not all, FBXW7 substrates, which is a prerequisite for FBXW7 recognition and subsequent substrate destruction (5). To determine whether GSK3β was required for SETD3 degradation, we transfected 293T cells with GSK3β and SETD3, and the SETD3 protein levels were measured. As shown in Fig. 3A, GSK3β overexpression significantly reduced SETD3 levels. This reduction is due to GSK3β expression but not due to variations in the transfected Setd3 plasmids, as co-transfected GFP expression was constant in each sample. Conversely, knockdown of GSK3β by two independent siRNA oligonucleotides increased levels of SETD3, as well as cyclin E1, a known GSK3β-dependent FBXW7 substrate (Fig. 3B). Moreover, cells treated with a novel GSK3β covalent inhibitor, PLS-02-061 (IC50 = 96 nm), or lithium chloride (LiCl) showed increased SETD3 levels in a dose-dependent manner, suggesting a role for the GSK3β kinase activity in SETD3 degradation (Fig. 3, C and D). Similar to previous results, treating cells with PLS-02-061 significantly prohibited SETD3 protein turnover even in the presence of CHX, confirming that SETD3 protein degradation relies on GSK3β activity (Fig. 3E). Most importantly, in vitro reconstituted ubiquitination assays showed that SETD3 can be polyubiquitinated in the presence of the SCF complex containing Fbw7β, as well as GSK3β, and lack of either the E1, E2, or E3 enzyme abolished polyubiquitination of SETD3 (Fig. 3F and supplemental Fig. S3A). Altogether, this evidence supported that GSK3β is required for SETD3 destruction.
Figure 3.

SETD3 is degraded in a GSK3β-dependent manner. A, GFP vector and SETD3-GFP construct were co-transfected with an empty vector or FLAG-GSK3β into 293T cells. Relative levels of SETD3-GFP were analyzed by Western blotting. B, knockdown of GSK3β increased endogenous SETD3 levels. The 293T cells were transfected with control siRNA or two validated siRNAs targeting GSK3β. Protein levels of SETD3 and cyclin E1 were examined. C and D, inhibition of GSK3β increased SETD3 levels. HeLa cells were treated with different doses of the GSK3β inhibitor for 48 h in C or with 40 mm LiCl for 48 h in D. The endogenous SETD3 levels were examined by Western blotting. The cyclin E1 levels served as a control. The levels of phosphorylated GSK3β at Ser-9 were used to indicate the inhibitory efficiency of GSK3β. E, cells were treated with DMSO or a GSK3β inhibitor PL-02-061 in the presence of CHX. SETD3 stability was monitored at the indicated time points. F, in vitro ubiquitination assays were performed using individual recombinant proteins incubated with recombinant GST-SETD3, and the polyubiquitinated SETD3 bands were immunoblotted with an α-ubiquitin antibody.
CDC4 phosphodegron is required for SETD3 degradation
Next, we identified the phosphorylation sites of SETD3 by GSK3β. Most FBXW7 substrates contain a conserved phosphorylation degron CPD (supplemental Fig. S3B). By searching the protein sequence of hSETD3, we found six putative CPDs similar to the typical sequence, which contains two Thr/Ser residues separated by three or four residues which usually include proline residues (Fig. 4A). To determine whether these CPDs are required for FBXW7β-mediated SETD3 degradation, we generated a single CPD mutant in which the Ser/Thr residues were changed to alanine. Our data showed that only the CPD1 and CPD3 mutations completely abolished SETD3 degradation mediated by FBXW7β and GSK3β (Fig. 4B and supplemental Fig. S3C). Intriguingly, only the CPD1 mutation, but not other mutations, markedly reduced FBXW7β binding to SETD3, indicating that CPD1 may be the major, if not the only, phosphorylation site of GSK3β on SETD3 (Fig. 4C). To further explore whether both CPD1 and CPD3 mutations contribute to GSK3β binding SETD3, the capacity of this double mutation in protein degradation and FBXW7β binding was examined. We found that this CPD1 + CPD3 double mutation prevented SETD3 degradation, and it only attenuated but did not abolish the association with FBXW7β, which is similar to the effect generated by a CPD1 single mutant (Fig. 4, D and E). These results indicated that FBXW7β-dependent SETD3 degradation might be regulated by both CPDs but predominantly by CPD1. In addition, recombinant wild-type SETD3 was efficiently phosphorylated by GSK3β in vitro, whereas phosphorylation of the CPD1 mutant was significantly decreased, further supporting the major role of CPD1 (Fig. 4F). Notably, the CPD1 of SETD3 (SETD3CPD1) is highly conserved in most vertebrates (supplemental Fig. S3D). Therefore, these biochemical results suggest that GSK3β-mediated phosphorylation and subsequent ubiquitination of SETD3 by FBXW7β are mainly regulated in a CPD1-dependent manner.
Figure 4.

CPD motif is required for SETD3 degradation. A, alignment of six putative CPD sequences in SETD3. The numbers represent the first amino acid position, and the amino acids in red indicate the ones mutated to alanine. B, wild-type or six putative CPD mutants of SETD3-GFP (named m1–m6) were co-transfected with either the empty vector or FBXW7β and GSKβ accompanied by the GFP vector in 293T cells. SETD3 protein levels were analyzed by Western blotting. C, wild-type or four CPD mutants (m1–m4) of SETD3-GFP were co-transfected with FLAG-FBXW7β in 293T cells. FBXW7β was subjected to IP with α-FLAG, and associated SETD3-GFP proteins were analyzed by immunoblotting with α-GFP. D and E, double CPD mutant of SETD3 (m1 + m3) was examined by protein stability (D) or binding with FBXW7β (E), compared with wild-type or single mutants. F, in vitro kinase assays were performed using recombinant wild-type (WT) or a CPD1 mutant (m1) of GST-SETD3 incubated with different doses of GSK3β protein in the presence or absence of 32P-labeled γ-ATP. The phosphorylation signals of SETD3 were examined by autoradiography. The amounts of SETD3 protein used in each reaction were monitored by Coomassie Brilliant Blue staining (CBB).
SETD3 promotes cellular proliferation and tumorigenesis in cancer cells
A recent report (33) suggested that SETD3 may be associated with lymphoma oncogenesis; thus, we determined whether SETD3 protein levels are related to carcinogenesis. First, the SETD3 levels in various human cell lines were examined, in which they varied in different cell lines. Interestingly, SETD3 levels were lower in the normal liver L02 cell line compared with the cancer liver BEL7402 cell line (supplemental Fig. S4A). In addition, we found that SETD3 protein levels were relatively lower in the liver tissues than those in other human tissues (supplemental Fig. S4B). These results indicated a positive correlation between SETD3 and liver carcinogenesis. Consistent with this hypothesis, Western blot analysis showed that SETD3 protein levels gradually increased along with an enhancement in the malignancy of the liver cells (Fig. 5A). Interestingly, we found that, in the presence of CHX, turnover rate of SETD3 in the L02 normal cell line was much faster than that in the two tumor cell lines, which may explain why SETD3 protein levels are different in distinct liver cell lines (Fig. 5B). To further determine whether SETD3 promotes liver cancer cell proliferation, we performed cell growth assays and colony formation assays using the indicated liver cell lines either stably expressing SETD3 or shRNA specifically targeting SETD3. L02 or HepG2 cells expressing SETD3 showed increased proliferation compared with that of the control cells, and they also formed ∼1.6-fold more colonies than did the control cells (supplemental Fig. S4, C and D). Conversely, knockdown of SETD3 in HepG2 or BEL7402 cells markedly reduced cell proliferation, which was assessed by two different approaches (cell proliferation assay versus the real-time cell analyzer system), compared with the control cells (Fig. 5C and supplemental Fig. S4E). BEL7402 or HepG2 cells with knockdown of SETD3 formed 60% or even fewer colonies than did the control cells (Fig. 5D and supplemental Fig. S4F). These data further indicated that SETD3 promotes cell proliferation in liver cancer cells.
Figure 5.

SETD3 promotes cellular proliferation and tumorigenesis in liver cancer cells. A, relative SETD3 protein levels in various liver cell lines were examined by Western blotting with β-actin as a loading control. B, SETD3 protein stability in the indicated liver cell lines treated with CHX was examined by Western blotting (left panel), and relative SETD3 protein levels were plotted (right panel). C–F, cellular proliferation and colony formation assays were performed in the indicated HepG2 cell lines of either knockdown SETD3 by shRNA (C and D) or overexpression of SETD3 and FBXW7β (E and F). Cell growth curves were plotted over several consecutive days in C and E. Colony formation assays using the indicated cell lines were performed in D and F. Quantifications of colony numbers from the representative images (3 × 3 cm) from three independent plates are shown in D and F (right panel). G and H, SETD3 and FBXW7β regulate tumorigenicity of the liver cancer cells in nude mice. Images show tumor formation in nude mice (G) and the dissected tumors 27 days after injection (H). Tumor masses for each group are shown. Data are presented as mean ± S.E. *, p < 0.05; ***, p < 0.001, n.s., not significant.
To determine whether the effect of SETD3 on cell growth was due to its dynamic protein levels, we established stable cell lines ectopically co-expressing SETD3 and FBXW7β in HepG2 cells and examined cellular growth. As expected, this cell line displayed significantly lower cell proliferation and colony formation abilities than the cells expressing SETD3 alone, confirming the correlation of SETD3 and FBXW7β (Fig. 5, E and F).
Subsequently, a xenograft tumor model in nude mice was employed to probe the oncogenic effect of SETD3 in vivo. The liver cancer cells (HepG2) stably expressing SETD3, FBXW7β, or a FBXW7βΔF-box mutant were injected into nude mice. The efficiency of tumorigenicity of the cancer cells in each group was assessed 27 days after the injection. HepG2 cells expressing exogenous SETD3 alone formed tumors of ∼2.5-fold larger size than those in the control group. Cancer cells with endogenous SETD3 knockdown formed tumors about 35% smaller than the controls, which again suggested an oncogenic effect of SETD3 protein. Remarkably, cancer cells expressing SETD3 with wild-type FBXW7β barely formed tumors. In contrast, cancer cells expressing SETD3 with the FBXW7βΔF-box mutant that impaired its ubiquitination activity formed tumors of similar sizes compared with those expressing SETD3 alone (Fig. 5, G and H, and supplemental Fig. S4G). This result highlighted the possibility that tumor suppression by co-overexpression of SETD3-FBXW7β might be largely mediated through the FBXW7β-facilitated poly-ubiquitination of SETD3.
SETD3 is up-regulated in human liver cancer
To determine whether the oncogenic effects of SETD3 expression on liver cells are representative of what occurs in human tissues, mRNA expressions of the Setd3 gene in liver tissues obtained from The Cancer Genome Atlas were analyzed. Unexpectedly, the expression levels of Setd3 in tumors were slightly lower than those in normal tissues. The same was true for the expression of FBXW7, suggesting that FBXW7 may only regulate SETD3 at the protein level (supplemental Fig. S5A). Thus, we collected 54 pairs of tumor and adjacent liver tissues and examined the protein levels of SETD3 in each pair. Western blot analysis showed that SETD3 levels were obviously up-regulated in cancer tissues compared with adjacent tissues among the 32 paired samples (Fig. 6A), and quantification data showed that the average SETD3 protein levels are roughly 2-fold higher in cancer tissues than those in adjacent tissues (Fig. 6B). To further elucidate the relationship between SETD3 protein levels and clinical diagnosis, we examined SETD3 expression in liver clinical specimens by immunohistochemical (IHC) staining of tissue microarrays, which contained 53 pairs of tumor and adjacent tissues. Based on staining intensity, we grouped the liver tumor specimens according to SETD3 expression level as negative/weak, moderate, and strong. Using levels in adjacent tissues as controls, we found that SETD3 was up-regulated in liver cancers (Fig. 6, C and D). Remarkably, SETD3 levels were positively correlated with clinical staging (Fig. 6, E and F). Thus, we concluded that high expression of SETD3 is associated with high malignancy in liver tumors. The results underscored the association between liver cancers and the dysregulation of SETD3 expression.
Figure 6.
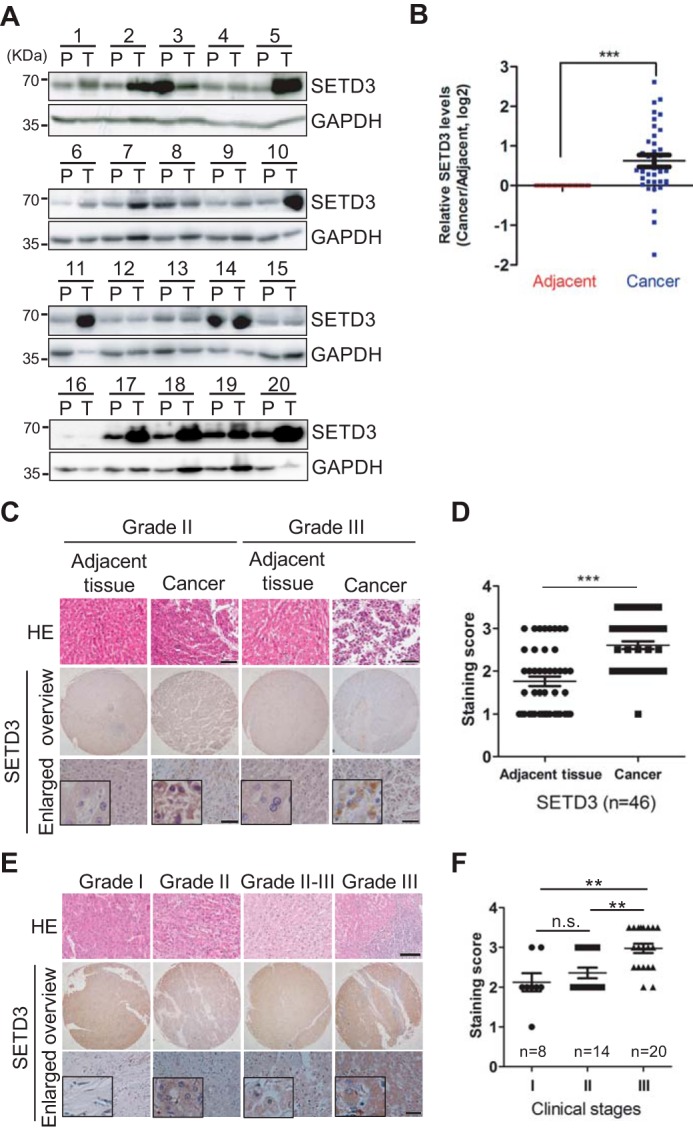
SETD3 protein levels are correlated with liver tumor. A and B, 54 pairs of human liver samples including adjacent tissues (P) and tumor tissues (T) were analyzed. 20 pairs of representative images using Western blot analysis examining SETD3 protein levels are shown in A. Arabic numerals represent individual patient case number. Average protein levels of SETD3 in the tumor tissues compared with the corresponding adjacent tissues were quantified in B. C and D, representative IHC staining images of SETD3 in human liver cancer and adjacent tissues obtained from hepatocellular carcinoma (HCC) tissue microarrays shown in C (53 pairs, 106 spots, each pair contains a cancer tissue and an adjacent tissue). We omitted seven pairs of spoiled dots in the microarray chips, and scores of the SETD3 staining in 46 pairs of human liver cancer and adjacent tissues were plotted in D. E and F, representative IHC staining images of SETD3 tissues with different clinicopathological stages (Grade I–III) obtained from the same tissue arrays shown in E. Scores of the staining of SETD3 in different stages of human liver cancer were plotted as the F. n, tested tissue numbers in individual clinical stages. Data are presented as the mean ± S.E. H&E (hematoxylin-eosin) staining results were provided by the Shanghai Outdo Biotech Co. Square frame, enlarged images. Scale bar, H&E staining, 100 μm; SETD3 staining, 50 μm.
Discussion
Here, we elucidated the dynamic regulation of SETD3 through the orchestrated post-translational modifications by a GSK3β- and FBXW7β-dependent pathway, and we indicated that SETD3 was up-regulated in liver cancer cells and tumor tissues. The down-regulation of SETD3 is a critical mechanism that defines the functional window of SETD3 during the cell cycle and has a potential role in suppression of liver tumorigenesis (Fig. 7).
Figure 7.
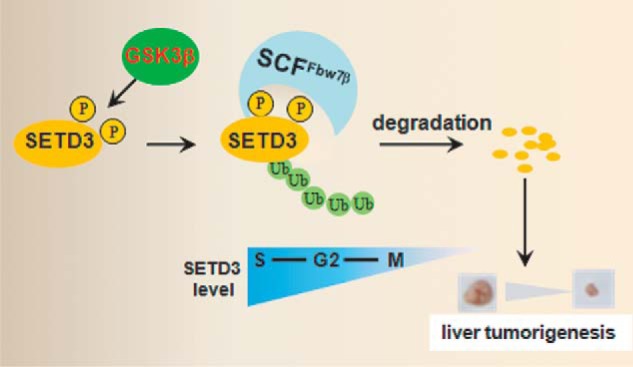
Model depicting how the GSK3β-CSFFBXW7β-SETD3 axis regulates cell cycle progression and attenuates liver tumorigenesis (see details in the text).
By synchronizing cells at different cell cycle stages, we found that SETD3 peaks in S phase and declines in M phase. SETD3 accumulates after the peak of cyclin E1, a critical regulator controlling G1/S transition, but before the induction of cyclin B1, a critical regulator of G2/M transition, implying that SETD3 functions from the S phase to G2 phase. Consistently, a recent study showed that SETD3 interacted with proliferating cell nuclear antigen, a key component of the DNA replication machinery, indicating its potential roles in DNA replication and DNA repair (36). It has been reported that both zebrafish and mouse homologs of SETD3 may catalyze H3K4 or H3K36 mono- and dimethylation in vitro (31, 32), and H3K4 and H3K36 methylations are associated with DNA replication and DNA repair (37–39). Therefore, it is speculated that hSETD3 might regulate these events via modification of these histone H3 methyl sites. However, knockdown or overexpression of hSETD3 in our experiments did not change the methyl status of both H3K4 and H3K36 in HeLa and 293T cells, suggesting that other unknown mechanisms may participate in these processes (supplemental Fig. S6 and data not shown). The role of SETD3 in these events is currently being investigated.
Besides SET8, SETD3 is the second methyltransferase that is tightly controlled during cell cycle progression. Using a candidate searching approach, we identified that the F-box protein FBXW7β is required for ubiquitination and degradation of SETD3. Several lines of evidence support our findings. First, each subunit of the SCF complex, including FBXW7β itself, interacted with endogenous SETD3 in vivo; second, alterations in FBXW7β levels inversely altered SETD3 levels; third, disrupting the interaction between SETD3 and FBXW7β or between FBXW7β and Skp1 abolished SETD3 degradation; and finally, formation of a Lys-48-conjugated ubiquitin chain was a prerequisite for FBXW7β-mediated SETD3 polyubiquitination (Fig. 2 and supplemental Fig. S2). Notably, we showed that the β-isoform, but not other isoforms, of FBXW7 regulates SETD3 degradation, indicating its specificity in recognizing substrates. Because each isoform of FBXW7 has a unique subcellular localization, FBXW7α in the nucleoplasm, FBXW7β in the cytoplasm, and FBXW7γ in the nucleolus, it was predicted that the cytoplasmically localized SETD3 protein is degraded by FBXW7β (6, 40). Interestingly, although nearly 20 different FBXW7 substrates have been identified thus far, only Mcl-1 and PGC-1α are degraded by the β-isoform of FBXW7 (41–43). Thus, identifying SETD3 as a new FBXW7β substrate expands our knowledge of the specificity of FBXW7-mediated protein proteolysis.
Dysregulation of cell cycle progression is a key event in cancer development. A large body of evidence indicates that aberrant destruction of cell cycle regulators, many of which have either tumor-suppressive or oncogenic functions, is tightly linked to carcinogenesis. Here, we examined the potential correlation of SETD3 with cancer. We show that increased SETD3 levels enhanced liver cellular proliferation and tumorigenesis in a xenograft mouse model, whereas overexpression of FBXW7β retarded cell growth, most likely by regulating SETD3 protein levels (Fig. 5). Using IHC staining, we observed that SETD3 levels were positively correlated with tumor malignancy (Fig. 6). However, no significant correlation was observed between FBXW7 IHC staining and clinical prognosis (supplemental Fig. S5B). Given that all commercial antibodies available could not specifically recognize the β-isoform of FBXW7, the detected signals in the IHC images were likely from three isoforms. Notably, as a well-established tumor suppressor, mutations in FBXW7 have been described in 6% of all primary human cancers (10). We have shown that several tumorous FBXW7β mutations located in the WD-40 domain region, including R385C, R399Q, and R425C, modestly affected SETD3 degradation, suggesting that SETD3 protein levels might be critical for cancer development.
It is interesting to note that the Setd3 gene is located in a region of chromosome 14 (14q32) that harbors several putative cancer risk genes, such as Akt (44). Akt1 functions in the PI3K-AKT pathway that inactivates GSK3β (5). Abnormal activity of Akt1 in cancers may inhibit SETD3 degradation. In addition, in multiple myeloma and lymphoma, the same region has been implicated in chromosomal translocation, concomitantly with elevated expression of many cancer-risk genes, including CCND1 and Setd3 (33, 45). Furthermore, it has been reported that high expression of a cluster of miRNAs, which map to the same chromosome locus, positively correlates with biomarkers of hepatocellular carcinoma (45). In this study, we found that elevated levels of SETD3 positively correlated with tumor malignancy in liver tissues, which suggests a potential role of SETD3 in tumorigenesis. Elucidation of the function and regulation of SETD3 will aid our understanding of its potential role in tissue-associated tumorigenesis.
Experimental procedures
Cell culture, cell synchronization, and transfection
HeLa, HeLa S3, 293T, and other various cell lines as indicated were all cultured in Dulbecco's modified Eagle's medium containing 10% FBS (Invitrogen), 1 mm l-glutamine, and antibiotics. Cells were synchronized at the G1/S boundary by a double-thymidine treatment (T/T; 18-h thymidine arrest and 8-h release followed by 18-h thymidine arrest) or at prometaphase by a thymidine-nocodazole arrest (T/N; 18-h thymidine arrest and 4-h release followed by 12-h nocodazole arrest) as described previously (46). Cells were synchronized at S phase by HU treatment for 24 h. siRNAs were synthesized by GenePharma Co. (Shanghai, China). siRNAs targeting FBXW7β were 5′-TATGGGTTTCTACGGCACATT-3′, 5′-CUGUGACAUACCUACCUGAdTdT-3′, and 5′-GAACACGGGGGCACAGAAdTdT-3′. siRNAs against GSK3β were 5′-CCCAAATGTCAAACTACCAAA-3′ and 5′-CCGATTGCGTTATTTCTTCTA-3′. The control siRNA was designed by GenePharma Co. Plasmids and RNAs were transfected using Lipofectamine 2000 (Invitrogen) following the manufacturer's protocols. Transfected cells were harvested between 36 and 48 h after transfection and analyzed by immunoprecipitation and Western blotting.
CHX assay
HeLa S3 cells were treated with 0.4 μg/ml CHX in the presence or absence of FBXW7β siRNA or a GSK3β inhibitor. Cells were collected at the indicated time points, and SETD3 protein stability was monitored by Western blotting probed with an α-SETD3 antibody, and the relative levels normalized with GAPDH were shown.
Ubiquitination assays
GFP-tagged SETD3 was immunoprecipitated from the cells expressing HA-ubiquitin treated with or without proteasome inhibitor MG132 (final concentration 20 μm). Cells were lysed in RIPA buffer (50 mm Tris-Cl, pH 7.4, 150 mm NaCl, 5 mm EDTA, 1% (v/v) Triton X-100, 0.1% SDS, and a protease inhibitor mixture (Biotool)). After the cell lysates were centrifuged at 18,000 × g at 4 °C for 15 min, the supernatants were subjected to IP experiments with GFP antibody, and the ubiquitinated SETD3 was detected by immunoblots with α-HA antibody.
For in vitro recombinant ubiquitination assays, GST-SETD3 protein was incubated with E1, UbcH3 (E2), each E3 component and Fbw7β, as well as GSK3β, in the presence of commercial ubiquitin in buffer (50 mm Tris, pH 7.4, 2.5 mm MgCl2, 0.5 mm DTT, 2 mm ATP) at 30 °C for 2 h. Reactions in the absence of either E1, E2, E3, or Fbw7β component served as negative controls. Reactions were quenched by adding SDS sample buffers, and samples were loaded onto 6% SDS-polyacrylamide gel. The polyubiquitination were detected by probing with an α-ubiquitin antibody.
In vitro kinase assay
Phosphorylation of SETD3 by GSK3β (New England Biolabs) was performed in a total volume of 20 μl of kinase reaction buffer (20 mm Hepes, pH 7.8, 15 mm KCl, 10 mm MgCl2, 1 mm EGTA, 0.1 mm ATP, and 0.1 mg/ml BSA) in the presence of 1 μCi of [γ-32P]ATP (PerkinElmer Life Sciences) for 1 h at 30 °C, using 5 or 10 units of GSK3β with 400 ng of either WT or the CPD1 mutant of GST-SETD3. Reactions were stopped by adding SDS sample buffer, and samples were loaded onto SDS-polyacrylamide gel and subjected to Coomassie Blue staining and autoradiography.
Cell proliferation assay
Cell proliferation assays using human liver L02, BEL7402, and HepG2 cells with stably expressing SETD3 and FBXW7β plasmids or knockdown of SETD3 were measured using WST-1 assay kit (Roche Applied Science) according to the manufacturer's instructions. Briefly, the cells were seeded onto 24-well plates at 2 × 103 cells per well and cultured for the indicated days before addition of 20 μl of WST-1 (5 mg/ml) to the culture medium in each well. After a 1-h incubation at 37 °C, absorbance values were read using a microplate reader (Bio-Tek Co., Winooski, VT) at the 450-nm wavelength. Each time point was repeated in three wells, and the experiments were independently performed three times. The effects of cell proliferation were characterized by a growth curve that was plotted by the value of A450.
Colony formation assay
The cells were plated onto a 6-well tissue culture plate (500 cells/well) and incubated at 37 °C for 8 days. The resulting colonies were rinsed with PBS, then fixed with methanol for 10 min, and stained with methylthionine chloride. The colonies in each sample were photographed and counted by FlowJo software.
Tumorigenesis study
The male BALB/c nude mice were randomly divided into five groups. A total of 1 × 107 HepG2 cells in 100 μl of phosphate-buffered saline mixed with an equal volume of Matrigel (BD Biosciences) were injected subcutaneously into the both oxters of the mice. Twenty seven days post-implantation, the mice were euthanized, and the tumors were surgically dissected, and the tumor weight was measured. All animal xenograft experiments were performed following the animal guidelines for the university and were approved by the Animal Experimentations Ethics Committee of Wuhan University.
IHC staining
Tissue microarray slides were de-paraffinized and heated to complete the antigen retrieval. Slides were then incubated with the α-SETD3 antibody at 1:1000, followed by biotin-conjugated goat anti-rabbit IgG antibody at 1:10,000. Detections were performed using the detection refine DAB kit (Biosharp). Nuclei were stained using hematoxylin (Beyotime Biotech Co.) before mounting.
Clinical specimens
The various human tissues were collected from patients in Xinhua Hospital, Medical College of Jiaotong University, Shanghai, China. The 54 pairs of liver cancer tissues were collected from patients in Hubei Cancer Hospital. The liver cancer tissue microarrays (OD-CT-DgLiv01-012), which contained samples from 53 cases of human hepatocellular carcinoma, were purchased from Shanghai Outdo Biotech. Informed consent was obtained from all subjects in accordance with the protocol approved by the individual institutional Ethics Committees.
Quantification and statistical analysis
For quantification of the Western blotting data, ImageJ software was used to measure the relative intensity of each band, and the relative SETD3 protein levels were normalized to the relative GAPDH levels. Unless otherwise indicated, data are presented as the mean ± S.E. from at least three biological replicates, and the differences between any two groups were compared by unpaired t tests using Prism 5 software. The relative IHC staining scores of human liver tissue microarrays were compared by paired t tests. *, p < 0.05; **, p < 0.01; ***, p < 0.001, and n.s. indicates “not significant.”
Author contributions
H.-N. D., D. H., and X. C. conceived the project. X. C., W. S., M. Z., C. Z., H.-N. D., and D. X. performed the experiments. Y. H., X. P., Y. W., P. Y., and D. H. collected the patient tissue samples. Z. P. provided a novel GSK3β inhibitor. H.-N. D., L. Y., X. C., C. Z., and G. Q. analyzed the data. H.-N. D. wrote the paper with the help from X. C.
Supplementary Material
Acknowledgments
We are grateful to members of the H.-N. Du laboratory for cooperation, reagent sharing, and discussions during the course of this investigation. We are grateful to Drs. Hong-Bing Shu, Ronggui Hu, Ping Wang, and Xiao-Dong Zhang for plasmids and to Drs. Yan Zhou, Wenhua Li, and Ling Zheng and Yu Sun for technical assistance. We also thank Drs. Xiang-Dong Fu and Bao-Liang Song for suggestions.
This work was supported by Major State Basic Research Development Program of China Grant 2013CB910700 (to H.-N. D), National Natural Science Foundation of China Grants 31271369 (to H.-N. D.) and 81401913 (to Y. W.), and Shenzhen Science and Technology Innovation Commission Grant JCYJ20150422150029096 (to H.-N. D). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Experimental procedures, Refs. 1–3, and Figs. S1–S6.
- APC
- anaphase-promoting complex
- CPD
- CDC4 phosphodegron
- SET
- SU(var), enhancer of zeste, and trithorax
- SCF
- Skp1-Cul1-F-box
- H3K36
- histone H3 lysine 36
- CHX
- cycloheximide
- IHC
- immunohistochemistry
- WST-1
- water-soluble tetrazolium salt
- IP
- immunoprecipitation
- HU
- hydroxyurea
- PI
- propidium iodide
- T/N
- thymidine-nocodazole
- SCF
- Skp, Cullin, F-box containing complex.
References
- 1. Tessarz P., and Kouzarides T. (2014) Histone core modifications regulating nucleosome structure and dynamics. Nat. Rev. Mol. Cell Biol. 15, 703–708 [DOI] [PubMed] [Google Scholar]
- 2. Kouzarides T. (2007) Chromatin modifications and their function. Cell 128, 693–705 [DOI] [PubMed] [Google Scholar]
- 3. Greer E. L., and Shi Y. (2012) Histone methylation: a dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 13, 343–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hochegger H., Takeda S., and Hunt T. (2008) Cyclin-dependent kinases and cell-cycle transitions: does one fit all? Nat. Rev. Mol. Cell Biol. 9, 910–916 [DOI] [PubMed] [Google Scholar]
- 5. Crusio K. M., King B., Reavie L. B., and Aifantis I. (2010) The ubiquitous nature of cancer: the role of the SCF(Fbw7) complex in development and transformation. Oncogene 29, 4865–4873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Davis R. J., Welcker M., and Clurman B. E. (2014) Tumor suppression by the Fbw7 ubiquitin ligase: mechanisms and opportunities. Cancer Cell 26, 455–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang R., Wang Y., Liu N., Ren C., Jiang C., Zhang K., Yu S., Chen Y., Tang H., Deng Q., Fu C., Wang Y., Li R., Liu M., Pan W., and Wang P. (2013) FBW7 regulates endothelial functions by targeting KLF2 for ubiquitination and degradation. Cell Res. 23, 803–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Inuzuka H., Shaik S., Onoyama I., Gao D., Tseng A., Maser R. S., Zhai B., Wan L., Gutierrez A., Lau A. W., Xiao Y., Christie A. L., Aster J., Settleman J., Gygi S. P., et al. (2011) SCF(FBW7) regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature 471, 104–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Welcker M., Singer J., Loeb K. R., Grim J., Bloecher A., Gurien-West M., Clurman B. E., and Roberts J. M. (2003) Multisite phosphorylation by Cdk2 and GSK3 controls cyclin E degradation. Mol. Cell 12, 381–392 [DOI] [PubMed] [Google Scholar]
- 10. Skaar J. R., Pagan J. K., and Pagano M. (2013) Mechanisms and function of substrate recruitment by F-box proteins. Nat. Rev. Mol. Cell Biol. 14, 369–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Feng Q., Wang H., Ng H. H., Erdjument-Bromage H., Tempst P., Struhl K., and Zhang Y. (2002) Methylation of H3-lysine 79 is mediated by a new family of HMTases without a SET domain. Curr. Biol. 12, 1052–1058 [DOI] [PubMed] [Google Scholar]
- 12. Nishioka K., Rice J. C., Sarma K., Erdjument-Bromage H., Werner J., Wang Y., Chuikov S., Valenzuela P., Tempst P., Steward R., Lis J. T., Allis C. D., and Reinberg D. (2002) PR-Set7 is a nucleosome-specific methyltransferase that modifies lysine 20 of histone H4 and is associated with silent chromatin. Mol. Cell 9, 1201–1213 [DOI] [PubMed] [Google Scholar]
- 13. Xiao B., Jing C., Kelly G., Walker P. A., Muskett F. W., Frenkiel T. A., Martin S. R., Sarma K., Reinberg D., Gamblin S. J., and Wilson J. R. (2005) Specificity and mechanism of the histone methyltransferase Pr-Set7. Genes Dev. 19, 1444–1454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Beck D. B., Oda H., Shen S. S., and Reinberg D. (2012) PR-Set7 and H4K20me1: at the crossroads of genome integrity, cell cycle, chromosome condensation, and transcription. Genes Dev. 26, 325–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Houston S. I., McManus K. J., Adams M. M., Sims J. K., Carpenter P. B., Hendzel M. J., and Rice J. C. (2008) Catalytic function of the PR-Set7 histone H4 lysine 20 monomethyltransferase is essential for mitotic entry and genomic stability. J. Biol. Chem. 283, 19478–19488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rice J. C., Nishioka K., Sarma K., Steward R., Reinberg D., and Allis C. D. (2002) Mitotic-specific methylation of histone H4 Lys 20 follows increased PR-Set7 expression and its localization to mitotic chromosomes. Genes Dev. 16, 2225–2230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pesavento J. J., Yang H., Kelleher N. L., and Mizzen C. A. (2008) Certain and progressive methylation of histone H4 at lysine 20 during the cell cycle. Mol. Cell. Biol. 28, 468–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jørgensen S., Elvers I., Trelle M. B., Menzel T., Eskildsen M., Jensen O. N., Helleday T., Helin K., and Sørensen C. S. (2007) The histone methyltransferase SET8 is required for S-phase progression. J. Cell Biol. 179, 1337–1345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tardat M., Murr R., Herceg Z., Sardet C., and Julien E. (2007) PR-Set7-dependent lysine methylation ensures genome replication and stability through S phase. J. Cell Biol. 179, 1413–1426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wu S., Wang W., Kong X., Congdon L. M., Yokomori K., Kirschner M. W., and Rice J. C. (2010) Dynamic regulation of the PR-Set7 histone methyltransferase is required for normal cell cycle progression. Genes Dev. 24, 2531–2542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang Z., Dai X., Zhong J., Inuzuka H., Wan L., Li X., Wang L., Ye X., Sun L., Gao D., Zou L., and Wei W. (2015) SCF(β-TRCP) promotes cell growth by targeting PR-Set7/Set8 for degradation. Nat. Commun. 6, 10185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yin Y., Yu V. C., Zhu G., and Chang D. C. (2008) SET8 plays a role in controlling G1/S transition by blocking lysine acetylation in histone through binding to H4 N-terminal tail. Cell Cycle 7, 1423–1432 [DOI] [PubMed] [Google Scholar]
- 23. Centore R. C., Havens C. G., Manning A. L., Li J. M., Flynn R. L., Tse A., Jin J., Dyson N. J., Walter J. C., and Zou L. (2010) CRL4(Cdt2)-mediated destruction of the histone methyltransferase Set8 prevents premature chromatin compaction in S phase. Mol. Cell 40, 22–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Oda H., Hübner M. R., Beck D. B., Vermeulen M., Hurwitz J., Spector D. L., and Reinberg D. (2010) Regulation of the histone H4 monomethylase PR-Set7 by CRL4(Cdt2)-mediated PCNA-dependent degradation during DNA damage. Mol. Cell 40, 364–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Abbas T., Shibata E., Park J., Jha S., Karnani N., and Dutta A. (2010) CRL4(Cdt2) regulates cell proliferation and histone gene expression by targeting PR-Set7/Set8 for degradation. Mol. Cell 40, 9–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Herz H. M., Garruss A., and Shilatifard A. (2013) SET for life: biochemical activities and biological functions of SET domain-containing proteins. Trends Biochem. Sci. 38, 621–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Binda O., Sevilla A., LeRoy G., Lemischka I. R., Garcia B. A., and Richard S. (2013) SETD6 monomethylates H2AZ on lysine 7 and is required for the maintenance of embryonic stem cell self-renewal. Epigenetics 8, 177–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen A., Feldman M., Vershinin Z., and Levy D. (2016) SETD6 is a negative regulator of oxidative stress response. Biochim. Biophys. Acta 1859, 420–427 [DOI] [PubMed] [Google Scholar]
- 29. Vershinin Z., Feldman M., Chen A., and Levy D. (2016) PAK4 methylation by SETD6 promotes the activation of the Wnt/β-catenin pathway. J. Biol. Chem. 291, 6786–6795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Levy D., Kuo A. J., Chang Y., Schaefer U., Kitson C., Cheung P., Espejo A., Zee B. M., Liu C. L., Tangsombatvisit S., Tennen R. I., Kuo A. Y., Tanjing S., Cheung R., Chua K. F., et al. (2011) Lysine methylation of the NF-κB subunit RelA by SETD6 couples activity of the histone methyltransferase GLP at chromatin to tonic repression of NF-κB signaling. Nat. Immunol. 12, 29–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kim D. W., Kim K. B., Kim J. Y., and Seo S. B. (2011) Characterization of a novel histone H3K36 methyltransferase setd3 in zebrafish. Biosci. Biotechnol. Biochem. 75, 289–294 [DOI] [PubMed] [Google Scholar]
- 32. Eom G. H., Kim K. B., Kim J. H., Kim J. Y., Kim J. R., Kee H. J., Kim D. W., Choe N., Park H. J., Son H. J., Choi S. Y., Kook H., and Seo S. B. (2011) Histone methyltransferase SETD3 regulates muscle differentiation. J. Biol. Chem. 286, 34733–34742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen Z., Yan C. T., Dou Y., Viboolsittiseri S. S., and Wang J. H. (2013) The role of a newly identified SET domain-containing protein, SETD3, in oncogenesis. Haematologica 98, 739–743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cohn O., Feldman M., Weil L., Kublanovsky M., and Levy D. (2016) Chromatin associated SETD3 negatively regulates VEGF expression. Sci. Rep. 6, 37115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Orlicky S., Tang X., Willems A., Tyers M., and Sicheri F. (2003) Structural basis for phosphodependent substrate selection and orientation by the SCFCdc4 ubiquitin ligase. Cell 112, 243–256 [DOI] [PubMed] [Google Scholar]
- 36. Cooper S. E., Hodimont E., and Green C. M. (2015) A fluorescent bimolecular complementation screen reveals MAF1, RNF7 and SETD3 as PCNA-associated proteins in human cells. Cell Cycle 14, 2509–2519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dorn E. S., and Cook J. G. (2011) Nucleosomes in the neighborhood: new roles for chromatin modifications in replication origin control. Epigenetics 6, 552–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rizzardi L. F., Dorn E. S., Strahl B. D., and Cook J. G. (2012) DNA replication origin function is promoted by H3K4 di-methylation in Saccharomyces cerevisiae. Genetics 192, 371–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li F., Mao G., Tong D., Huang J., Gu L., Yang W., and Li G. M. (2013) The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSα. Cell 153, 590–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Welcker M., Orian A., Grim J. E., Grim J. A., Eisenman R. N., and Clurman B. E. (2004) A nucleolar isoform of the Fbw7 ubiquitin ligase regulates c-Myc and cell size. Curr. Biol. 14, 1852–1857 [DOI] [PubMed] [Google Scholar]
- 41. Xu W., Taranets L., and Popov N. (2016) Regulating Fbw7 on the road to cancer. Semin. Cancer Biol. 36, 62–70 [DOI] [PubMed] [Google Scholar]
- 42. Trausch-Azar J. S., Abed M., Orian A., and Schwartz A. L. (2015) Isoform-specific SCF(Fbw7) ubiquitination mediates differential regulation of PGC-1α. J. Cell. Physiol. 230, 842–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ekholm-Reed S., Goldberg M. S., Schlossmacher M. G., and Reed S. I. (2013) Parkin-dependent degradation of the F-box protein Fbw7β promotes neuronal survival in response to oxidative stress by stabilizing Mcl-1. Mol. Cell. Biol. 33, 3627–3643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kamnasaran D., and Cox D. W. (2002) Current status of human chromosome 14. J. Med. Genet. 39, 81–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tian E., Sawyer J. R., Heuck C. J., Zhang Q., van Rhee F., Barlogie B., and Epstein J. (2014) In multiple myeloma, 14q32 translocations are nonrandom chromosomal fusions driving high expression levels of the respective partner genes. Genes Chromosomes Cancer 53, 549–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Seki A., Coppinger J. A., Du H., Jang C. Y., Yates J. R. 3rd., Fang G. (2008) Plk1- and β-TrCP-dependent degradation of Bora controls mitotic progression. J. Cell Biol. 181, 65–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.