

Abstract
Pathogenic Acinetobacter species, including Acinetobacter baumannii and Acinetobacter nosocomialis, are opportunistic human pathogens of increasing relevance worldwide. Although their mechanisms of drug resistance are well studied, the virulence factors that govern Acinetobacter pathogenesis are incompletely characterized. Here we define the complete secretome of A. nosocomialis strain M2 in minimal medium and demonstrate that pathogenic Acinetobacter species produce both a functional type I secretion system (T1SS) and a contact-dependent inhibition (CDI) system. Using bioinformatics, quantitative proteomics, and mutational analyses, we show that Acinetobacter uses its T1SS for exporting two putative T1SS effectors, an Repeats-in-Toxin (RTX)-serralysin-like toxin, and the biofilm-associated protein (Bap). Moreover, we found that mutation of any component of the T1SS system abrogated type VI secretion activity under nutrient-limited conditions, indicating a previously unrecognized cross-talk between these two systems. We also demonstrate that the Acinetobacter T1SS is required for biofilm formation. Last, we show that both A. nosocomialis and A. baumannii produce functioning CDI systems that mediate growth inhibition of sister cells lacking the cognate immunity protein. The Acinetobacter CDI systems are widely distributed across pathogenic Acinetobacter species, with many A. baumannii isolates harboring two distinct CDI systems. Collectively, these data demonstrate the power of differential, quantitative proteomics approaches to study secreted proteins, define the role of previously uncharacterized protein export systems, and observe cross-talk between secretion systems in the pathobiology of medically relevant Acinetobacter species.
Keywords: bacterial pathogenesis, bacterial toxin, biofilm, protein secretion, proteomics, Acinetobacter, contact-dependent inhibition system, type I secretion system
Introduction
Medically relevant Acinetobacter species are nosocomial pathogens accounting for ∼2% of all healthcare-associated infections in the United States (1). More alarming has been the increasing rate of multidrug resistance (MDR)7 phenotypes associated with Acinetobacter-induced infections; so much so that the CDC has listed Acinetobacter as a severe threat, and last-line therapeutic options like carbapenems, tigecycline, and colistin are now the recommended treatment options for the majority of Acinetobacter infections (2, 3). Furthermore, recent epidemiological data indicate that Acinetobacter spp. appear to be acquiring MDR phenotypes faster than almost all other Gram-negative bacteria over the last decade (4).
For decades, the causative agent behind Acinetobacter infections was almost exclusively reported as Acinetobacter baumannii; however, Acinetobacter taxonomy has undergone major modifications over the last several years. Specifically, we now know that medically relevant Acinetobacter spp. predominately come from a single group, designated the Acinetobacter baumannii (Ab) group, given their close genetic relatedness and indistinguishability at the phenotypic level when examined in clinical laboratories. Members of the Ab group include A. baumannii, Acinetobacter nosocomialis, Acinetobacter pittii, Acinetobacter seifertii, and Acinetobacter dijkshoorniae, all of which act as opportunistic pathogens and acquire MDR phenotypes (5–7). Nevertheless, A. baumannii still remains the most comprehensively studied member of the Ab group, with A. nosocomialis a distant second.
Although our understanding of the mechanisms that mediate MDR phenotypes is quite comprehensive (8), those that mediate Acinetobacter pathogenesis are only beginning to be defined. The role of secretion systems in Acinetobacter virulence has been receiving more and more attention because of their importance in other clinically relevant pathogens. Multiple secretion systems, including a type II secretion system (T2SS), a type V secretion system (T5SS), and a T6SS, have all been characterized and attributed specific roles in the pathobiology of Acinetobacter (9–13). Both the T2SS and T5SS have clear roles in pathogenesis, as specific exported factors have been directly linked to virulence, including the type II effector CpaA and the type V autotransporter Ata (11, 14). The T6SS, not yet linked directly to eukaryotic pathology, is responsible for killing bacteria and commonly found to be active in clinical isolates extracted from polymicrobial infections, indicating that it may impart a fitness advantage in vivo (15). Furthermore, plasmid-borne conjugation systems similar to a T4SS have also been identified in A. baumannii (16), but detailed molecular analyses describing these systems are lacking. Lastly, type IV pili (T4P), bacterial grappling hooks evolutionarily related to the T2SS, have also been characterized in Acinetobacter for their role in natural transformation, motility, and biofilm formation (17, 18).
Given the extensive genetic variation of Acinetobacter spp. and our lack of a thorough understanding of their molecular pathogenesis, we hypothesized that other unrecognized secreted factors may be contributing toward Acinetobacter fitness and virulence. To this end, we bioinformatically identified, in Ab group member genomes, a T1SS and contact-dependent inhibition (CDI) systems. The T1SS of Acinetobacter is homologous to the prototypical TolC-HlyD-HlyB system from Escherichia coli for export of the hemolysin HlyA (19). This three-component secretion system requires an inner membrane ATP-binding cassette (ABC) transporter, a periplasmic adaptor protein (PAP), and an outer membrane protein channel (20).
In E. coli, TolC acts as the outer membrane protein and forms a homotrimer that embeds into the outer membrane, extending far into the periplasmic space, where it can interact with the periplasmic adaptor protein HlyD (21). The periplasmic adaptor protein HlyD, anchored by a transmembrane domain within the inner membrane (22), not only interacts with the outer membrane protein TolC and the ABC transporter HlyB but can also interact with the T1SS substrate via an N-terminal cytoplasmic region (23). The ABC transporter HlyB, also embedded within the inner membrane, hydrolyzes ATP to provide the energy required for export of a specific T1SS substrate (22). Together, these proteins act to facilitate the secretion of unfolded effectors directly from the cytoplasm to the extracellular environment.
CDI systems, first discovered in 2005 (24), constitute a tripartite mechanism for inhibiting the growth of neighboring bacteria independent from T6SS. Both CdiB and CdiA are considered to be T5SS, where the outer membrane pore-forming CdiB protein facilities the export of a CdiA toxin to the cell surface. CdiA toxins are polymorphic, usually containing multiple filamentous hemagglutinin domains in the N-terminal region and a C-terminal toxin domain. The third component, the CdiI protein, acts as an immunity protein preventing autoinhibition. Immunity proteins commonly contain domains associated with the SUKH superfamily (25). A fourth component, the toxin receptor, is also required for CDI killing; however, these are not intuitively identifiable and are not associated with cdiBAI loci. To date, only a handful of other CDI systems (Dickeya dadantii (26), Burkholderia (26), Enterobacter cloacae (27), Pseudomonas aeruginosa (28), and Neisseria meningitidis (29)) have been functionally characterized. Here, utilizing a quantitative, differential proteomics technique paired with bioinformatics, we report the complete secretome of A. nosocomialis strain M2 and simultaneously identify two previously unrecognized secretion systems across medically relevant Acinetobacter species.
Results
Identification of a T1SS-associated locus and its role in the secreted protein profile of A. nosocomialis strain M2
Previously, we demonstrated that A. nosocomialis strain M2 (formerly designated A. baumannii strain M2 (30)) has a functioning T2SS that secretes multiple effectors, including the metalloprotease CpaA (10). Furthermore, given that strain M2 also encodes for a functioning T6SS (12), we hypothesized that other secretion systems not yet characterized may also be contributing to the secreted protein profile. Using a BLASTp search, we identified homologs of the T1SS components tolC, hlyB, and hlyD from E. coli (Fig. 1A). To determine whether this putative system was contributing toward the secreted protein profile in strain M2, we constructed a tolC-hlyB-hlyD::kan mutant (T1SS mutant). Using one-dimensional SDS-PAGE analysis, we found that the T1SS mutant exhibited an altered secreted protein profile, as evidenced by the absence of high-molecular-weight proteins at 70, 130, and 250 kDa compared with the parent (Fig. 1B). Importantly, the complemented strain displayed a secreted protein profile nearly identical to the parental strain, indicating that the absence of the tolC-hlyB-hlyD gene cluster was responsible for the observed phenotype (Fig. 1B). Next, we sought to further characterize these differences and simultaneously define the complete secretome of strain M2 in minimal medium.
Figure 1.

Acinetobacter nosocomialis strain M2 encodes for a functional T1SS. A, genetic arrangement and locus identifiers for the T1SS locus in strain M2. Boxes below each gene contain the relevant protein family domain information associated with the respective amino acid sequence for TolC, HlyB, and HlyD, as determined by BLASTp. B, Coomassie-stained, one-dimensional SDS-PAGE analysis of the secreted protein profiles from the wild-type M2, the T1SS mutant (tolC-hlyD::kan), and the complemented T1SS mutant. Secreted proteins were enriched from bacteria grown overnight in rich medium. C, the most abundant high-confidence secreted proteins from strain M2 grown to mid-log in minimal medium. High-confidence proteins were defined as proteins with ≥20 unique identified peptides across replicates. The export column details the export/secretion mechanism for the each protein. The asterisk beside T1SS denotes putative secretion by the TolC-HlyB-HlyD system, as determined by the presence of the type I secretion C-terminal target domain (VC_A0848) for M215_09430 and M215_01695 and the absence of these proteins in our differential proteomics analysis comparing secreted proteins from the wild type and the T1SS mutant, described in detail in Fig. 2.
The complete secretome of A. nosocomialis strain M2 reveals T1SS-, T2SS-, T6SS-, and CDI-secreted candidates
To define the complete secretome and simultaneously identify differentially secreted proteins between strain M2 and the T1SS mutant, we utilized a quantitative proteomics approach. Briefly, we enriched for secreted proteins from each strain using bacteria grown to mid-log in M9 minimal medium supplemented with 0.2% casamino acids. This method eliminated the use of rich medium containing high concentrations of yeast proteins as well as limited cellular lysis contaminants found in stationary-phase bacterial cultures. Using dimethyl labeling of enzymatically digested peptides obtained from the secreted protein fractions of each strain, we were then able to compare the relative quantities of all secreted proteins in pairwise fashions. Four biological samples were prepared for each strain and processed as described under “Experimental Procedures” prior to LC/MS analysis. Isotopically labeled samples were then pooled, and the four biological dimethylation triplex experiments were analyzed by LC/MS. A sample of each replicate for each strain was run on an SDS-PAGE gel, silver-stained, and found to have similar loadings and reflected the overall secretion trend observed in Fig. 1 (supplemental Fig. 1).
First we compiled a list of the top secreted proteins from the wild-type M2. Our list was ranked based on proteins with the highest number of unique identified peptides (arbitrarily cut off at 20 for Fig. 1C). The complete list of proteins can be found in the supplemental Mass Spectrometry Table. We identified components of the T6SS and the T2SS, which was expected given previous characterizations of these systems in pathogenic Acinetobacter spp. Furthermore, we identified proteins putatively associated with the T1SS and a CDI system, both of which are discussed in detail below.
RTX and Bap are secreted via a T1SS
Using the Log2 ratio of high-confidence proteins (defined as proteins with ≥13 unique identified peptides) observed across replicates, we were able to identify multiple proteins that were significantly and differentially secreted by the wild type compared with the T1SS mutant (Fig. 2A). As expected, the top differentially secreted protein between the parent and the T1SS mutant was a predicted T1SS protein based on the presence of the type I secretion C-terminal target domain (VC_A0849) in the C terminus of the M215_09430 open reading frame. The M215_09430 locus encodes for a protein containing multiple peptidase M10 serralysin C-terminal domains, a Ca2+-binding protein RTX toxin-related domain, and the type I secretion C-terminal target domain (VC_A0849 subclass). For simplicity, we will refer to M215_09430 as the RTX protein. We also identified a second protein containing a type I secretion C-terminal target domain, M215_01695, which was significantly and differentially secreted between the wild type and T1SS mutant. The M215_1695 locus encodes for a large protein predicted to contain multiple group 3 bacterial Ig-like domains (pfam13754) and a type I secretion C-terminal target domain (VC_A0848). Given its homology to biofilm-associated proteins (Baps) in A. baumannii, this protein was designated the Bap ortholog in A. nosocomialis strain M2.
Figure 2.

Quantitative proteomics analysis comparing secreted proteins from strain M2, the T1SS mutant, and the complemented strain demonstrates global changes to the secretome. A, heatmap of quantitative proteomics results of high-confidence proteins (defined as proteins with ≥13 unique identified peptides). The Log2 ratio of high-confidence proteins observed across replicates demonstrates the loss of multiple proteins in response to disruption of T1SS (low ΔtolC-hlyD/WT demethylation ratios). Upon restoration of T1SS in the form of the complemented strain, secreted protein profiles return to near wild-type levels (complement/WT demethylation ratios of ∼0). B, scatterplot of quantitative proteomics results showing the −log10 (p values) compared with the average ΔtolC-hlyD/WT ratios of alterations observed within the secretome of ΔtolC-hlyD biological replicates compared with WT replicates. Statistically significant changes (p < 0.05) are highlighted in red.
The second most differentially secreted protein between the wild type and the T1SS mutant was M215_02910 (Fig. 2, A and B), a protein predicted to contain multiple group 3 bacterial Ig-like domains (pfam13754) similar to Bap; however, the protein annotation for M215_02910 lacks the type I secretion C-terminal target domain commonly found in T1SS proteins. Recent molecular, in silico, and PCR analyses of bap genes from A. baumannii demonstrate that the bap repetitive elements are prone to alignment errors in genome sequences, resulting in premature truncations and chromosomal rearrangements (31–33). Together, this may indicate that M215_02910 is part of the same open reading frame as Bap. Conversely, it is also possible that this open reading frame has been incorrectly aligned and is, in fact, part of the RTX protein, given its similarities to large repetitive RTX proteins (34). Regardless, the presence of peptides from this protein in the secreted fraction of both the wild type and complemented strain strongly correlate with its secretion with the T1SS.
Mutation of the T1SS reveals cross-talk between other secretion systems
Analysis of our proteomics results indicated that proteins without the type I C-terminal targeting domain were also differentially secreted between the wild type and the T1SS mutant. Specifically, multiple T6SS-associated proteins, including three VgrG orthologs and Hcp, were all found at significantly lower levels in the T1SS mutant. Although not nearly as significant, multiple known T2SS-associated proteins, including CpaA, LipH, a rhombosortase, and a rhombotarget, were also found in lower quantities in the T1SS mutant compared with the wild type. Last, a predicted CDI-associated protein was also identified as significantly reduced in the T1SS mutant (discussed in detail below). The full list of differentially secreted proteins can be found in the supplemental Mass Spectrometry Table. For the majority of instances, including the predicted T1SS-associated proteins described above, the dimethylation ratios comparing the secreted proteins from the complemented strain with the parent strain were quite similar, again indicating that the absence of the tolC-hlyB-hlyD genes was responsible for the differential secretion profile in the mutant.
Deletion of the T1SS systems represses T6SS activity in minimal medium only
It has been shown previously that deletion of tolC can have pleiotropic effects on membrane integrity and cellular physiology in E. coli (35). Therefore, we initially hypothesized that the observed differences between the secreted proteins of the parent and T1SS mutant were due to membrane stress associated with the absence of the TolC protein. To gain insight into the molecular mechanism behind the diminished protein profile of the T1SS mutant and more intricately probe the role of tolC, hlyB, and hlyD in the observed phenotypes, we constructed in-frame unmarked mutations in each gene as well as the entire locus.
First we validated the quantitative proteomics results by Western blotting across the entire mutant panel in minimal medium. As expected, Hcp expression was found to be similar across all strains tested; however, Hcp secretion was not detected in any of the mutants analyzed when secreted proteins were collected from strains grown in minimal medium (Fig. 3A). Interestingly, when the mutant panel was tested for Hcp secretion from strains grown in rich medium, no differences were observed in Hcp expression or secretion (Fig. 3B).
Figure 3.

Mutation of any component of the T1SS abrogates T6SS activity, as determined by Hcp secretion in minimal medium only. A and B, Western blot analysis probing for Hcp expression and secretion from the wild type and T1SS mutant panel and complemented strains grown in minimal medium (A) or rich medium (B). Hcp expression in whole cells from strains grown in either minimal or rich medium was equal. However, Hcp secretion was found to be suppressed in strains with mutations in any component of the T1SS in minimal medium. Importantly, complemented strains had Hcp secretion levels approximately equivalent to the wild type, with the exception of the hlyB complement, which had an intermediate phenotype. As a loading and lysis control, RNA polymerase expression was probed for across all conditions. C, whole cells; S, secreted proteins.
Next we sought to determine whether the cross-talk between the T1SS system and the T6SS was specific or whether disrupting other secretions systems would also abrogate T6SS activity. To this end, we probed for Hcp expression and secretion in a pilD mutant and complemented strain. PilD is the pre-pilin peptidase required for processing of both pilins and pseudopilins for the T4P system and T2SS, respectively. Thus, the absence of pilD results in a T4P- and T2SS-negative phenotype (10). The pilD mutant displayed no differences in Hcp expression or secretion compared with the parental strain and the complemented strain. Furthermore, this phenotype held true for both rich and minimal medium (Fig. 4, A and B).
Figure 4.

Mutation of both the T2SS and T4P system does not abrogate T6SS. A and B, Western blot analysis probing for Hcp expression and secretion from wild-type M2, a pilD mutant, and the pilD complemented strain grown in minimal medium (A) or rich medium (B). Both Hcp expression and secretion were equivalent across strains grown in minimal medium or rich medium. As a loading and lysis control, RNA polymerase (RNAP) expression was probed for across all conditions.
The T1SS system is required for biofilm formation
Given that Bap was one of the most differentially secreted proteins between the wild type and the T1SS full locus mutant, we hypothesized that our unmarked individual T1SS mutants would have impaired ability to form biofilms. Using a microtiter plate biofilm assay, we observed that mutants lacking any or all components of the T1SS have severely reduced ability to form biofilms, as determined by crystal violet staining both 3 and 6 h after inoculation (Fig. 5, A and B, respectively). Importantly, we performed biofilm formation assays in rich medium to avoid confounding results from altered secretion of non-T1SS components, like Hcp secretion, as shown in Fig. 3. Nevertheless, the attenuated biofilm phenotype was observed in both rich and minimal medium (data not shown). The biofilm defect was not due to alterations in Csu pili or T4P expression and surface presentation either. Specifically, the major pilin subunits PilA of the T4P and CsuA/B of Csu pili were equally expressed in whole cells and on the surface of all mutants and complemented strains (supplemental Fig. 2). Last, the biofilm phenotype was not due to differences in the second most abundantly secreted protein, M215_12375 (CdiA), as a cdi locus mutant had the same level of biofilm as the parent strain (supplemental Fig. 3).
Figure 5.
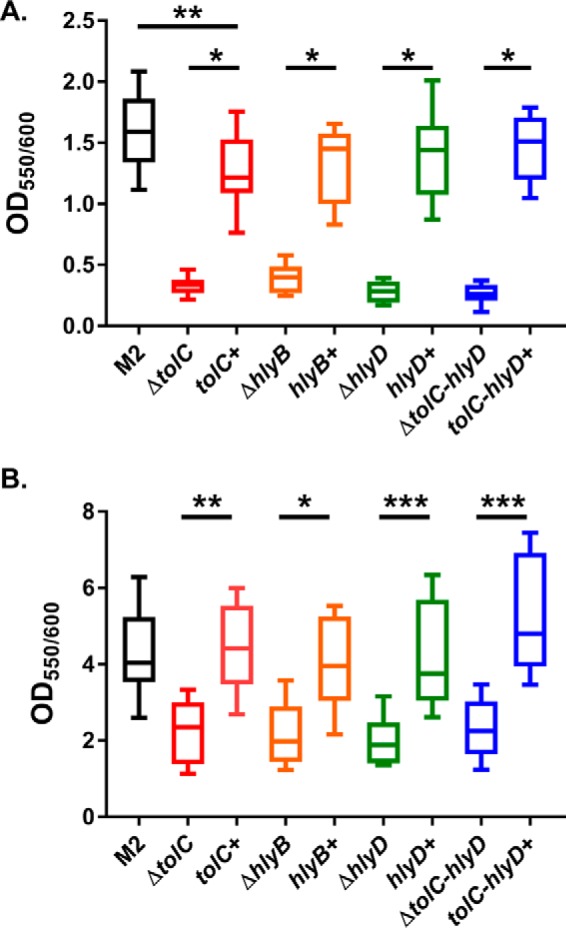
The Acinetobacter T1SS is required for biofilm formation. A and B, biofilm formation was assessed by crystal violet staining. Plates were statically incubated at 37 °C for 3 h (A) or 6 h (B). After the designated time, an aliquot of the bacterial culture was removed for A600 readings. Wells were subsequently washed and stained for 10 min with 0.1% crystal violet. Excess crystal violet was removed by washing and solubilized by addition of 30% acetic acid. Biofilm formation was assessed by comparing the absorbance ratio at 550 nm for crystal violet and 600 nm for bacteria. As seen in both panels, T1SS mutants had significantly reduced ability to form biofilms; however, complemented strains had biofilm phenotypes similar to wild-type M2. For statistical analyses, one-way ANOVA with Tukey's correction for multiple comparisons was performed. *, p = 0.01; **, p = 0.001; ***, p = 0.0001.
T1SS mutants appear to have attenuated virulence phenotypes in a Galleria mellonella infection model
The greater wax moth, G. mellonella, is a commonly used model system for measuring Acinetobacter virulence and correlates well with in vivo virulence in mammalian systems (36). To assess the role of the T1SS in virulence, we individually infected larvae with the wild type, a T1SS mutant, or its respective complemented strain and monitored larva viability over 24 h. Larvae were injected with 10 μl of a bacterial suspension determined previously to be the LD50 for the wild-type M2 (10). Although not statistically significant, a clear virulence attenuation trend was observed in all T1SS mutants compared with the parental or complemented strains, indicating that the T1SS contributes to the pathogenicity of medically relevant Acinetobacter spp. (Fig. 6, A–D). Although the T1SS mutant has an altered T6SS phenotype in minimal medium only, we have demonstrated previously that T6SS mutants are equally as virulent as wild-type A. baumannii (13).
Figure 6.

T1SS have an attenuated virulence phenotype in the G. mellonella infection model. A–D, groups of G. mellonella larvae were injected with 10 μl of either the parent strain, the tolC mutant (A), the hlyB mutant (B), the hlyD mutant (C), the full locus mutant (D), or the respective complemented strains at an inoculum previously determined to be the equivalent of the LD50 for wild-type M2. Larvae were checked for viability as determined by melanin accumulation and motility. Although not statistically significant as determined by Mantel-Cox comparison of survival curves, a clear attenuation trend was observed for all mutants tested.
Pathogenic Acinetobacter spp. utilize a contact-dependent inhibition system for bacterial killing
Two of the most abundantly secreted proteins, based on observed peptides within our datasets, were the RTX protein and M215_12375. Both proteins show profound alterations in their secretion between the T1SS mutant and the wild-type strain. As mentioned above, the RTX protein is an RTX serralysin-like protein, a common class of toxins secreted by T1SS (37), whereas the M215_12375 locus encodes for a polymorphic toxin similar to those found in CDI systems (Fig. 7A). Given its homology to CDI systems and the data presented below, we have designated this gene cluster the cdiBAIM2 system (Fig. 7A).
Figure 7.

Pathogenic Acinetobacter species encode for functional CDI systems. A, genetic arrangement and locus identifiers for the CDI locus in strain M2. Boxes below each gene contain the relevant protein family domain information associated with the respective amino acid sequence for CdiB, CdiA, and CdiI, as determined by BLASTp. B, growth inhibition assays for strain M2 cdi mutants. Strain M2 is able to inhibit the growth of the cdi mutant at a statistically significant level; however, co-incubation between the cdi mutant and the cdi mutant complemented with the immunity protein alone, or between the wild type and the cdi mutant complemented with the immunity protein, did not show statistically significant differences. CDI-mediated growth inhibition was independent of the T6SS, as shown in the last column. C and D, growth inhibition assays in A. baumannii 19606 (C) and the A. baumannii 1225 (D) mutant panel. Both 19606 and 1225 encode for two cdi loci, and both are able to inhibit the growth of mutants lacking the cognate immunity proteins. For statistical analyses, one-way ANOVA with Tukey's correction for multiple comparisons was performed for B and C, whereas a non-parametric Kruskal-Wallis test with Dunn's correction for multiple comparisons was utilized for D. The CDI loci for 19606 can be found with the locus tags F911_RS14340 to F911_RS14350 for cdi_1 and F911_RS17415 to F911_RS17425 for cdi_2.
To determine whether the cdiBAIM2 system was active in strain M2, the entire locus was replaced with a kanamycin cassette, generating the M2Δcdi mutant. Furthermore, we generated a variant of the cdi mutant expressing just the cdiI immunity gene in trans. Next we performed inhibition assays in pairwise fashions. As seen in Fig. 7B, M2 is able to inhibit the growth of the cdi mutant by ∼1 log compared with co-incubations between the cdi mutant and the cdi mutant expressing just the immunity protein. Furthermore, M2 is not able to inhibit the growth of the cdi mutant expressing just the immunity protein, indicating that CdiI is indeed the immunity protein for this cdi system. Last, CDI killing was found to be T6SS-independent, as an hcp mutant co-incubated with a double hcp/cdi mutant had an indistinguishable phenotype from wild-type M2 co-incubated with the cdi mutant (Fig. 7B).
CDI systems are conserved in medically relevant Acinetobacter
To expand our findings to the majority of medically relevant Acinetobacter spp., we bioinformatically identified CDI systems in A. baumannii 19606 and 1225, two clinical isolates representing a type strain as well as newer, more relevant clinical isolate, respectively. Interestingly, both A. baumannii 19606 and 1225 encode for two predicted cdi loci, a trait common to P. aeruginosa (28). CDI loci from 19606 and 1225 were similar in gene architecture to A. nosocomialis strain M2 with a putative transporter protein, the polymorphic toxin, and an immunity protein. For both strains, each cdi locus was individually mutated, generating cdi_1 and cdi_2 mutants for both 19606 and 1225. Subsequently, the cdi_1 and cdi_2 mutants were individually co-incubated with their respective isogenic parent strains for determination of growth inhibition. Similarly to the strain M2 co-incubations, 19606 and 1225 mutants lacking their respective cdi systems had their growth inhibited by ∼1 log compared with co-incubations between the wild-type strain and a cdi mutant complemented with the respective immunity protein (Fig. 7, C and D). In each case, the difference between the parent strain and a cdi mutant was statistically significant, whereas co-incubations between a parent strain and a cdi mutant expressing the immunity protein closely reflected the growth of the cdi mutant in the absence of an inhibitor strain (Fig. 7, C and D, last columns).
Discussion
Pathogenic Acinetobacter spp., including A. baumannii and A. nosocomialis, are opportunistic pathogens with an unmatched ability to acquire MDR phenotypes. Although the mechanisms behind drug resistance are well documented, the full slate of virulence factors that mediate virulence are not yet defined. In this work, we applied quantitative proteomics techniques to identify previously uncharacterized, secreted Acinetobacter proteins. This powerful approach, coupled with previous secretion system characterizations, facilitated the assignment of the complete secretome of A. nosocomialis strain M2 in minimal medium. Importantly analysis of the top secreted candidates revealed the presence of a functional T1SS and identified two putative T1SS effectors. Last, we found functional CDI systems in both A. nosocomialis strain M2 and two A. baumannii clinical isolates, 19606 and 1225.
Bacterial interactions with the extracellular environment are commonly mediated through secreted proteins; however, the full arsenal of Acinetobacter secretion systems compared with other classical pathogens like P. aeruginosa is significantly undercharacterized. Nevertheless, a broad collection of publically available sequenced Acinetobacter genomes has allowed the in silico identification of previously characterized secretion systems important for pathogenesis in other bacteria. Using this approach, we were able to identify a T1SS locus in A. nosocomialis strain M2, homologous to the E. coli prototype T1SS containing tolC, hlyB, and hlyD genes (19). Unlike E. coli, however, the tolC, hlyB, and hlyD genes of strain M2 are in a three-gene cluster and are most likely in an operon, given that the open reading frame for hlyB overlaps with both tolC and hlyD. Furthermore, this gene arrangement is commonly found across medically relevant Acinetobacter spp., including A. baumannii, with 88% identity conserved to the nucleotide level for commonly used laboratory strains like A. baumannii ATCC 17978, AB5075, AYE, ACICU, and AB307-0294. Furthermore, homologs of both bap and the RTX protein are also found in these strains, highlighting their probable secretion by the T1SS.
The Acinetobacter T1SS is related to other ABC transporters, like the resistance-nodulation-cell division (RND) efflux pumps. Many RND pumps, such as AdeABC, AdeFGH, and AdeIJK, have been extensively characterized in A. baumannii; however, their roles in the pathobiology of Acinetobacter have been linked directly with the efflux of small molecules, particularly antibiotics, and not protein substrates (38–40). However, Yoon et al. (41) demonstrated that overexpression of the RND pumps AdeABC and AdeIJK alters the composition of the Acinetobacter outer membrane, as evidenced by the repression of Csu pili, OmpA, and some metabolism-associated proteins, demonstrating the importance of ABC transporters in outer membrane homeostasis.
In a related fashion, we observed that mutations in the T1SS altered the secretion profile compared with wild-type M2. Specifically, our analysis identified that the T6SS is affected by alterations to the T1SS, but only under nutrient limitation. Furthermore, this abrogation is not due to membrane stress associated with mutations in tolC, as mutants lacking the periplasmic adaptor protein HlyD or the ATPase HlyB also displayed the same phenotype. Even more intriguing is that this reduction in T6SS activity is specific for mutations in the T1SS, as mutations that alter both T2SS and T4P functionality had not effect on Hcp secretion. The fact that T6SS is only abrogated under nutrient limitation in T1SS mutants suggests a regulatory cross-talk that favors the functional activity of these two systems simultaneously, which has yet to be fully explained. These results have parallels with cross-talk between secretion systems in other bacteria. Specifically, others have shown that the T2SS and T6SS of P. aeruginosa are co-regulated by quorum sensing (42, 43), and transcription factors co-regulate the T3SS and T6SS of Burkholderia cenocepacia (44). The regulatory mechanism behind the Acinetobacter Hcp secretion repression in T1SS mutants has yet to be determined but is not dependent on the previously described plasmid regulation system found in A. baumannii 17978 (15), given that M2 does not carry a homologous plasmid. Nevertheless, the levels of Hcp expression in whole cells were equivalent across strains. Further, hcp is predicted to be co-transcribed with the complete T6SS biogenesis machinery in a single operon. Based on these observations, we hypothesize that the down-regulation of T6SS in the T1SS-deficient strains takes place at the posttranscriptional level. Several other Acinetobacter isolates also express Hcp but do not constitutively secrete Hcp (13). Additional work is needed to understand T6SS regulation in Acinetobacter and its connections with other cellular processes.
Two of the most abundantly secreted proteins, the RTX protein and Bap, relied on the T1SS for export, as evidenced by their relative absence in the T1SS mutant and the presence of a type I C-terminal target domain (VC_A0849) in both proteins. Little is known about the role of RTX domain-containing proteins in the pathobiology of Acinetobacter; however, De Gregorio et al. (33) have bioinformatically found that some Acinetobacter RTX domain-containing proteins also have bacterial Ig-like domains found in Bap orthologs. These proteins, like ABAYE0821 from A. baumannii AYE, also contain the same peptidase M10 serralysin C-terminal domain found in the M2 RTX protein. Interestingly, all or some of these domains are also commonly found in other well studied large repetitive RTX proteins, like LapA from Pseudomonas fluorescens and Pseudomonas putida as well as SiiE from Salmonella enterica (34). As shown in our proteomics data, the second most differentially secreted protein between the wild type and the T1SS mutant was the M215_02910 protein, which also contained multiple bacterial Ig-like domains but not a type I C-terminal target domain. It is tempting to speculate that these two proteins (RTX and M215_02910) are actually a single protein constituting a new member of the large repetitive RTX protein family; however, multiple unsuccessful attempts were made to clone and resequence the RTX gene. We hypothesize that, because of the presence of extensive repetitive sequences in these genes, the original genome sequence may be incorrect. Resequencing the genome of this strain will be necessary to properly assemble these genes.
Although little is known about Acinetobacter large repetitive RTX proteins, Bap orthologs have been studied in detail for their role in Acinetobacter virulence and biofilm formation (45–47). From a molecular standpoint, Bap has been shown to be localized on the surface of A. baumannii (45) and, as we have shown, was readily identified in the culture supernatant. We have also shown that Bap is likely secreted by the T1SS. These results are congruent with those found for secretion of LapA, whereby a T1SS-like system, designated LapEBC, is responsible for its secretion (48). Nevertheless, molecular tagging experiments are required to definitively link Acinetobacter Bap secretion with the T1SS. From a phenotypic perspective, though, it is likely that the attenuated biofilm formation was due to the absence of Bap, given its previously characterized role in higher-order biofilm structure and maintenance (33).
During the course of our characterization of the T1SS, we identified a CdiAM2 polymorphic toxin. CDI systems, composed of an outer membrane pore-forming protein, the polymorphic toxin, and an immunity protein, deliver toxic effectors to neighboring bacteria to inhibit growth. CdiA toxins are usually large, repetitive proteins with multiple N-terminal hemagglutinin repeats and a polymorphic C-terminal toxin domain (49). To date, CDI systems in Acinetobacter have been mentioned minimally in published reports, essentially just in bioinformatics comparisons of other previously well characterized systems (50–52), and none have shown any functional demonstration of activity. Although we demonstrated that CDI systems are functional, we did not identify the toxin receptor or the proposed mechanism of killing. However, the CdiAM2 toxin contains domains associated with DNase/tRNase activity, indicating that it may function as a cytoplasmic toxin degrading nucleic acids. Aside from the assigned toxic activities, novel signaling roles have been assigned to CDI systems, including signaling to coordinate cooperation between bacteria that carry the same cdi alleles (53). Given that CDI systems appear widespread in Acinetobacter and that Acinetobacter is commonly associated with polymicrobial infections, these systems may also function as signaling molecules to coordinate killing activity in vivo.
Recently, a new contact-dependent killing system, designated the Cdz system, was discovered in Caulobacter crescentus and was also bioinformatically identified in A. baumannii (54). These systems share similarities with CDI systems but rely on a T1SS for export of the surface-associated toxins. Further, the authors were able to identify the putative toxins in A. baumannii. Unfortunately, a BLASTp analysis revealed that there are no orthologs in A. nosocomialis M2 for these putative A. baumannii Cdz toxins. Nevertheless, it is likely that, in other Acinetobacter strains encoding for Cdz toxins, a T1SS is responsible for their secretion.
Collectively, we have demonstrated the functionality of two previously uncharacterized secretion systems in pathogenic Acinetobacter: a T1SS that secretes two putative effectors and a CDI system that mediates bacterial growth inhibition of sister cells. Furthermore, using quantitative proteomics, we were also able to define the secretome of M2 in minimal medium. These findings provide the foundation for future work aimed at deciphering the role of each system and other uncharacterized secreted proteins in the pathobiology of Acinetobacter, ultimately providing a framework of Acinetobacter virulence.
Experimental procedures
Strains, plasmids, and growth conditions
All strains used in this study are listed in supplemental Table 1. Bacteria were grown at 37 °C on L-agar, in LB broth, or in M9 salts supplemented with 0.2% casamino acids, 2 mm magnesium sulfate, and 100 μm calcium chloride. When appropriate, the following antibiotic concentrations were used: for E. coli, 100 μg of ampicillin/ml and 20 μg of kanamycin/ml. For Acinetobacter species, 200 μg of carbenicillin/ml, 7.5 μg of kanamycin/ml, and 12.5 μg of chloramphenicol/ml. Sucrose was used at 10% for counterselection.
Mutant and complemented strain construction
Both marked and unmarked mutants were constructed according to our protocols published previously (55). The complete list of primers utilized for mutagenesis can be found in supplemental Table 2. Briefly, for strain M2 derivatives, kanamycin-sacB-interrupted gene cassettes were generated using In-Fusion HD EcoDry cloning kits (Takara Bio). Kanamycin-sacB cassettes were introduced into strain M2 via natural transformation, and transformants were selected on L-agar supplemented with 7.5 μg of kanamycin/ml. Unmarked mutants were generated by transiently introducing and expressing the flp recombinase from pFLP2 via a triparental mating strategy described previously (55). For A. baumannii 1225 and 19606, unmarked in-frame mutations were generated according to the protocol of Tucker et al. (18).
Complemented mutants in the M2 background were generated by introducing a mini-Tn7 element via a four-parental mating strategy described previously with the following modification (55): exconjugants were selected on L-agar supplemented with 200 μg of carbenicillin/ml and 12.5 μg of chloramphenicol/ml. Complemented mutants in 19606 and 1225 were generated by cloning the respective cdi immunity gene into pSH1. The plasmid pSH1 was constructed by replacing the multiple cloning site and promoter of pBAVMCS with the araC gene, PBAD promoter, and Shine-Dalgarno from pMLBAD using the In-Fusion HD EcoDry cloning kit with the primers listed in supplemental Table 2.
Secreted protein one dimensional SDS-PAGE analysis
Secreted proteins for one-dimensional SDS-PAGE analysis were enriched from bacteria grown in LB medium overnight (∼16 h) at 37 °C and 225 rpm. Briefly, overnight cultures were normalized to A600 = 5.0, and 1.5-ml aliquots were removed and pelleted. One milliliter of supernatant was removed, and TCA was precipitated according to methods published previously (15). Precipitated protein was resuspended in 25 μl and used for SDS-PAGE analysis followed by Coomassie staining.
Secreted protein enrichment for quantitative proteomics analysis
A. nosocomialis strain M2, the ΔtolC-hlyD::kan mutant, and its respective complement were grown overnight in 5 ml of M9 minimal medium supplemented with 0.2% casamino acids at 37 °C and 225 rpm. The following morning, each strain was inoculated into 50 ml of minimal medium at A600 = 0.05. Bacteria were grown at 37 °C with 225 rpm for 3 h (approximately mid-log). Cultures were removed from the incubator and centrifuged at 15,000 × g for 2 min, and supernatants were removed. Supernatants were filter-sterilized with Steriflip vacuum-driven filtration devices (Millipore) and concentrated using an Amicon Ultra (Millipore) concentrator with a 10-kDa molecular weight cutoff. Supernatants were concentrated to ∼150 μl, flash-frozen in liquid nitrogen, and then lyophilized. Lyophilized protein samples were then processed for mass spectrometry analysis as described below. For each strain, four individual 50-ml cultures were prepared, providing four biological replicates for each strain.
Digestion and dimethyl labeling of secretomes
Lyophilized secretomes from identical culture volumes were resuspended in 6 m urea, 2 m thiourea, and 40 mm NH4HCO3 and reduced/alkylated prior to digestion with Lys-C (1/200 w/w) and then trypsin (1/50 w/w) overnight as described previously (56). Digested samples were acidified to a final concentration of 0.5% formic acid and desalted using C18 stage tips (57, 58) prior to reductive dimethylation labeling. C18 stage tips were conditioned with buffer B (80% ACN, 0.1% formic acid) and washed with 10 volumes of buffer A* (0.1% TFA, 2% acetonitrile (ACN)). The sample was loaded, the column was washed with 10 volumes of buffer A*, and bound peptides were eluted with buffer B and then dried. Dimethylation was performed according to the protocol of Parker et al. (59). Briefly, peptide samples were resuspended in 30 μl of 100 mm triethylammonium bicarbonate by bath sonication and 30 μl of 200 mm formaldehyde, followed by addition of 3 μl of 1 m cyanoborohydride to initiate amine labeling. For secretome samples derived from wild-type strains, “light” combinations of formaldehyde and cyanoborohydride were utilized (CH2O and NaBH3CN), whereas the T1SS mutant and complemented strain were labeled with “medium” (CD2O and NaBH3CN) and “heavy” (13CD2O and NaBD3CN) reagents, respectively. The samples were then incubated at room temperature in the dark for 1 h, and two rounds of dimethyl labeling were undertaken to ensure complete labeling. Labeling reactions were quenched with 3 m NH4Cl and acidified with buffer A* prior to mixing. Combined dimethylated samples were desalted with C18 stage tips prior to LC/MS analysis. A total of four biological replicates of dimethylated secretomes were analyzed by LC/MS.
LC/MS
C18 stage tip-purified peptides were resuspended in buffer A* and loaded onto an in-house-packaged 35-cm, 75-μm inner diameter, 360-μm outer diameter, 1.7-μm, 130-Å CSH C18 (Waters, Manchester, UK) reverse-phase analytical column with an integrated HF-etched nano-electrospray ionization (nESI) tip. Samples were loaded directly onto the column using an Acquity ultra performance liquid chromatography (UPLC) M-class system (Waters) at 400 nl/min for 35 min with buffer A (0.1% formic acid) and eluted at 300 nl/min using a gradient altering the concentration of buffer B (99.9% ACN, 0.1% formic acid) from 0% to 32% B over 100 min, then from 32% to 40% B in the next 10 min, then increased to 80% B over an 8-min period, held at 100% B for 2 min, and then dropped to 0% B for another 20 min. reverse phase (RP)-separated peptides were infused into a Q-Exactive (Thermo Scientific, San Jose, CA) mass spectrometer, and data were acquired using data-dependent acquisition. One full precursor scan (resolution, 70,000; 350–2000 m/z; automatic gain control (AGC) target of 3 × 106), followed by 10 data-dependent higher-energy collisional dissociation (HCD) MS-MS events (resolution, 17.5 k automatic gain control (AGC) target of 1 × 105 with a maximum injection time of 200 ms, NCE 28 with 20% stepping), were allowed, with 35-s dynamic exclusion enabled.
Data analysis
MaxQuant (v1.5.3.1) (60) was used for identification and quantification of the experiments, with the resulting biological replicates searched together to ensure a global false discovery rate of less than 1% in accordance with the work of Schaab et al. (61). Database searching was carried out against the A. nosocomialis M2 database (NCBI whole genome sequence (WGS) entry AWOW00000000, downloaded July 3, 2016) with the following search parameters: carbamidomethylation of cysteine as a fixed modification, oxidation of methionine, acetylation of protein N termini, trypsin/P cleavage with a maximum of two missed cleavages. A multiplicity of three was used, with each multiplicity denoting one of the dimethylation labeling combinations (light, medium, and heavy, respectively). The precursor mass tolerance was set to 50 ppm for the first search and 10 ppm for the main search, with a maximum false discovery rate of 1.0% set for protein identifications. To enhance the identification of peptides between fractions and replicates, the Match between Runs option was enabled, with a precursor match window set to 2 min and an alignment window of 10 min. A minimum of two unique peptides was required for protein quantification. The resulting protein group output was processed within the Perseus (v1.4.0.6) (62) analysis environment to remove reverse matches and common protein contaminants prior to visualization of the data with Matlab R2016a. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD005881 (63).
Western blotting for Hcp secretion
Hcp Western blotting was performed as described previously (15). Briefly, cells were grown to mid-log and then normalized to A600 = 0.5. One milliliter of culture was then centrifuged to collect supernatants. Secreted proteins were precipitated using TCA. Precipitated proteins were resuspended in 50 μl of Laemmli buffer, and 10 μl was loaded onto polyacrylamide gels. The anti-Hcp (13) and anti-RNA polymerase (Biolegend, San Diego, CA) antibodies were both used at a concentration of 1:1000. Secondary IRdye antibodies from Licor were used at 1:10,000. All blots were blocked in TBS blocking buffer (Licor).
Biofilm assay
96-well plate biofilm/crystal violet retention assays were performed according to the following protocol. Bacteria were grown overnight in 5 ml of LB medium at 37 °C and 225 rpm. The following day, each culture was normalized to A600 = 1.0 and diluted 1:100 into 200 μl of fresh LB broth. Plates were statically incubated at 37 °C for 3 or 6 h. After the designated time, an aliquot of the bacterial culture was removed for A600 readings. Wells were subsequently washed and stained for 10 min with 0.1% crystal violet. Excess crystal violet was removed by washing and solubilized by addition of 30% acetic acid. Biofilm formation was assessed by comparing the absorbance ratio at 550 nm for crystal violet and 600 nm for bacteria. Biofilm assays were performed three separate times with three technical replicates per biological replicate.
G. mellonella infection model
The G. mellonella infection experiment was performed as described in Harding et al. (10). Three groups of 10 larvae were injected with the same LD50 established previously, and larva viability was scored approximately every 5 h based on response to physical stimuli and melanin accumulation.
CDI assay
Bacterial inhibition assays were performed utilizing a modified protocol from Weber et al. (15) for killing associated with type VI secretion systems. Briefly, CDI Assays were set up using overnight cultures normalized to A600 = 1.0. Strains grown with antibiotics or 0.2% arabinose were washed prior to co-incubation. Inhibitor and target strains were mixed at a 1:10 ratio, and a 5-μl suspension was spotted on a dry LB agar plate or a 1% arabinose LB agar plate. After 4 h, the spot was cut out, resuspended in 1 ml of LB, and serially diluted on LB plates with the appropriate antibiotics for colony-forming units (CFU) enumeration (either kanamycin, 20 μg/ml for M2 cdi mutants and complemented strains, or rifampicin, 150 μg/ml for A. baumannii 19606 and 1225 cdi mutants and complemented strains).
Statistical analyses
GraphPad Prism7 software was used for statistical analyses of biofilm formation, G. mellonella infection experiments, and CDI assays. For normally distributed datasets, parametric one-way ANOVA was performed with Tukey's correction for multiple comparisons. For non-normally distributed datasets, non-parametric Kruskal-Wallis test with Dunn's correction for multiple comparisons was used.
Author contributions
N. E. S. performed the quantitative mass spectrometry experiments and data analysis. A. I. W. provided mass spectrometry access. R. L. K. performed the G. mellonella infection experiments. G. D. V. performed the contact-dependent growth inhibition experiments. M. R. P. performed all other experiments. C. M. H. designed the experiments and wrote the manuscript. M. F. F. and J. P. helped to coordinate the study and provided editorial criticism of the written manuscript.
Supplementary Material
The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Figs. 1–3, Tables 1 and 2, and a Mass Spectrometry Table.
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD005881.
- MDR
- multidrug resistance
- Ab
- Acinetobacter baumannii
- T2SS
- type II secretion system(s)
- T4P
- type IV pili
- CDI
- contact-dependent inhibition
- ABC
- ATP-binding cassette
- Bap
- biofilm-associated protein
- RND
- resistance-nodulation-cell division
- ANOVA
- analysis of variance
- RTX
- repeat-in-toxin.
References
- 1. Magill S. S., Edwards J. R., Bamberg W., Beldavs Z. G., Dumyati G., Kainer M. A., Lynfield R., Maloney M., McAllister-Hollod L., Nadle J., Ray S. M., Thompson D. L., Wilson L. E., Fridkin S. K., Emerging Infections Program Healthcare-Associated Infections and Antimicrobial Use Prevalence Survey Team (2014) Multistate point-prevalence survey of health care-associated infections. N. Engl. J. Med. 370, 1198–1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Center for Disease Control and Prevention (2013) Antibiotic Resistance Threats in the United States, 2013 [Google Scholar]
- 3. Peleg A. Y., and Hooper D. C. (2010) Hospital-acquired infections due to Gram-negative bacteria. N. Engl. J. Med. 362, 1804–1813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Giammanco A., Cala C., Fasciana T., and Dowzicky M. J. (2017) Global assessment of the activity of tigecycline against multidrug-resistant Gram-negative pathogens between 2004 and 2014 as part of the tigecycline evaluation and surveillance trial. mSphere 2, e00310–e00316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nemec A., Krizova L., Maixnerova M., Sedo O., Brisse S., and Higgins P. G. (2015) Acinetobacter seifertii sp. nov., a member of the Acinetobacter calcoaceticus-Acinetobacter baumannii complex isolated from human clinical specimens. Int. J. Syst. Evol. Microbiol. 65, 934–942 [DOI] [PubMed] [Google Scholar]
- 6. Marí-Almirall M., Cosgaya C., Higgins P. G., Van Assche A., Telli M., Huys G., Lievens B., Seifert H., Dijkshoorn L., Roca I., and Vila J. (2016) MALDI-TOF/MS identification of species from the Acinetobacter baumannii (Ab) group revisited: inclusion of the novel A. seifertii and A. dijkshoorniae species. Clin. Microbiol. Infect. 23, 210.e1–210.e9 [DOI] [PubMed] [Google Scholar]
- 7. Cosgaya C., Marí-Almirall M., Van Assche A., Fernández-Orth D., Mosqueda N., Telli M., Huys G., Higgins P. G., Seifert H., Lievens B., Roca I., and Vila J. (2016) Acinetobacter dijkshoorniae sp. nov., a member of the Acinetobacter calcoaceticus-Acinetobacter baumannii complex mainly recovered from clinical samples in different countries. Int. J. Syst. Evol. Microbiol. 66, 4105–4111 [DOI] [PubMed] [Google Scholar]
- 8. Roca I., Espinal P., Vila-Farrés X., and Vila J. (2012) The Acinetobacter baumannii oxymoron: commensal hospital dweller turned pan-drug-resistant menace. Front. Microbiol. 3, 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Johnson T. L., Waack U., Smith S., Mobley H., and Sandkvist M. (2015) Acinetobacter baumannii is dependent on the type II secretion system and its substrate LipA for lipid utilization and in vivo fitness. J. Bacteriol. 198, 711–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Harding C. M., Kinsella R. L., Palmer L. D., Skaar E. P., and Feldman M. F. (2016) Medically relevant Acinetobacter species require a type II secretion system and specific membrane-associated chaperones for the export of multiple substrates and full virulence. PLoS Pathog. 12, e1005391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bentancor L. V., Camacho-Peiro A., Bozkurt-Guzel C., Pier G. B., and Maira-Litrán T. (2012) Identification of Ata, a multifunctional trimeric autotransporter of Acinetobacter baumannii. J. Bacteriol. 194, 3950–3960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Carruthers M. D., Nicholson P. A., Tracy E. N., and Munson R. S. Jr. (2013) Acinetobacter baumannii utilizes a type VI secretion system for bacterial competition. PLoS ONE 8, e59388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Weber B. S., Miyata S. T., Iwashkiw J. A., Mortensen B. L., Skaar E. P., Pukatzki S., and Feldman M. F. (2013) Genomic and functional analysis of the type VI secretion system in Acinetobacter. PLoS ONE 8, e55142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tilley D., Law R., Warren S., Samis J. A., and Kumar A. (2014) CpaA a novel protease from Acinetobacter baumannii clinical isolates deregulates blood coagulation. FEMS Microbiol. Lett. 356, 53–61 [DOI] [PubMed] [Google Scholar]
- 15. Weber B. S., Ly P. M., Irwin J. N., Pukatzki S., and Feldman M. F. (2015) A multidrug resistance plasmid contains the molecular switch for type VI secretion in Acinetobacter baumannii. Proc. Natl. Acad. Sci. U.S.A. 112, 9442–9447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu C. C., Kuo H. Y., Tang C. Y., Chang K. C., and Liou M. L. (2014) Prevalence and mapping of a plasmid encoding a type IV secretion system in Acinetobacter baumannii. Genomics 104, 215–223 [DOI] [PubMed] [Google Scholar]
- 17. Harding C. M., Tracy E. N., Carruthers M. D., Rather P. N., Actis L. A., and Munson R. S. Jr. (2013) Acinetobacter baumannii strain M2 produces type IV pili which play a role in natural transformation and twitching motility but not surface-associated motility. MBio 4, e00360–e00313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tucker A. T., Nowicki E. M., Boll J. M., Knauf G. A., Burdis N. C., Trent M. S., and Davies B. W. (2014) Defining gene-phenotype relationships in Acinetobacter baumannii through one-step chromosomal gene inactivation. MBio 5, e01313–01314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Thomas S., Holland I. B., and Schmitt L. (2014) The type 1 secretion pathway: the hemolysin system and beyond. Biochim. Biophys. Acta 1843, 1629–1641 [DOI] [PubMed] [Google Scholar]
- 20. Holland I. B., Peherstorfer S., Kanonenberg K., Lenders M., Reimann S., and Schmitt L. (2016) Type I protein secretion: deceptively simple yet with a wide range of mechanistic variability across the family. EcoSal Plus 7, ESP-0019–2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Koronakis V., Sharff A., Koronakis E., Luisi B., and Hughes C. (2000) Crystal structure of the bacterial membrane protein TolC central to multidrug efflux and protein export. Nature 405, 914–919 [DOI] [PubMed] [Google Scholar]
- 22. Wang R. C., Seror S. J., Blight M., Pratt J. M., Broome-Smith J. K., and Holland I. B. (1991) Analysis of the membrane organization of an Escherichia coli protein translocator, HlyB, a member of a large family of prokaryote and eukaryote surface transport proteins. J. Mol. Biol. 217, 441–454 [DOI] [PubMed] [Google Scholar]
- 23. Balakrishnan L., Hughes C., and Koronakis V. (2001) Substrate-triggered recruitment of the TolC channel-tunnel during type I export of hemolysin by Escherichia coli. J. Mol. Biol. 313, 501–510 [DOI] [PubMed] [Google Scholar]
- 24. Aoki S. K., Pamma R., Hernday A. D., Bickham J. E., Braaten B. A., and Low D. A. (2005) Contact-dependent inhibition of growth in Escherichia coli. Science 309, 1245–1248 [DOI] [PubMed] [Google Scholar]
- 25. Zhang D., Iyer L. M., and Aravind L. (2011) A novel immunity system for bacterial nucleic acid degrading toxins and its recruitment in various eukaryotic and DNA viral systems. Nucleic Acids Res. 39, 4532–4552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Aoki S. K., Diner E. J., de Roodenbeke C. T., Burgess B. R., Poole S. J., Braaten B. A., Jones A. M., Webb J. S., Hayes C. S., Cotter P. A., and Low D. A. (2010) A widespread family of polymorphic contact-dependent toxin delivery systems in bacteria. Nature 468, 439–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Beck C. M., Morse R. P., Cunningham D. A., Iniguez A., Low D. A., Goulding C. W., and Hayes C. S. (2014) CdiA from Enterobacter cloacae delivers a toxic ribosomal RNase into target bacteria. Structure 22, 707–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mercy C., Ize B., Salcedo S. P., de Bentzmann S., and Bigot S. (2016) Functional characterization of Pseudomonas contact dependent growth inhibition (CDI) systems. PLoS ONE 11, e0147435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Arenas J., Schipper K., van Ulsen P., van der Ende A., and Tommassen J. (2013) Domain exchange at the 3′ end of the gene encoding the fratricide meningococcal two-partner secretion protein A. BMC Genomics 14, 622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Carruthers M. D., Harding C. M., Baker B. D., Bonomo R. A., Hujer K. M., Rather P. N., and Munson R. S. Jr. (2013) Draft genome sequence of the clinical isolate Acinetobacter nosocomialis strain M2. Genome Announc. 1, e00906–e00913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kodori M., Douraghi M., Yaseri M., and Rahbar M. (2017) The impact of primer sets on detection of the gene encoding biofilm-associated protein (Bap) in Acinetobacter baumannii: in silico and in vitro analysis. Lett. Appl. Microbiol. 64, 304–308 [DOI] [PubMed] [Google Scholar]
- 32. Goh H. M., Beatson S. A., Totsika M., Moriel D. G., Phan M. D., Szubert J., Runnegar N., Sidjabat H. E., Paterson D. L., Nimmo G. R., Lipman J., and Schembri M. A. (2013) Molecular analysis of the Acinetobacter baumannii biofilm-associated protein. Appl. Environ. Microbiol. 79, 6535–6543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. De Gregorio E., Del Franco M., Martinucci M., Roscetto E., Zarrilli R., and Di Nocera P. P. (2015) Biofilm-associated proteins: news from Acinetobacter. BMC Genomics 16, 933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Satchell K. J. (2011) Structure and function of MARTX toxins and other large repetitive RTX proteins. Annu. Rev. Microbiol. 65, 71–90 [DOI] [PubMed] [Google Scholar]
- 35. Zgurskaya H. I., Krishnamoorthy G., Ntreh A., and Lu S. (2011) Mechanism and function of the outer membrane channel TolC in multidrug resistance and physiology of Enterobacteria. Front. Microbiol. 2, 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Peleg A. Y., Jara S., Monga D., Eliopoulos G. M., Moellering R. C. Jr, and Mylonakis E. (2009) Galleria mellonella as a model system to study Acinetobacter baumannii pathogenesis and therapeutics. Antimicrob. Agents Chemother. 53, 2605–2609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Delepelaire P. (2004) Type I secretion in Gram-negative bacteria. Biochim. Biophys. Acta 1694, 149–161 [DOI] [PubMed] [Google Scholar]
- 38. Magnet S., Courvalin P., and Lambert T. (2001) Resistance-nodulation-cell division-type efflux pump involved in aminoglycoside resistance in Acinetobacter baumannii strain BM4454. Antimicrob. Agents Chemother. 45, 3375–3380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Coyne S., Rosenfeld N., Lambert T., Courvalin P., and Périchon B. (2010) Overexpression of resistance-nodulation-cell division pump AdeFGH confers multidrug resistance in Acinetobacter baumannii. Antimicrob. Agents Chemother. 54, 4389–4393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Damier-Piolle L., Magnet S., Brémont S., Lambert T., and Courvalin P. (2008) AdeIJK, a resistance-nodulation-cell division pump effluxing multiple antibiotics in Acinetobacter baumannii. Antimicrob. Agents Chemother. 52, 557–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yoon E. J., Courvalin P., and Grillot-Courvalin C. (2013) RND-type efflux pumps in multidrug-resistant clinical isolates of Acinetobacter baumannii: major role for AdeABC overexpression and AdeRS mutations. Antimicrob. Agents Chemother. 57, 2989–2995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lesic B., Starkey M., He J., Hazan R., and Rahme L. G. (2009) Quorum sensing differentially regulates Pseudomonas aeruginosa type VI secretion locus I and homologous loci II and III, which are required for pathogenesis. Microbiol-Sgm 155, 2845–2855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schuster M., Lostroh C. P., Ogi T., and Greenberg E. P. (2003) Identification, timing, and signal specificity of Pseudomonas aeruginosa quorum-controlled genes: a transcriptome analysis. J. Bacteriol. 185, 2066–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sun G. W., Chen Y., Liu Y., Tan G. Y., Ong C., Tan P., and Gan Y. H. (2010) Identification of a regulatory cascade controlling type III secretion system 3 gene expression in Burkholderia pseudomallei. Mol. Microbiol. 76, 677–689 [DOI] [PubMed] [Google Scholar]
- 45. Loehfelm T. W., Luke N. R., and Campagnari A. A. (2008) Identification and characterization of an Acinetobacter baumannii biofilm-associated protein. J. Bacteriol. 190, 1036–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Brossard K. A., and Campagnari A. A. (2012) The Acinetobacter baumannii biofilm-associated protein plays a role in adherence to human epithelial cells. Infect. Immun. 80, 228–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Luo T. L., Rickard A. H., Srinivasan U., Kaye K. S., and Foxman B. (2015) Association of blaOXA-23 and bap with the persistence of Acinetobacter baumannii within a major healthcare system. Front. Microbiol. 6, 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hinsa S. M., Espinosa-Urgel M., Ramos J. L., and O'Toole G. A. (2003) Transition from reversible to irreversible attachment during biofilm formation by Pseudomonas fluorescens WCS365 requires an ABC transporter and a large secreted protein. Mol. Microbiol. 49, 905–918 [DOI] [PubMed] [Google Scholar]
- 49. Jamet A., and Nassif X. (2015) New players in the toxin field: polymorphic toxin systems in bacteria. MBio 6, e00285–00215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ruhe Z. C., Low D. A., and Hayes C. S. (2013) Bacterial contact-dependent growth inhibition. Trends Microbiol. 21, 230–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Garcia E. C., Anderson M. S., Hagar J. A., and Cotter P. A. (2013) Burkholderia BcpA mediates biofilm formation independently of interbacterial contact-dependent growth inhibition. Mol. Microbiol. 89, 1213–1225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hayes C. S., Koskiniemi S., Ruhe Z. C., Poole S. J., and Low D. A. (2014) Mechanisms and biological roles of contact-dependent growth inhibition systems. Cold Spring Harb. Perspect. Med. 4, 10.1101/cshperspect.a010025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Garcia E. C., Perault A. I., Marlatt S. A., and Cotter P. A. (2016) Interbacterial signaling via Burkholderia contact-dependent growth inhibition system proteins. Proc. Natl. Acad. Sci. U.S.A. 113, 8296–8301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. García-Bayona L., Guo M. S., and Laub M. T. (2017) Contact-dependent killing by Caulobacter crescentus via cell surface-associated, glycine zipper proteins. eLife 6, e24869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Harding C. M., Nasr M. A., Kinsella R. L., Scott N. E., Foster L. J., Weber B. S., Fiester S. E., Actis L. A., Tracy E. N., Munson R. S. Jr, and Feldman M. F. (2015) Acinetobacter strains carry two functional oligosaccharyltransferases, one devoted exclusively to type IV pilin, and the other one dedicated to O-glycosylation of multiple proteins. Mol. Microbiol. 96, 1023–1041 [DOI] [PubMed] [Google Scholar]
- 56. Scott N. E., Parker B. L., Connolly A. M., Paulech J., Edwards A. V., Crossett B., Falconer L., Kolarich D., Djordjevic S. P., Hojrup P., Packer N. H., Larsen M. R., and Cordwell S. J. (2011) Simultaneous glycan-peptide characterization using hydrophilic interaction chromatography and parallel fragmentation by CID, higher energy collisional dissociation, and electron transfer dissociation MS applied to the N-linked glycoproteome of Campylobacter jejuni. Mol. Cell Proteomics 10, M000031MCP000201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ishihama Y., Rappsilber J., and Mann M. (2006) Modular stop and go extraction tips with stacked disks for parallel and multidimensional peptide fractionation in proteomics. J. Proteome Res. 5, 988–994 [DOI] [PubMed] [Google Scholar]
- 58. Rappsilber J., Mann M., and Ishihama Y. (2007) Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2, 1896–1906 [DOI] [PubMed] [Google Scholar]
- 59. Parker R., Melathopoulos A. P., White R., Pernal S. F., Guarna M. M., and Foster L. J. (2010) Ecological adaptation of diverse honey bee (Apis mellifera) populations. PLoS ONE 5, e11096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Cox J., and Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 61. Schaab C., Geiger T., Stoehr G., Cox J., and Mann M. (2012) Analysis of high accuracy, quantitative proteomics data in the MaxQB database. Mol. Cell. Proteomics 11, M111 014068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tyanova S., Temu T., Sinitcyn P., Carlson A., Hein M. Y., Geiger T., Mann M., and Cox J. (2016) The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740 [DOI] [PubMed] [Google Scholar]
- 63. Vizcaíno J. A., Csordas A., Del-Toro N., Dianes J. A., Griss J., Lavidas I., Mayer G., Perez-Riverol Y., Reisinger F., Ternent T., Xu Q. W., Wang R., and Hermjakob H. (2016) 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 44, 11033. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.