

Abstract
Hrd1 is the core structural component of a large endoplasmic reticulum membrane-embedded protein complex that coordinates the destruction of folding-defective proteins in the early secretory pathway. Defining the composition, dynamics, and ultimately, the structure of the Hrd1 complex is a crucial step in understanding the molecular basis of glycoprotein quality control but has been hampered by the lack of suitable techniques to interrogate this complex under native conditions. In this study we used genome editing to generate clonal HEK293 (Hrd1.KI) cells harboring a homozygous insertion of a small tandem affinity tag knocked into the endogenous Hrd1 locus. We found that steady-state levels of tagged Hrd1 in these cells are indistinguishable from those of Hrd1 in unmodified cells and that the tagged variant is functional in supporting the degradation of well characterized luminal and membrane substrates. Analysis of detergent-solubilized Hrd1.KI cells indicates that the composition and stoichiometry of Hrd1 complexes are strongly influenced by Hrd1 expression levels. Analysis of affinity-captured Hrd1 complexes from these cells by size-exclusion chromatography, immunodepletion, and absolute quantification mass spectrometry identified two major high-molecular-mass complexes with distinct sets of interacting proteins and variable stoichiometries, suggesting a hitherto unrecognized heterogeneity in the functional units of Hrd1-mediated protein degradation.
Keywords: E3 ubiquitin ligase, endoplasmic reticulum (ER), endoplasmic-reticulum-associated protein degradation (ERAD), ubiquitin, ubiquitin ligase, Hrd1
Introduction
Approximately one-third of all proteins in eukaryotes are targeted to the secretory pathway (1, 2). To attain functionality, these proteins must fold into their specific three-dimensional conformation, a process that, although assisted by abundant molecular chaperones and folding enzymes, is under constant challenge by molecular crowding, genetic mutations, protein synthesis errors, and environmental stress (3). Production of folding-defective proteins in the secretory pathway is associated with a broad range of diseases including cystic fibrosis, α1-antitrypsin deficiency, Gaucher's disease, neurodegenerative diseases, and cancer (4). To relieve the burden of aberrant proteins in the ER,4 cells have evolved ER quality control mechanisms that detect and selectively remove these potentially toxic proteins via a process termed ER-associated degradation (ERAD) (5). ERAD is also important for regulating the abundance of some native proteins including metabolic enzymes and ER-resident ion channels (6, 7).
ERAD is capable of recognizing and degrading both secreted and membrane-spanning integral membrane proteins of all possible topologies and membrane orientations. Once recognized, substrates must, in the case of secreted proteins, be fully dislocated across the ER membrane to be destroyed in a ubiquitin-dependent fashion by 26S proteasomes in the cytoplasm (5). This enormous substrate diversity, together with the inherent topological constraint of having substrates initially on the opposite side of the ER membrane from the protease that degrades them, is reflected by the existence of a highly complex multiprotein system traversing the ER membrane. Intense genetic and biochemical dissection of the ERAD system in yeast and mammals has identified a large set of proteins organized around Hrd1, a membrane-integrated ubiquitin E3 ligase that forms the main structural element of the channel through which ERAD substrates are dislocated across the ER membrane (8–10). Hrd1 acts as a membrane hub that, via its obligate partner SEL1L, coordinates substrate recognition in the ER lumen with membrane dislocation and, via its ubiquitin ligase activity, recruits the cytoplasmic AAA ATPase p97/VCP to facilitate substrate extraction and hand-off to 26S proteasomes.
Proteomic interrogation of the Hrd1 interaction network in yeast (11) and mammals (12) suggests that Hrd1 exists within a highly elaborate modular protein network containing over 80 distinct nodes (12). However, despite this progress and recent application of functional genomic analyses (13–15), scant progress has been achieved toward either a structural understanding of the composition and stoichiometry of Hrd1 complexes or their dynamics, which must be highly coordinated to manage substrate diversity to achieve both fidelity and efficient coupling between substrate recognition and degradation.
Knowledge of the Hrd1 network comes mostly from studies using ectopic expression of various ERAD components by transient or stable gene expression from engineered promoters (12, 16–22). Importantly, several reports have found that overexpression of wild-type Hrd1 or its interacting partners can dominantly impair the degradation of ERAD substrates (12, 17, 23, 24). In this study we used genome editing to generate a cell line with a small tandem-affinity purification (TAP) tag knocked in to the endogenous Hrd1 locus, replacing all genomic copies of Hrd1 with a tagged variant that is expressed at normal endogenous steady-state levels. We have used this cell line to begin to biochemically dissect the Hrd1 multiprotein complex. Quantitative proteomics, together with multidimensional biochemical fractionation of Hrd1 indicates that Hrd1 complexes can be resolved into two distinct macromolecular species of different compositions and sizes. The implications of these findings in terms of the organization of ERAD are discussed.
Results
Generation of clonal cell lines expressing tandem-affinity-tagged Hrd1 from its native locus
We used genome editing to generate a clonal HEK293 cell line (Hrd1.KI) harboring a TAP tag “knocked in” to the genomic locus encoding the C terminus of Hrd1 (supplemental Fig. S1A). This TAP tag consists of a tandem Strep tag and S-epitope tag separated by a PreScission protease cleavage site. Steady-state levels of Hrd1-TAP in Hrd1.KI cells were comparable with those of Hrd1 in unmodified HEK293 cells and ∼2.7-fold lower than in a transgenic line, Hrd1.OE, that stably expresses Hrd1 bearing a variant tag, TAP*, which differs from the tag in Hrd1.KI cells by the presence of His6 instead of Strep tag. To determine whether TAP-tagged Hrd1 in Hrd1.KI cells is functional in ERAD, we performed translational shut-off assays to monitor the stabilities of two different ERAD substrates known to have a strong dependence on Hrd1: the core-glycosylated form of CD147, an endogenous type I membrane protein that assembles inefficiently (21) (Fig. 1B) and the null Hong Kong variant of α1-antitrypsin (NHK), an ectopically expressed folding-defective luminal protein (20) (Fig. 1C). Knockdown of endogenous Hrd1 (shHrd1) depleted immunodetectable Hrd1 (Fig. 1A) and strongly stabilized both CD147 and NHK. Importantly, no difference was observed in the rate of degradation of either substrate in Hrd1.KI compared with unmodified HEK293 or Hrd1.OE cells. These results establish that Hrd1.KI cells harbor a homozygous, in-frame TAP tag insertion into the endogenous Hrd1 locus and are functional in degrading two known Hrd1-dependent ERAD substrates.
Figure 1.

Genome edited Hrd1-TAP tag knock-in cells are functional in glycoprotein ERAD. A, immunoblot analysis of cell lysates from indicated HEK 293 cell lines probed with antibodies against Hrd1 or tubulin. Control, unmodified cells expressing untagged wild-type Hrd1; Hrd1.KI, Hrd1-TAP knock-in cells; shHrd1, Hrd1 knockdown cells; Hrd1.OE, Hrd1-TAP* overexpression cells. The TAP tag consists of Strep and S tags separated by a PreScission protease cleavage site, and the TAP* tag consists of His6 and S tags separated by a PreScission protease cleavage site. B, effect of Hrd1 levels and tag insertion on degradation of endogenous CD147. The indicated cell lines were treated with 25 μm emetine to inhibit protein synthesis for the indicated times. Upper panel, cell lysates were analyzed by immunoblotting with antibodies to the indicated endogenous proteins and the band intensities were quantified. M, mature; CG, core-glycosylated. Lower panel, the amount of core-glycosylated CD147 remaining at each time point was normalized to the tubulin signal, and the ratio was plotted relative to untreated. Two biological replicates for each condition are plotted. C, effect of Hrd1 levels and tag insertion on degradation of α1-antitrypsin null Hong Kong variant. Indicated cell lines were transfected with NHK-HA expressing plasmid. After 24 h, the cells were treated with 25 μm emetine for the indicated times, and the cell lysates were analyzed by immunoblotting (upper panel) and plotted (lower panel) as in A. The data represent a single experiment.
Hrd1 overexpression alters the composition of the Hrd1 complex
To analyze the effect of Hrd1 overexpression on the relative abundance of associated proteins, we used S-affinity capture to isolate Hrd1-containing protein complexes from Hrd1.OE and Hrd1.KI cells following lysis in 1% digitonin and analysis on SDS-PAGE (Fig. 2A). Although the overall pattern of major proteins from the two cell lines was grossly similar when examined by silver staining (Fig. 2A), analysis by immunoblotting (Fig. 2B) revealed notable differences in the relative abundance of proteins co-purifying with Hrd1 from Hrd1.KI cells compared with Hrd1.OE cells. Specifically, we found that SEL1L, OS9.1, and XTP3B were reduced relative to Hrd1 by a factor of ∼2, indicating that modest overexpression can alter the stoichiometry of Hrd1 with its interaction partners. Similar stoichiometries were seen when we compared the relative spectral counts of Hrd1 interaction partners obtained by LC-MS/MS analysis of Hrd1.KI cells with our previously reported proteomic data from a stable transgenic line expressing C-terminal S-tagged Hrd1 (12). This comparison shows that most Hrd1 interactors, including SEL1L, Derlin-2, Ube2g2, Ube2j1, XTP3B, and BiP, were present in affinity-captured material from cells that modestly overexpress Hrd1 at half or less the abundance (relative to Hrd1) than in Hrd1.KI cells (Fig. 2C and supplemental Table S1). Some interactors, including HERP1 and HERP2, were consistently detected in Hrd1.KI cells but were undetectable in Hrd1 overexpressing cells (Fig. 2C, asterisks). Others, notably Erlin1, Erlin2 (elevated 4× and 26×; supplemental Table S1), and VCP (elevated 2.3×; Fig. 2C and supplemental Table S1), were dramatically elevated in relative abundance upon Hrd1 overexpression. We failed to detect proteasome-derived peptides in Hrd1.KI cell line, even though we robustly captured the entire 26S proteasome in our previous published proteomic Hrd1 interactome (12). Hrd1 also had a 20–40-fold greater propensity to form high-molecular-mass species on non-reducing SDS-PAGE, suggesting that overexpressed Hrd1 can form non-native disulfide-linked multimers (Fig. 2D). Taken together, these data suggest that modest overexpression of Hrd1 may favor Hrd1 oligomerization over interactions with native binding partners.
Figure 2.

Hrd1 complex stoichiometry is altered upon Hrd1 overexpression. A, SDS-PAGE analysis of affinity captured Hrd1-TAP protein complexes. Hrd1-TAP and associated proteins were isolated by S-affinity capture from 1% digitonin lysates of either Hrd1-TAP* overexpression cells (Hrd1.OE) or Hrd1-TAP knock-in cells (Hrd1.KI). This experiment was repeated three times with similar results. B, immunoblot analysis of affinity captured Hrd1-TAP protein complexes. Hrd1-TAP and associated proteins were isolated by S-affinity capture from 1% digitonin lysates of indicated cell lines and analyzed by immunoblotting for the indicated proteins. The band intensities were quantified by LiCOR and normalized to Hrd1. The dotted graph on the right plots the ratio of Hrd1-normalized band intensity for the indicated interactors in Hrd1.OE cells compared with that in Hrd1.KI cells. Individual dots represent two independent biological replicates, and the line represents the mean. C, Hrd1 complex stoichiometry is altered by overexpression. Hrd1-associated proteins were isolated by S-affinity capture from 1% digitonin lysates of Hrd1.KI cells and analyzed by LC-MS/MS. The graph plots the ratio of bait-normalized TSCs for the indicated interactors in Hrd1-overexpressing cells (12) compared with the bait-normalized TSCs in Hrd1.KI cells. The data are calculated from the averages of three independent experiments for Hrd1.KI and four independent experiments for Hrd1-overexpressing cells. The raw data and calculations are presented in supplemental Table S1, A and B. Similar LC-MS/MS capture profiles were observed for three other independently isolated Hrd1 knock-in clones (clones 10, 15, and 20 in supplemental Fig. S1B; data not shown). Asterisks indicate proteins that were not detected in Hrd1-overexpressing cells. D, overexpression promotes Hrd1 oligomerization. Immunoblot analysis of Hrd1-TAP complexes captured from 1% digitonin lysates of the indicated cell lines by S-protein affinity capture and analyzed under non-reducing (lanes 1–3) or reducing (lanes 4–6) conditions. The blot was probed with S-tag antibody. Numerals in blue indicate different oligomeric status. Band intensities were quantified by LiCOR and graphed as a fold increase in Hrd1 level in non-reducing condition in Hrd1.OE cell line compared with the Hrd1.KI cell line. The data plotted are the means of two biological replicates with individual data points displayed as dots.
Hrd1 forms heterogeneous high-molecular-mass complexes with distinct sets of interacting proteins
To analyze native Hrd1 complexes, we used size-exclusion chromatography (SEC) to fractionate S-affinity-captured Hrd1-associated proteins isolated from detergent lysates of Hrd1.KI cells (Fig. 3, A and B). Although digitonin has been widely used to solubilize protein complexes from the ER, we found that it strongly inhibited PreScission protease activity, thereby interfering with our primary elution strategy (supplemental Fig. S2A). We therefore screened a panel of zwitterionic or non-ionic detergents and selected N-dodecyl-β-d-maltoside (DDM) for this study based on its compatibility with PreScission protease (supplemental Fig. S2A), its ability to solubilize Hrd1 from the ER membrane (supplemental Fig. S2B), and its capacity to preserve the interactions between Hrd1 and known binding partners (supplemental Fig. S2C). Hrd1 complexes were isolated from DDM lysates of Hrd1.KI cells by S-affinity capture and eluted with PreScission protease prior to Superose 6 SEC fractionation (Fig. 3B). SEC fractions were concentrated by a second round of affinity capture with strep-Tactin, and elution in SDS was followed by separation on SDS-PAGE and detection by silver staining (Fig. 3C) or immunoblotting (Fig. 3D). Hrd1 eluted in a broad profile ranging from close to the void volume of the column (∼6 MDa) to ∼100 kDa, near the predicted mass of a Hrd1 monomer (∼68 kDa), assuming a ∼50-kDa micelle molecular mass (25). The majority (95%) of Hrd1 eluted in 8.5–16 ml, corresponding roughly to 0.15 to >2 MDa and could be resolved into three distinct peaks, which we designated peak I (Ve = 8.5–12 ml, molecular mass = 0.6 to >2 MDa), peak II (Ve = 12.5–14 ml, molecular mass = 0.4–0.6 MDa), and peak III (Ve = 14.5–16 ml, molecular mass = 0.15–0.4 MDa) (Fig. 3, C and D). Known Hrd1 interactors exhibited distinct elution profiles that corresponded to one or more of these peaks (Fig. 3, C–F). SEL1L co-eluted with OS9 in a broad asymmetric, monophasic profile with a maximum at 13.5 ml, corresponding to peak II in the Hrd1 elution profile, consistent with the well established association among these three proteins (20). Reinjection of fractions from peak II (Ve = 13 ml) into a second round of SEC gave a nearly identical elution profile with no appreciable loss of protein, confirming the stability of DDM-solublized complexes (data not shown). XTP3B also eluted with peak II, consistent with its interaction with SEL1L (20), but its elution profile was distinctly more symmetric and right-shifted with respect to OS9 by ∼0.5–1 ml, suggesting that these two lectins participate in distinct complexes with the membrane-integrated Hrd1-SEL1L machinery. Interestingly, Fam8A1 and Derlin-2 exhibited biphasic elution profiles, co-fractionating with peaks I and III, complementary to the elution profile observed for SEL1L/OS9/XTP3B. Fam8A1 is a hydrophobic protein of unknown function that we previously identified as a Hrd1 interactor (12). HERP1, an ER-resident protein that is highly regulated by ER stress and known to associate with Hrd1 (26, 27), co-eluted with Fam8A1 in peak I, and a minor fraction was present in peaks II-III (Fig. 3, C–G).
Figure 3.

Native Hrd1 forms high-molecular-mass complexes. A, schematic diagram of Hrd1-TAP. Hrd1.KI cells harbor a genome-edited version of endogenous Hrd1 containing an in-frame addition of 38 amino acids consisting of a TAP tag, composed of a Strep tag and a PreScission proteaseTM cleavage site followed by an S tag and a stop codon. B, workflow for Hrd1 complex purification. Hrd1 complexes were isolated from detergent-solubilized lysates of Hrd1.KI cells by S-affinity capture and eluted with PreScission protease prior to fractionation on a Superose 6 SEC column. SEC fractions were concentrated by a second round of affinity capture with Strep-Tactin and analyzed by SDS-PAGE or LC-MS/MS. C, DDM-soluble cell lysates from Hrd1.KI cells were subjected to S-affinity capture, followed by PreScission protease elution and fractionation by Superose 6 SEC into 48 fractions of 0.5 ml. Hrd1 complexes were captured from each fraction using Strep-Tactin affinity purification, eluted in SDS sample buffer, and analyzed by SDS-PAGE and silver staining. Elution volumes of column calibration standards and their theoretical molecular masses are indicated at the top. The arrowhead between 12 and 12.5 ml indicates where images of two separate gels (prepared and run in parallel) were digitally spliced together. Asterisks indicate nonspecific background bands that are also present in the control SEC elution profile obtained with wild-type HEK293 cells harboring unmodified Hrd1 (supplemental Fig. S2D). This experiment was repeated three times with similar results. D, DDM-soluble proteins associated with purified Hrd1-TAP were captured and fractionated as described for A and analyzed by immunoblotting for the indicated proteins. The arrowhead between 12 and 12.5 ml indicates where images of two separate immunoblots prepared in parallel were digitally spliced together. The data are representative of two (OS9, XTP3B, and HERP1) or four (Hrd1, SEL1L, Fam8A1, and Der2) biological replicates. Derlin-2 was abbreviated to Der2 throughout the figures. E, elution profiles of affinity-captured Hrd1-interacting proteins. Band intensities in B were quantified by LiCOR, normalized to the peak fraction, and graphed as percentages of the total for each fraction (n = 4, ± S.E.). The raw data and calculations are in supplemental Table S2. Major peaks are indicated by Roman numerals. F, heat map of hierarchically clustered, normalized protein levels in SEC fractions obtained from data in D and supplemental Table S2. Band intensities in each blot of D were normalized to the peak fraction and are indicated in the color-intensity scale. G, heat map of hierarchically clustered normalized ion currents obtained for SEC fractions from LC-MS/MS analysis of S-protein affinity-captured Hrd1-TAP protein complexes fractionated by Superose 6 SEC (24 fractions of 1 ml) prior to Strep-Tactin affinity purification and digestion with trypsin. Ion currents of peptides mapping to known Hrd1-interacting proteins, normalized to the maximum signal across the SEC fractions, are indicated in the color intensity scale. The data are derived from a single experiment, and the calculations are in supplemental Table S3A.
The relationships among the different Hrd1 interactors are more clearly visualized by hierarchical cluster analysis of the mean normalized protein levels determined from immunoblots of four biological replicates (Fig. 3F and supplemental Table S2). In this analysis, SEL1L, OS9, and XTP3B co-elute with Hrd1 in peak II, corresponding to an apparent molecular mass of ∼400–600 kDa, whereas HERP1, Fam8A1, and Derlin-2 elute with distinctly different profiles that are largely distinct from the core complex enriched in SEL1L and the ER lectins.
These elution profiles of the core Hrd1 complex components described above were confirmed by performing LC-MS/MS analysis on SEC fractions, an approach that also enabled assessment of elution profiles for Hrd1 complex components that were not amenable to quantification by immunoblotting. Total ion currents for selected peptides corresponding to previously identified ERAD components were determined (supplemental Table S3A), and their elution profiles were plotted (supplemental Fig. S3A). Hierarchical cluster analysis of these data (Fig. 3G) revealed excellent concordance with the immunoblot data and permitted analysis of several additional Hrd1 interactors including GRP94, BiP, Ube2j1, UBXD8, ubiquitin, Erlin2, and AUP1, some of which exhibited unique elution patterns. All the known components of the “core” glycan-sensing ERAD machinery, including SEL1L, OS9, XTP3B, the ubiquitin-conjugating enzyme Ube2j1, and BiP, co-elute with Hrd1 in a relatively tight cluster (peak II) of ∼400–600 kDa. A substantial fraction of Hrd1 also elutes at considerably higher molecular mass (∼0.6–3 MDa; peak I) where it is associated with Fam8A1, Erlin2, and HERP1. Finally, we also noted that the low-molecular-mass fractions (peak III) were strikingly enriched in the core-glycosylated form of the endogenous Hrd1-dependent ERAD substrate, CD147(CG).
Effect of ubiquitin-proteasome system inhibitors on SEC fractionation of substrate-bound Hrd1 complexes
To investigate the dynamics of Hrd1-substrate complexes, we evaluated the effect of inhibitors of the ubiquitin proteasome system on the elution profiles of Hrd1, SEL1L, and CD147(CG) (Fig. 4). Inhibition of either the ubiquitin-activating enzyme E1 (C1 (28)) or VCP/p97 (CB-5083 (29, 30)) but not the proteasome (MG132) led to a substantial decrease in CD147(CG) abundance in peak III and a concomitant increase in its presence in higher-molecular-mass fractions (Fig. 4, A and B). C1 and CB-5083, but not MG132, also modestly influenced the elution of Hrd1 and SEL1L, reducing the fraction eluting in peak II with corresponding increases in peak I (Fig. 4A). Treatment with MG132 or CB-5083 led to the appearance of a ladder of bands that were detected with CD147 antibodies in both peak II and peak III fractions (Fig. 4B). This pattern, characteristic of ubiquitylation, is also consistent with our detection of ubiquitin by LC-MS/MS in these fractions (Fig. 3G and supplemental Fig. S3) and with the absence of the ladder pattern following inhibition of the ubiquitin-activating enzyme E1 (Fig. 4B). Considering the low molecular masses of protein complexes eluting in peak III (<400 kDa) and the calculated molecular mass of Hrd1 (∼68 kDa), it is likely that peak III is comprised of largely dissociated Hrd1 complexes. The persistence of a type I ERAD-M substrate with Hrd1 in these low-molecular-mass fractions and the shift toward higher molecular masses observed with inhibitors of ubiquitylation and VCP/p97 are consistent with a role for ubiquitin in substrate dissociation from Hrd1 and suggest a model in which VCP/p97 catalyzed substrate dissociation is a rate-limiting step in ERAD.
Figure 4.

Effect of ubiquitin-proteasome system inhibitors on SEC fractionation of substrate-bound Hrd1 complexes. A, LiCOR quantification of immunoblot band intensities from SEC fractions, normalized to the peak fraction, and plotted as the percentage of the total. B, CD147 immunoblot analysis of SEC fractionated Hrd1 complexes isolated from DDM solubilized lysates of Hrd1.KI cells left untreated or treated for 2 h with either 10 μm MG132, 5 μm CB-5083, or 10 μm C1. CG, core-glycosylated. The arrowhead between 12 and 12.5 ml indicates where images of two separate immunoblots prepared in parallel were digitally spliced together.
Hrd1 forms heterogeneous high-molecular-mass complexes with distinct stoichiometries
We used immunodepletion of affinity-purified and eluted Hrd1 from Hrd1.KI cells with antibodies to Fam8A1 and SEL1L to assess the subunit composition of Hrd1 complexes across the SEC spectrum (Fig. 5). Immunoblot analysis confirmed that we were able to deplete ∼95% of total SEL1L and ∼99% of Fam8A1 from affinity-purified Hrd1 complex (Fig. 5A). Depletion of SEL1L resulted in a loss of 28 ± 11% of Hrd1, indicating that a majority of Hrd1 is not complexed with SEL1L under these conditions. Consistent with this observation, depletion of SEL1L caused a shift of Hrd1 away from the largest (peak I) and smallest (peak III) fractions in favor of the “core” complex (peak II) (Fig. 5, B and C, and supplemental Table S4). In sharp contrast, depletion of Fam8A1 resulted in a loss of 71 ± 12% of Hrd1, indicating that the majority of Hrd1 is associated with Fam8A1. Accordingly, Fam8A1 depletion resulted in a pronounced shift of Hrd1 toward “core” (peak II) complexes (Fig. 5, B and C, and supplemental Table S4). Simultaneous depletion of both SEL1L and Fam8A1 led to additive losses of Hrd1 from larger toward intermediate sized (peak II) complexes. Together these data suggest that the high-molecular-mass Hrd1 complexes contain both Fam8A1 and SEL1L, a conclusion that is supported by the observation that depletion of SEL1L and Fam8A1, individually, also caused significant rightward shifts of the elution profile of the reciprocal protein (Fig. 5, B and C). We conclude that, under these solubilization conditions, Hrd1 is broadly distributed into heterogeneous protein complexes that contain variable stoichiometries of its major interaction partners, SEL1L and Fam8A1, as well as distinct cohorts of other interaction partners.
Figure 5.

Dissection of Hrd1 complex stoichiometry by immunodepletion. A, efficiency of Fam8A1 and SEL1L immunodepletion. Left panel, DDM-soluble Hrd1-TAP complexes captured by S-protein affinity purification were untreated (lane 1) or mock immunodepleted without antibody (mock, lane 2) or with antibodies against SEL1L (lane 3), Fam8A1 (lane 4), or both SEL1L and Fam8A1 (lane 5). The remaining material was analyzed by immunoblotting for the indicated proteins. Right panel, immunoblot band intensities were quantified using LiCOR and normalized relative to the mock sample. The data represent means ± S.E. of four independent experiments (for SEL1L and Fam8A1 immunodepletion) or two experiments (for co-depletion of SEL1L and Fam8A1). B, SEC elution profiles of Hrd1, Fam8A1, and SEL1L following immunodepletion of the indicated proteins. S-affinity-captured Hrd1 complexes isolated from DDM solubilized Hrd1.KI lysates were immunodepleted with antibodies against SEL1L, Fam8A1, or both SEL1L and Fam8A1 prior to SEC fractionation. Hrd1 complexes remaining in each SEC fraction were reisolated using Strep-Tactin affinity purification and analyzed by immunoblotting for the indicated proteins. Lanes marked Pre and Post denote S-protein affinity-captured Hrd1 complexes prior to and following immunodepletion, respectively. The arrowhead between 12 and 12.5 ml indicates where images of two separate immunoblots prepared in parallel were digitally spliced together. C, immunoblot band intensities in B were quantified by LiCOR, normalized to the peak intensity, and plotted as the percentages of the total of each indicated protein for each depletion condition. The raw data and calculations for Fig. 4 are in supplemental Table S4.
AQUA-MS analysis
We used AQUA-MS (31) to determine the absolute levels of Hrd1 and its major interaction partners. In unfractionated tandem affinity-purified complexes, Hrd1 was present in stoichiometric excess over its binding partners ranging from ∼2-fold for Fam8A1 to ∼5- and ∼8-fold for OS9 and SEL1L, respectively (Tables 1 and 2). The profile of absolute levels of Hrd1 and its interactors following SEC fractionation (Fig. 6) closely paralleled the distributions observed in immunoblotting (Fig. 3E), spectral counting (supplemental Fig. S3B and Table S3B) or ion current (Fig. 3G and (supplemental Fig. S3A and Table S3A) measurements. However, we found that the absolute molar ratio of Hrd1 to its interactors varies considerably across the elution profile, with SEL1L being strikingly disenriched in the highest- (peak I) and lowest-molecular-mass (peak III) SEC fractions. OS9, which is known to bind directly to SEL1L (20), exhibits a similar elution profile and for the most part is present throughout the SEC fractions at a level approximately twice that of SEL1L, reflecting the possibility that SEL1L has two binding sites for OS9. By contrast, Fam8A1 is strongly disenriched in complexes isolated from peak II and peak III, reflecting that the vast majority of this protein is associated with highest-molecular-mass Hrd1 complexes where it is present at approximately half the level of Hrd1.
Table 1.
Analysis of Hrd1 complex stoichiometry by AQUA-MS to determine the protein amount in tandem affinity-captured Hrd1 complexes
The absolute amounts of the indicated proteins within unfractionated Hrd1 complexes and the ratio between interactors are shown. The data represent the averages ± S.E., combining measurements of all peptide standards per target across three biological replicates.
| Protein | Absolute amount | Ratio (Hrd1:X) |
|---|---|---|
| fmol | ||
| Hrd1 | 637 ± 133 | 1.0 |
| Fam8A1 | 273 ± 54 | 2.3 |
| SEL1L | 78 ± 19 | 8.1 |
| OS9 | 130 ± 22 | 4.9 |
Table 2.
Analysis of Hrd1 complex stoichiometry by AQUA-MS to determine the amount of Hrd1 relative to interactor
Stoichiometry of Hrd1 relative to the indicated proteins within the indicated SEC fractions is shown.
| Interactor | SEC elution volume |
|||||||
|---|---|---|---|---|---|---|---|---|
| 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | |
| ml | ||||||||
| Fam8A1 | 2.0 | 1.6 | 2.0 | 3.4 | 8.1 | 4.4 | 1.1 | 0.6 |
| SEL1L | 24.3 | 33.1 | 20.0 | 9.9 | 8.6 | 5.2 | 50.9 | 54.3 |
| OS9 | 6.6 | 11.6 | 9.5 | 4.5 | 3.6 | 3.6 | 33.7 | 16.9 |
Figure 6.
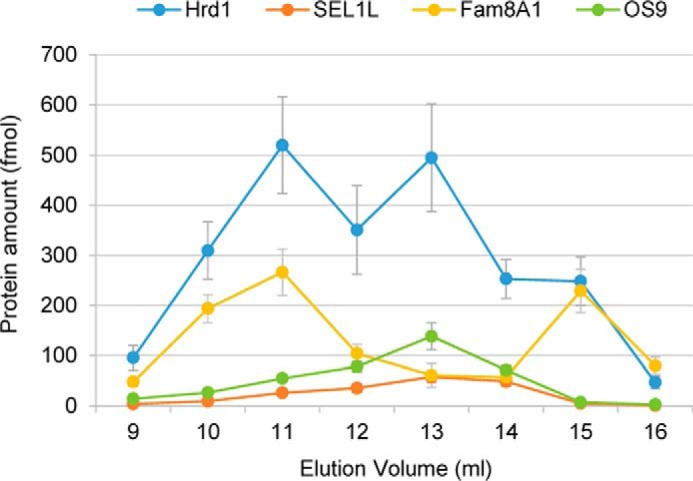
Stoichiometry of Hrd1 complexes determined by AQUA-MS. Absolute quantification of the indicated proteins within the indicated SEC fractions. The data represent the means ± S.E. derived from combining measurements from two reference peptides (for Hrd1, SEL1L, and Fam8A1) or one reference peptide (for OS9) across three biological replicates.
Discussion
Hrd1 is the central element of a large protein complex that coordinates the processes of quality-control protein degradation in the ER. In addition to functioning to scaffold and organize substrate recognition and ubiquitylation at the luminal and cytoplasmic sides of the ER membrane, respectively, Hrd1 is likely to constitute the key structural member of the transmembrane pore though which glycoproteins are conducted to be destroyed by cytoplasmic proteasomes (5, 8–10, 32–36). Understanding the structure and composition of native Hrd1 protein complexes is therefore central to our understanding of the molecular mechanism of ERAD. Although a large number of Hrd1 interactors have been identified from high-throughput (12) and conventional (37–39) biochemical analyses that have relied largely on protein overexpression, the number, size, dynamics, and stoichiometries of mammalian Hrd complexes and the effect of overexpression on these parameters have remained largely uninvestigated.
In this study we used genome editing to generate a human cell line in which we have replaced all copies of endogenous Hrd1 with a variant containing a TAP tag at the C terminus. This knock-in does not alter the genomic organization of the endogenous Hrd1 locus. We demonstrate that steady-state levels of tagged Hrd1 in these cells are indistinguishable from those of Hrd1 in unmodified cells and that the tagged variant is fully functional as judged by observing normal kinetics of degradation of two different ERAD substrates. These cells allow Hrd1 to be efficiently isolated from detergent lysates by sequential affinity capture, establishing them as a useful resource for biochemical and structural analysis of Hrd1 protein complexes.
Because of its central role in mediating ERAD, Hrd1 has been the focus of several prior biochemical and proteomic analyses (11, 40–42), including our systematic analysis of the Hrd1 interactome (12). However, nearly all previous interaction studies have relied on overexpression of affinity-tagged Hrd1 and often overexpression of the interaction partner(s) as well. The data reported herein show that even modest (2–3-fold) overexpression alters the stoichiometry with nearly all of the interaction partners of Hrd1. We find that SEL1L and its partners, XTP3B and OS9, are reduced relative to Hrd1 by a factor of ∼2. Other established members of the Hrd1 complex, including UBXD8, Derlin-2, Ube2J1, and HERP1, are reduced by more than 3-fold upon Hrd1 overexpression. It is possible that this apparent discrepancy could be trivially explained as a simple artifact of the fact that the data were normalized to Hrd1 levels. A simple dilution model would predict that all interactors would be subject to a similar reduction in level upon overexpression. However, we found that several proteins, notably Erlin1, Erlin2, and VCP, are strongly over-represented in Hrd1 captured material from overexpressing cells. Finally the complete absence of 26S proteasome subunits, which were prominently identified in our previously published Hrd1 interactome study (12) and were not detectably associated with Hrd1.KI at endogenous levels, does not support a trivial dilution argument. Our finding of non-native, disulfide-linked Hrd1 oligomers in overexpressing cells leads us to propose that homo-oligomerization of overexpressed Hrd1 displaces other interactors and promotes autoubiquitylation of the cytoplasmically exposed portions of Hrd1, thereby recruiting polyubiquitin binding proteins such as the 26S proteasome and VCP. These findings underscore the need to avoid even modest overexpression to preserve native subunit stoichiometries.
SEC fractionation of affinity-purified Hrd1 complexes identified two distinct high molecular complexes centered at ∼2 and 0.5 MDa that differ most notably in the relative content of the two major Hrd1 interaction partners, Fam8A1 and SEL1L (along with proteins such as OS9 and XTP3B, which bind to the complex via SEL1L). Previous studies using sucrose density gradient fractionation to analyze the size distribution of digitonin-solubilized, overexpressed mammalian Hrd1 complexes reported the identification of a 27S fraction containing Hrd1, OS9, SEL1L, and VCP, along with overexpressed XTP3B (18). However, in addition to differences caused by overexpression and detergent solubilization, SEC and density gradient sedimentation are influenced by fundamentally different physical properties including size, shape, and matrix interactions, thereby limiting direct comparisons of estimated size distributions obtained in previous studies. Comparisons between our data and previous analyses (26, 43, 44) of mammalian Hrd1 complexes are further complicated because previous work interrogated different subsets of Hrd1-interacting proteins (aside from the aforementioned SEL1L and associated proteins). Ours is the first study to use unbiased mass spectrometric identification to analyze the composition of SEC fractionated mammalian Hrd1 complexes. Of note, one previous study (11) applied similar unbiased metrics to analyze sucrose density gradient-fractionated, chromosomally tagged Hrd1 in Saccharomyces cerevisiae, identifying a complex with estimated molecular mass of ∼500 kDa composed of the yeast orthologs of the same core components (Hrd3/SEL1L, Yos9/OS9) present in peak II in this study.
The finding in the present study of two high-molecular-mass peaks of Hrd1 in our SEC fractionation experiment could reflect an intrinsic heterogeneity in the number and size of stable endogenous Hrd1 complexes (perhaps with different substrate ranges or localization), a dynamic balance between multiple complexes, or the consequence of dissociation of a single, large complex upon cell lysis in DDM. The strong enrichment in peak I of Fam8A1, a very hydrophobic protein of unknown function that has no apparent ortholog in yeast, and its 1:2 stoichiometric relationship with Hrd1 suggests that Fam8A1 likely contributes to the structure and organization of a high-molecular-mass Hrd1 complex.
SEL1L (and its ortholog in yeast, Hrd3) is an obligate partner of Hrd1 and is required for its stability (45). It is essential for Hrd1-mediated degradation of proteins with luminal conformational lesions (i.e. ERAD-L) in both fungi and mammals (46, 47). The very strong disenrichment of SEL1L from peak I is therefore difficult to reconcile with the conclusion that this high-molecular-mass complex is functional, at least for ERAD-L. Our data do not exclude the possibility that Hrd1 may participate, together with Fam8A1, Erlin2, and HERP, in a SEL1L-deficient high-molecular-mass complex dedicated to a specific class of ERAD substrates. For example, Hrd3 is not required for degradation of some integral membrane proteins in yeast upon Hrd1 overexpression and is dispensable for retrotranslocation of ERAD-L substrates in vitro (10). Perhaps the ∼500-kDa (peak II) Hrd1 complex, which contains a complement of proteins similar to those present in the core yeast complex of similar molecular mass (11), is specialized for degradation of ERAD-M type glycoprotein substrates. Finally, our data do not exclude the possibility that mammalian Hrd1 normally exists as an ensemble of very-high-molecular-mass complex that mediates both ERAD-L and ERAD-M but dissociates upon cell lysis into the smaller complexes that we observe upon SEC fractionation in this study. Further study will be needed to distinguish between these possibilities.
Materials and methods
TALEN and targeting vector construction
TALEN genomic binding sites were chosen to be 19 bp in length separated by a 16-bp spacer. The Hrd1 genomic sequences for TALEN binding are as follows: TALEN-L binding site: 5′-CCTGTTGCCCACTGACACT-3′ and TALEN-R binding site: 5′-GCTCAAAAGAGCAGAGGCT-3′. TALE repeats were constructed using the two-step Golden Gate assembly method described previously (48). The final arrays of repeat variable di-residues were assembled into the MR015 plasmid (a kind gift from Dr. Matthew Porteus, Stanford University), which enables expression of FLAG-tagged TALE fused to a nuclear localization signal and the FokI nuclease domain. pDTA-TK vector (49) was used as the backbone to construct the Hrd1 targeting vector for homologous recombination. The vector was modified to contain a TAP tag and a puromycin-resistant gene under the control of the phosphoglycerate kinase promoter. The amino-acid sequence for the TAP tag, designed as immediately preceding the C terminus of Hrd1, is as follows: linker (LE), Strep tag II (WSHPQFEK), PreScission protease cleavage site (LEVLFQGP), linker (LTGRT), and S tag (KETAAAKFERQHMDS). A 519-bp 5′ homologous arm (using the primer pairs, 5′-GTCGACATCATTTGGCCCTTGAGTCC-3′ and 5′-CTCGAGGTGGGCAACAGGAGACTCCA-3′) and a 516-bp 3′ homologous arm (using the primer pairs, 5′-ATCGATTGACACTGCCCCAGCCCA-3′ and 5′-GCTAGCCCACCACCACAAGAGGCTTC-3′) were amplified by PCR from HEK293 genomic DNA and then cloned into the modified pDTA-TK vector.
Cell culture and transfection
HEK293 cells were cultured in DMEM containing 4.5 g/liter glucose and l-glutamine (Mediatech, Manassas, VA), and supplemented with 10% FetalPlex animal serum complex (Gemini Bio-Products, West Sacramento, CA) at 37 °C and 5% CO2. The pair of TALENs recognizing Hrd1 loci and the targeting vector of Hrd1 fused to TAP tag were co-transfected into HEK293 cells using a standard calcium-phosphate co-precipitation technique (50). Stable cell lines were selected in 10 μg/ml puromycin (Sigma-Aldrich), cloned by limiting dilution, and thereafter maintained in 1 μg/ml puromycin. pSUPER shRNA expression construct targeting Hrd1 has been described previously (12). HEK293 cells overexpressing Hrd1-TAP or Hrd1-TAP* were generated using a standard calcium-phosphate co-precipitation technique followed by puromycin or G418 selection, respectively. Stable expression of Hrd1-TAP* was driven by a CMV promoter in pcDNA3.1. The TAP* sequence immediately preceding the C terminus of Hrd1 is as follows: linker (GTG), His6 tag (HHHHHH), PreScission protease cleavage site (LEVLFQGP), linker (GGGT), and S tag (KETAAAKFERQHMDS). The plasmid encoding HA-tagged NHK was previously described (20) and was transfected and expressed for 48 h prior to performing emetine-chase assays.
Emetine-chase assay
Cells were treated with emetine (Sigma-Aldrich) at a final concentration of 25 μm for the indicated time. The cells were collected and washed in PBS before lysis in 1% SDS lysis buffer (50 mm Tris, pH 7.4, 150 mm NaCI, 1% SDS). Lysates were analyzed by immunoblotting.
Antibodies and inhibitors
The following rabbit polyclonal antibodies were used for immunoblotting at the indicated concentrations: anti-Hrd1 (a gift from R. Wojcikiewicz, SUNY Upstate Medical University; 1:100, Proteintech Group, Chicago, IL; and 1:1,000, Novus Biologicals, Littleton, CO; 1:1,000), anti-SEL1L and anti-Derlin-2 (gifts from H. Ploegh, Whitehead Institute for Biomedical Research; 1:2,000), anti-OS-9 (1:500) and anti-XTP3-B (1:500) (12), anti-Fam8A1 (1:10,000) (12), and anti-HERP1 (a gift from L. Hendershot, St Jude's Children's Research Hospital; 1:1,000). The following mouse monoclonal antibodies were used at the indicated concentrations: anti-CD147 clone A-12 (Santa Cruz Biotechnology, Santa Cruz, CA; 1:500), anti-S-peptide (EMD Millipore, Billerica, MA; 1:5,000), and anti-tubulin clone T6199 (Sigma-Aldrich; 1:10,000). Secondary antibodies include: IRDye800CW goat anti-mouse and rabbit IgG and IRDye680RD goat anti-mouse and rabbit IgG (LI-COR Biosciences, Lincoln, NE; 1:10,000). Anti-rabbit and anti-mouse secondary antibodies conjugated to HRP were from GE Healthcare Life Sciences (1:10,000). MG132 was purchased from Enzo Life Sciences (Farmingdale, NY). CB-5083 was generously provided by Cleave Biosciences (Burlingame, CA). C1 (compound I) was provided by Takeda Oncology (Cambridge, MA).
Immunoblotting
Samples suspended in Laemmli buffer containing 2-mercaptoethanol 2% (v/v) were heated at 65 °C for 10 min, separated by SDS-PAGE and transferred to PVDF (GE Healthcare Life Sciences). Laemmli buffer lacking 2-mercaptoethanol was used for non-reducing SDS-PAGE presented in Fig. 2D. PVDF membranes were blocked with 5% nonfat milk in PBS to reduce nonspecific antibody binding and incubated with primary antibodies diluted in PBS containing 0.2% Tween 20 and 3% BSA. Membranes were washed in PBS containing 0.2% Tween 20 and incubated with HRP-conjugated secondary antibodies. Immunoreactivity was detected using ECL+ chemiluminescence reagents (GE Healthcare Life Sciences) for immunoblots presented in Fig. 1B and supplemental Figs. S1 and S2. Fluorescent IRDye secondary antibodies were used for all the remaining immunoblots and scanned by Odyssey imaging (LI-COR Biosciences). Band intensities were quantified by Image Studio Life software (LI-COR Biosciences).
Cell lysis and affinity purifications
HEK293 cells were harvested by scraping and washed in ice-cold PBS before lysis in buffer containing 50 mm Tris-HCl (pH 7.4), 150 mm NaCl, 2.5 mm EDTA, protease inhibitor cOmplete EDTA-free tablet (Roche Applied Science), and 1% (w/v) DDM (Anatrace, Maumee, OH) for 30 min at 4 °C. The lysates were cleared by centrifugation at 20,000 × g for 15 min at 4 °C. Protein concentration was measured by BCA protein assay (Pierce). Normalized lysates were incubated with S protein-agarose (Novagen) for 4–12 h at 4 °C with mixing and washed four to five times in lysis buffer containing 0.2% DDM.
For tandem-affinity purification, protein complexes bound to S protein-agarose were incubated with GST-tagged Prescission protease (GE Healthcare Life Sciences) in 50 mm Tris-HCl (pH 7.4), 150 mm NaCl, 0.2% DDM, and 2 mm DTT for 4–12 h at 4 °C. Prescission protease was removed from the cleavage reaction by incubation with glutathione-Sepharose 4B (GE Healthcare Life Sciences) for 2–3 h at 4 °C. Unbound supernatant was filtered with a 0.45-μm PVDF syringe filter (Thermo Fisher Scientific). The resulting eluted affinity-purified protein complexes were analyzed by immunoblotting or subjected to SEC.
For antibody depletion assays, after removal of GST-tagged prescission protease, the affinity-purified protein complexes were incubated with anti-SEL1L, anti-Fam8A1, or no antibody with gentle rocking for 3 h at 4 °C, followed by incubation with protein A/G Plus agarose bead (Pierce) for 1 h at 4 °C. The unbound material was then analyzed by immunoblotting or subjected to size-exclusion chromatography.
Size-exclusion chromatography
Affinity-purified protein complexes were applied to a Superose 6 10/300 GL column (GE Healthcare Life Sciences) equilibrated with buffer contained 50 mm Tris (pH 7.4), 150 mm NaCI, and 0.2% DDM. Either 0.5- or 1-ml fractions were collected at a flow rate of 0.4 ml/min depending on the downstream analysis. Size determination was carried out with thyroglobulin (669 kDa), ferritin (440 kDa), aldolase (158 kDa), conalbumin (75 kDa) (GE Healthcare), purified 40S ribosome (∼1.3 MDa) (a gift from J. Puglisi, Stanford University), and purified 26S and 20S proteasome (∼800 kDa) (56) as calibration proteins. Collected fractions were incubated with MagStrep type-2 beads (IBA, Goettingen, Germany) for 3–4 h at 4 °C with mixing to reisolate Hrd1 containing complexes. The beads were washed two to three times in lysis buffer containing 0.2% DDM and eluted in Laemmli buffer at 65 °C for 10 min for immunoblotting or silver staining (Pierce) or prepared for mass spectrometry.
Sample preparation for MS and AQUA-MS
For LC-MS/MS analysis, the affinity-purified protein complexes bound to MagStrep type 2 beads were washed three times with 50 mm ammonium bicarbonate (pH 8.0), eluted in 0.1% RapiGest SF (Waters, Milford, MA) in 50 mm ammonium bicarbonate (pH 8.0) at 37 °C, and incubated with sequencing grade trypsin (Promega) for 16 h at 37 °C. Samples were acidified with hydrochloric acid to pH 2, centrifuged at 20,000 × g for 10 min, and analyzed using a linear ion-trap mass spectrometer as previously described (20).
AQUA peptide standards were synthesized and quantified by JPT (Berlin, Germany) or Sigma-Aldrich corresponding to tryptic peptides (stable isotope-labeled amino acids indicated by asterisks) for Hrd1 (ALEGHER*, HQFYPTV*VYLTK), SEL1L (LMTAYNSYK*, LTEEGSPK*), Fam8A1 (TAAGISTPAPVALGL*GPR, APHVQASVR*), and OS9 (IVRPWAEGTEEGAR*). Lyophilized peptide standards were resuspended in 10% formic acid, vortexed, and diluted to 5 pmol/μl in 0.1% formic acid before being combined as a master mix. 125 fmol of each peptide standard were spiked into affinity-purified protein complexes subjected to SEC (as described above) and bound to MagStrep type-2 beads in the presence of 50 mm ammonium bicarbonate, 6.8 m urea, and 5 mm tris-(2-carboxyethyl) phosphine (Pierce) and incubated for 30 min at room temperature. Samples were alkylated with 10 mm 2-chloroacetamide for 30 min in the dark at room temperature. 300 ng of mass spectrometry grade lysyl endopeptidase (Lys-C; Wako Chemicals) was added, and samples were incubated for 4 h at 37 °C before dilution to 1 m urea with 50 mm ammonium bicarbonate and addition of 300 ng of sequencing grade trypsin (Sigma-Aldrich). The samples were further incubated for 12 h at 37 °C before acidification with HCl to pH 2 and desalted with the stage-tip method (51).
Combined discovery and targeted (AQUA-MS) instrumentation and data analysis
Samples were analyzed by online capillary nano-LC-MS/MS. Peptide mixtures were separated on an in-house made 20-cm reversed-phase column (100-μm inner diameter, packed with ReproSil-Pur C18-AQ 3.0-μm resin (Dr. Maisch GmbH)) equipped with a laser-pulled nanoelectrospray emitter tip. Peptides were eluted at a flow rate of 400 nl/min using a two-step linear gradient including 3–25% buffer B in 70 min and 25–40% B in 20 min (buffer A: 0.2% formic acid and 5% DMSO in water; buffer B: 0.2% formic acid and 5% DMSO in acetonitrile) in a Dionex UltiMate 3000 HPLC system (Thermo Fisher Scientific). Peptides were then analyzed using a LTQ Orbitrap Velos mass spectrometer (Thermo Fisher Scientific). Data acquisition was executed in data-dependent mode with full MS scans acquired in the Orbitrap mass analyzer with a resolution of 60000 and m/z scan range of 340–2000. A global parent mass list was enabled including the m/z of all the AQUA peptides and their corresponding light peptides, as well as the retention time windows (the time window was set to 4 min and centered around the retention time obtained from a previous regular data-dependent run). The top two most abundant ions from the list and top eight most abundant ions from MS1 with intensity threshold above 500 counts and charge states 2 and above were selected for fragmentation using collision-induced dissociation with an isolation window of 2 m/z, a collision energy of 35%, an activation Q of 0.25, and an activation time of 5 ms. The collision-induced dissociation fragments were analyzed in the ion trap with rapid scan rate. Dynamic exclusion was enabled with repeat count of 3 in 30 s and exclusion duration of 20 s. The AGC target was set to 500,000 and 1000 for full FTMS scans and ITMSn scans. The maximum injection time was set to 500 and 100 s for full FTMS scans and ITMSn scans.
The resulting spectra were searched against a “target-decoy” sequence database (52) consisting of the Uniprot human database (downloaded June 11, 2013 and containing 88,902 entries) and the corresponding reversed sequences using the SEQUEST algorithm (SEQUEST version 28, revision 12). The parent mass tolerance was set to 20 ppm, and the fragment mass tolerance was set to 0.6 Da. Enzyme specificity was set to trypsin, and the maximum number of internal cleavages was set to 3. Oxidation of methionines and acetylation of the protein N terminus were set as variable modification. Carbamidomethylation of cysteines was set as a static modification. The data were filtered to a 1% peptide and 5% protein false discovery rate using a linear discriminator analysis (53). The AQUA peptides were manually identified using Xcalibur. Peak areas were generated for each pair of heavy and light peptides by extracting-ion chromatograms within 10 ppm of the observed m/z for the monoisotopic peaks, as previously described (54).
MS instrumentation and data analysis
Other LC-MS/MS analyses were performed on a LTQ-Orbitrap model XL (ThermoFisher) using identical or similar settings to those described above. Peptide chromatographic separations were performed either in nanoflow mode on a 750-μm inner diameter × 10-cm BioBasic C18 column (New Objective, Inc.) or in microflow mode on a 300-μm inner diameter × 15-cm C18 column (CVI, Inc.). Peptides were eluted with a linear gradient of 5 to 45% acetonitrile in the presence of 0.1% formic acid. The data were analyzed manually in Xcalibur Qual browser software by extracting parent ion masses of standards and analytes using a mass window of 6 mTh after assuring that the mass measurements were obtained with adequate mass accuracy. Relative peak intensities for standards and analytes were measured on extracted, smoothed chromatographic peaks. Database searches were performed using Byonic software (Protein Metrics Inc.) using a variable modification set consisting of methionine oxidation, deamidation of asparagine and glutamine, pyroglutamate for N-terminal glutamine and glutamic acid, carbamidomethylation of cysteine, mono- and di-methylation of lysine residues on trypsin, and the appropriate heavy-isotope modifications on the proteins that had isotope-labeled standards. Searches were against a recent human protein database containing reverse-sequence decoys. Relative intensities of relevant peptides from proteins of interest were extracted from extracted-ion chromatograms as described previously (55).
Heat map analysis
The hierarchically clustered heat maps representing immunoblot and MS data were generated using MultiExperimental Viewer v4.7. Quantified protein levels from Immunoblot (Fig. 3D and supplemental Table S2) or ion current intensity (supplemental Table S3A), and TSCs (supplemental Table S3B) from MS were normalized to the peak SEC fraction. The heat map scale of normalized intensities is from 0 (white) to 1 (red).
Author contributions
J.H., C. P. W., and R. R. K. conceived and coordinated the study and wrote the paper. J. H. and C. P. W. designed, performed, and analyzed the experiments except as noted. J. H. and J. A. O. designed and performed the experiments shown in supplemental Fig. S1. T. A. S. contributed to Figs. 2B and 3G and supplemental Fig. S3. C. P. W., L. Z., and J. E. E. performed the experiments in Fig. 6 and Tables 1 and 2.
Supplementary Material
Acknowledgments
We thank members of the Kopito lab and Susan Michaelis (Johns Hopkins University) for helpful discussion and critical comments on the manuscript. We thank Ethan Greenblatt for generating the Hrd1-TAP* stable cell line. We thank Fiona McAllister for initial analysis and interpretation of mass spectrometry data. We also thank Hidde Ploegh (MIT) John C. Christianson (University of Oxford), Linda M. Hendershot (St. Jude Children's Research Hospital), and Yihong Ye (National Institutes of Health) for providing, respectively, antibodies to SEL1L, HERP1, and Derlin-2.
This work was supported by National Institutes of Health Grant R01GM074874 (to R. R. K.) and a Ruth L. Kirschstein National Research Service Award F32GM113378 (to C. P. W.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Tables S1–S4 and Figs. S1–S3.
- ER
- endoplasmic reticulum
- ERAD
- ER-associated degradation
- SEC
- size-exclusion chromatography
- AQUA
- absolute quantification
- DDM
- N-dodecyl-β-d-maltoside
- TAP
- tandem-affinity purification
- TSC
- total spectral counts.
References
- 1. Ghaemmaghami S., Huh W. K., Bower K., Howson R. W., Belle A., Dephoure N., O'Shea E. K., and Weissman J. S. (2003) Global analysis of protein expression in yeast. Nature 425, 737–741 [DOI] [PubMed] [Google Scholar]
- 2. Kanapin A., Batalov S., Davis M. J., Gough J., Grimmond S., Kawaji H., Magrane M., Matsuda H., Schönbach C., Teasdale R. D., Yuan Z., RIKEN GER Group, and GSL Members (2003) Mouse proteome analysis. Genome Res. 13, 1335–1344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Balchin D., Hayer-Hartl M., and Hartl F. U. (2016) In vivo aspects of protein folding and quality control. Science 353, aac4354. [DOI] [PubMed] [Google Scholar]
- 4. Guerriero C. J., and Brodsky J. L. (2012) The delicate balance between secreted protein folding and endoplasmic reticulum-associated degradation in human physiology. Physiol. Rev. 92, 537–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Olzmann J. A., Kopito R. R., and Christianson J. C. (2013) The mammalian endoplasmic reticulum-associated degradation system. Cold Spring Harb. Perspect. Biol. 5, a013185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ward C. L., Omura S., and Kopito R. R. (1995) Degradation of CFTR by the ubiquitin-proteasome pathway. Cell 83, 121–127 [DOI] [PubMed] [Google Scholar]
- 7. Stevenson J., Huang E. Y., and Olzmann J. A. (2016) Endoplasmic reticulum-Associated degradation and lipid homeostasis. Annu. Rev. Nutrition 36, 511–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Baldridge R. D., and Rapoport T. A. (2016) Autoubiquitination of the Hrd1 ligase triggers protein retrotranslocation in ERAD. Cell 166, 394–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stein A., Ruggiano A., Carvalho P., and Rapoport T. A. (2014) Key steps in ERAD of luminal ER proteins reconstituted with purified components. Cell 158, 1375–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Carvalho P., Stanley A. M., and Rapoport T. A. (2010) Retrotranslocation of a misfolded luminal ER protein by the ubiquitin-ligase Hrd1p. Cell 143, 579–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Carvalho P., Goder V., and Rapoport T. A. (2006) Distinct ubiquitin-ligase complexes define convergent pathways for the degradation of ER proteins. Cell 126, 361–373 [DOI] [PubMed] [Google Scholar]
- 12. Christianson J. C., Olzmann J. A., Shaler T. A., Sowa M. E., Bennett E. J., Richter C. M., Tyler R. E., Greenblatt E. J., Harper J. W., and Kopito R. R. (2011) Defining human ERAD networks through an integrative mapping strategy. Nat. Cell Biol. 14, 93–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Timms R. T., Menzies S. A., Tchasovnikarova I. A., Christensen L. C., Williamson J. C., Antrobus R., Dougan G., Ellgaard L., and Lehner P. J. (2016) Genetic dissection of mammalian ERAD through comparative haploid and CRISPR forward genetic screens. Nat. Commun. 7, 11786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Travers K. J., Patil C. K., Wodicka L., Lockhart D. J., Weissman J. S., and Walter P. (2000) Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 101, 249–258 [DOI] [PubMed] [Google Scholar]
- 15. Grotzke J. E., Lu Q., and Cresswell P. (2013) Deglycosylation-dependent fluorescent proteins provide unique tools for the study of ER-associated degradation. Proc. Natl. Acad. Sci. U.S.A. 110, 3393–3398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Iida Y., Fujimori T., Okawa K., Nagata K., Wada I., and Hosokawa N. (2011) SEL1L protein critically determines the stability of the HRD1-SEL1L endoplasmic reticulum-associated degradation (ERAD) complex to optimize the degradation kinetics of ERAD substrates. J. Biol. Chem. 286, 16929–16939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mueller B., Klemm E. J., Spooner E., Claessen J. H., and Ploegh H. L. (2008) SEL1L nucleates a protein complex required for dislocation of misfolded glycoproteins. Proc. Natl. Acad. Sci. U.S.A. 105, 12325–12330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hosokawa N., Wada I., Nagasawa K., Moriyama T., Okawa K., and Nagata K. (2008) Human XTP3-B forms an endoplasmic reticulum quality control scaffold with the HRD1-SEL1L ubiquitin ligase complex and BiP. J. Biol. Chem. 283, 20914–20924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Klemm E. J., Spooner E., and Ploegh H. L. (2011) Dual role of ancient ubiquitous protein 1 (AUP1) in lipid droplet accumulation and endoplasmic reticulum (ER) protein quality control. J. Biol. Chem. 286, 37602–37614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Christianson J. C., Shaler T. A., Tyler R. E., and Kopito R. R. (2008) OS-9 and GRP94 deliver mutant α1-antitrypsin to the Hrd1-SEL1L ubiquitin ligase complex for ERAD. Nat. Cell Biol. 10, 272–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tyler R. E., Pearce M. M., Shaler T. A., Olzmann J. A., Greenblatt E. J., and Kopito R. R. (2012) Unassembled CD147 is an endogenous endoplasmic reticulum-associated degradation substrate. Mol. Biol. Cell 23, 4668–4678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Huang C. H., Chu Y. R., Ye Y., and Chen X. (2014) Role of HERP and a HERP-related protein in HRD1-dependent protein degradation at the endoplasmic reticulum. J. Biol. Chem. 289, 4444–4454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Plemper R. K., Bordallo J., Deak P. M., Taxis C., Hitt R., and Wolf D. H. (1999) Genetic interactions of Hrd3p and Der3p/Hrd1p with Sec61p suggest a retro-translocation complex mediating protein transport for ER degradation. J. Cell Sci. 112, 4123–4134 [DOI] [PubMed] [Google Scholar]
- 24. Hosokawa N., and Wada I. (2016) Association of the SEL1L protein transmembrane domain with HRD1 ubiquitin ligase regulates ERAD-L. FEBS J. 283, 157–172 [DOI] [PubMed] [Google Scholar]
- 25. Rosevear P., VanAken T., Baxter J., and Ferguson-Miller S. (1980) Alkyl glycoside detergents: a simpler synthesis and their effects on kinetic and physical properties of cytochrome c oxidase. Biochemistry 19, 4108–4115 [DOI] [PubMed] [Google Scholar]
- 26. Kny M., Standera S., Hartmann-Petersen R., Kloetzel P. M., and Seeger M. (2011) Herp regulates Hrd1-mediated ubiquitylation in a ubiquitin-like domain-dependent manner. J. Biol. Chem. 286, 5151–5156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schulze A., Standera S., Buerger E., Kikkert M., van Voorden S., Wiertz E., Koning F., Kloetzel P. M., and Seeger M. (2005) The ubiquitin-domain protein HERP forms a complex with components of the endoplasmic reticulum associated degradation pathway. J. Mol. Biol. 354, 1021–1027 [DOI] [PubMed] [Google Scholar]
- 28. Chen J. J., Tsu C. A., Gavin J. M., Milhollen M. A., Bruzzese F. J., Mallender W. D., Sintchak M. D., Bump N. J., Yang X., Ma J., Loke H. K., Xu Q., Li P., Bence N. F., et al. (2011) Mechanistic studies of substrate-assisted inhibition of ubiquitin-activating enzyme by adenosine sulfamate analogues. J. Biol. Chem. 286, 40867–40877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Anderson D. J., Le Moigne R., Djakovic S., Kumar B., Rice J., Wong S., Wang J., Yao B., Valle E., Kiss von Soly S., Madriaga A., Soriano F., Menon M. K., Wu Z. Y., Kampmann M., et al. (2015) Targeting the AAA ATPase p97 as an approach to treat cancer through disruption of protein homeostasis. Cancer Cell 28, 653–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhou H. J., Wang J., Yao B., Wong S., Djakovic S., Kumar B., Rice J., Valle E., Soriano F., Menon M. K., Madriaga A., Kiss von Soly S., Kumar A., Parlati F., Yakes F. M., et al. (2015) Discovery of a first-in-class, potent, selective, and orally bioavailable inhibitor of the p97 AAA ATPase (CB-5083). J. Med. Chem. 58, 9480–9497 [DOI] [PubMed] [Google Scholar]
- 31. Gerber S. A., Rush J., Stemman O., Kirschner M. W., and Gygi S. P. (2003) Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS. Proc. Natl. Acad. Sci. U.S.A. 100, 6940–6945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hirsch C., Gauss R., Horn S. C., Neuber O., and Sommer T. (2009) The ubiquitylation machinery of the endoplasmic reticulum. Nature 458, 453–460 [DOI] [PubMed] [Google Scholar]
- 33. Kostova Z., Tsai Y. C., and Weissman A. M. (2007) Ubiquitin ligases, critical mediators of endoplasmic reticulum-associated degradation. Semin. Cell Dev. Biol. 18, 770–779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hoseki J., Ushioda R., and Nagata K. (2010) Mechanism and components of endoplasmic reticulum-associated degradation. J. Biochem. 147, 19–25 [DOI] [PubMed] [Google Scholar]
- 35. Vembar S. S., and Brodsky J. L. (2008) One step at a time: endoplasmic reticulum-associated degradation. Nat. Rev. Mol. Cell Biol. 9, 944–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bagola K., Mehnert M., Jarosch E., and Sommer T. (2011) Protein dislocation from the ER. Biochim. Biophys. Acta 1808, 925–936 [DOI] [PubMed] [Google Scholar]
- 37. Sayeed A., and Ng D. T. (2005) Search and destroy: ER quality control and ER-associated protein degradation. Crit. Rev. Biochem. Mol. Biol. 40, 75–91 [DOI] [PubMed] [Google Scholar]
- 38. Nakatsukasa K., and Brodsky J. L. (2008) The recognition and retrotranslocation of misfolded proteins from the endoplasmic reticulum. Traffic 9, 861–870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hebert D. N., and Molinari M. (2007) In and out of the ER: protein folding, quality control, degradation, and related human diseases. Physiol. Rev. 87, 1377–1408 [DOI] [PubMed] [Google Scholar]
- 40. Denic V., Quan E. M., and Weissman J. S. (2006) A luminal surveillance complex that selects misfolded glycoproteins for ER-associated degradation. Cell 126, 349–359 [DOI] [PubMed] [Google Scholar]
- 41. Gauss R., Sommer T., and Jarosch E. (2006) The Hrd1p ligase complex forms a linchpin between ER-lumenal substrate selection and Cdc48p recruitment. EMBO J. 25, 1827–1835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gardner R. G., Swarbrick G. M., Bays N. W., Cronin S. R., Wilhovsky S., Seelig L., Kim C., and Hampton R. Y. (2000) Endoplasmic reticulum degradation requires lumen to cytosol signaling: transmembrane control of Hrd1p by Hrd3p. J. Cell Biol. 151, 69–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hagiwara M., Ling J., Koenig P. A., and Ploegh H. L. (2016) Posttranscriptional regulation of glycoprotein quality control in the endoplasmic reticulum is controlled by the E2 Ub-conjugating enzyme UBC6e. Mol. Cell 63, 753–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ballar P., Zhong Y., Nagahama M., Tagaya M., Shen Y., and Fang S. (2007) Identification of SVIP as an endogenous inhibitor of endoplasmic reticulum-associated degradation. J. Biol. Chem. 282, 33908–33914 [DOI] [PubMed] [Google Scholar]
- 45. Sun S., Shi G., Han X., Francisco A. B., Ji Y., Mendonça N., Liu X., Locasale J. W., Simpson K. W., Duhamel G. E., Kersten S., Yates J. R. 3rd, Long Q., and Qi L. (2014) Sel1L is indispensable for mammalian endoplasmic reticulum-associated degradation, endoplasmic reticulum homeostasis, and survival. Proc. Natl. Acad. Sci. U.S.A. 111, E582–E591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mueller B., Lilley B. N., and Ploegh H. L. (2006) SEL1L, the homologue of yeast Hrd3p, is involved in protein dislocation from the mammalian ER. J. Cell Biol. 175, 261–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bernasconi R., Galli C., Calanca V., Nakajima T., and Molinari M. (2010) Stringent requirement for HRD1, SEL1L, and OS-9/XTP3-B for disposal of ERAD-LS substrates. J. Cell Biol. 188, 223–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sanjana N. E., Cong L., Zhou Y., Cunniff M. M., Feng G., and Zhang F. (2012) A transcription activator-like effector toolbox for genome engineering. Nat. Protoc. 7, 171–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nam H. S., and Benezra R. (2009) High levels of Id1 expression define B1 type adult neural stem cells. Cell Stem Cell 5, 515–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kingston R. E., Chen C. A., and Okayama H. (2001) Calcium phosphate transfection. In Current Protocols in Immunology, Chapter 10, Unit 10.13, John Wiley & Sons, Inc., New York: [DOI] [PubMed] [Google Scholar]
- 51. Rappsilber J., Mann M., and Ishihama Y. (2007) Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2, 1896–1906 [DOI] [PubMed] [Google Scholar]
- 52. Elias J. E., and Gygi S. P. (2007) Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214 [DOI] [PubMed] [Google Scholar]
- 53. Huttlin E. L., Jedrychowski M. P., Elias J. E., Goswami T., Rad R., Beausoleil S. A., Villén J., Haas W., Sowa M. E., and Gygi S. P. (2010) A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143, 1174–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kim W., Bennett E. J., Huttlin E. L., Guo A., Li J., Possemato A., Sowa M. E., Rad R., Rush J., Comb M. J., Harper J. W., and Gygi S. P. (2011) Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol. Cell 44, 325–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wang W., Zhou H., Lin H., Roy S., Shaler T. A., Hill L. R., Norton S., Kumar P., Anderle M., and Becker C. H. (2003) Quantification of proteins and metabolites by mass spectrometry without isotopic labeling or spiked standards. Anal. Chem. 75, 4818–4826 [DOI] [PubMed] [Google Scholar]
- 56. Hipp M.S., Patel C. N., Bersuker K., Riley B. E., Kaiser S.E., Shaler T. A., Brandeis M., and Kopito R. R. (2012) Indirect inhibition of 26S proteasome activity in a cellular model of Huntington's disease. J. Cell Biol. 196, 573–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.