

Abstract
2-Alkylquinolone (2AQ) alkaloids are pharmaceutically and biologically important natural products produced by both bacteria and plants, with a wide range of biological effects, including antibacterial, cytotoxic, anticholinesterase, and quorum-sensing signaling activities. These diverse activities and 2AQ occurrence in vastly different phyla have raised much interest in the biosynthesis pathways leading to their production. Previous studies in plants have suggested that type III polyketide synthases (PKSs) might be involved in 2AQ biosynthesis, but this hypothesis is untested. To this end, we cloned two novel type III PKSs, alkyldiketide-CoA synthase (ADS) and alkylquinolone synthase (AQS), from the 2AQ-producing medicinal plant, Evodia rutaecarpa (Rutaceae). Functional analyses revealed that collaboration of ADS and AQS produces 2AQ via condensations of N-methylanthraniloyl-CoA, a fatty acyl-CoA, with malonyl-CoA. We show that ADS efficiently catalyzes the decarboxylative condensation of malonyl-CoA with a fatty acyl-CoA to produce an alkyldiketide-CoA, whereas AQS specifically catalyzes the decarboxylative condensation of an alkyldiketide acid with N-methylanthraniloyl-CoA to generate the 2AQ scaffold via C–C/C–N bond formations. Remarkably, the ADS and AQS crystal structures at 1.80 and 2.20 Å resolutions, respectively, indicated that the unique active-site architecture with Trp-332 and Cys-191 and the novel CoA-binding tunnel with Tyr-215 principally control the substrate and product specificities of ADS and AQS, respectively. These results provide additional insights into the catalytic versatility of the type III PKSs and their functional and evolutionary implications for 2AQ biosynthesis in plants and bacteria.
Keywords: biosynthesis, crystal structure, enzyme, polyketide, secondary metabolism, Evodia rutaecarpa, alkaloid, alkyldiketide synthase, alkylquinolone synthase, type III polyketide synthase
Introduction
The 2-alkylquinolones (2AQs)4 comprise a subclass of alkaloids with a wide range of biological effects, such as antibacterial (1), cytotoxic (1), and anticholinesterase (2) activities, as well as quorum-sensing signaling (3). Gram-negative bacteria, Pseudomonas aeruginosa (3), and the medicinal Rutaceae family plants, Evodia rutaecarpa (1), Zanthoxylum schinifolium (4), and Esenbeckia leiocarpa (2), produce 2AQs. In the biosynthesis of 2-heptylquinolinone produced by P. aeruginosa, a previous study suggested that a β-ketoacyl carrier protein synthase III (FabH)-like enzyme, PqsD, catalyzes the condensation of anthraniloyl-CoA and malonyl-CoA (1) to produce 2-aminobenzoyldiketide-CoA (Fig. 1A). Subsequently, the thioesterase PqsE converts 2-aminobenzoyldiketide-CoA to the corresponding 2-aminobenzoyldiketide acid, which is then accepted by the FabH-like enzymes, the PqsB and PqsC heterodimeric enzymes, as the extender substrate with octanoyl-CoA (2) as the starter substrate to generate 2-heptyl-4-quinolone (5–7). In contrast, the heterologously expressed type III polyketide synthase 3 (HsPKS3) from the Lycopodiaceae family plant, Huperzia serrata, yields 2AQ by catalyzing four reaction steps, consisting of the condensation reaction of 1 and fatty acyl-CoA to produce alkyldiketide-CoA (AD-CoA), the hydrolysis reaction of AD-CoA to produce the alkyldiketide acid (AD acid), the coupling reaction of N-methylanthraniloyl-CoA (3) and AD acid to produce the linear N-methylanthraniloyldiketide intermediate, and the subsequent C–N bond formation to generate the 2AQ scaffold in the in vitro enzyme reaction (8). For example, HsPKS3 reportedly generated 2-heptyl-N-methyl-4-quinolone (4) from the condensation of 2, 3, and 1 via the formations of octanoyldiketide-CoA (5) and octanoyldiketide acid (6) (Fig. 1B). However, because there are no reports about the isolation of 2AQs from H. serrata, the 2AQ synthesis by HsPKS3 is predicted to be a non-physiological function of the enzyme limited to the in vitro enzyme reactions. In contrast, in the biosynthesis of curcumin in the turmeric Curcuma longa, feruloyldiketide-CoA is produced by diketide-CoA synthase (ClDCS), a type III polyketide synthase (PKS), from the condensation of feruloyl-CoA with 1 (Fig. 1C). Subsequently, the feruloyldiketide acid, the hydrolyzed product of feruloyldiketide-CoA, is accepted by the other type III PKS, named curcumin synthase (ClCURS1), and curcumin is generated from the decarboxylative condensation of the diketide acid with feruloyl-CoA (Fig. 1D) (9). Therefore, it is most likely that two type III PKSs are involved in the biosynthesis of the 2AQ alkaloids in E. rutaecarpa, in a manner similar to the curcumin biosynthesis in turmeric.
Figure 1.

Enzymatic formation of 2AQs and curcuminoids. A, 2-heptyl-4-quinolone formed by PqsB, -C, -D, and -E; B, 2-heptyl-N-methyl-4-quinolone (4) formed by HsPKS3; C, feruloyldiketide-CoA formed by ClDCS; D, curcumin formed by ClCURS1; E, decanoyldiketide-CoA (21) formed by ADS; F, 2-nonyl-N-methyl-4-quinolone (17) formed by AQS.
Herein, we report two unique type III PKSs from the buds of E. rutaecarpa, named alkyldiketide-CoA synthase (ADS) and alkylquinolone synthase (AQS). The functional analyses of ADS and AQS demonstrated that collaboration of ADS and AQS is required for the generation of the 2AQ scaffold in E. rutaecarpa. Furthermore, our functional analyses indicated that ADS produced AD-CoA from fatty acyl-CoA and 1 as the substrates, whereas AQS generated 2AQ from 3 and AD acid as the substrates. Remarkably, unlike the previously reported 2AQ-producing HsPKS3, both enzymes lacked the ability to catalyze the one-pot formation of 2AQ by a single enzyme. The X-ray crystal structures of ADS and AQS and the structure-based site-directed mutagenesis studies revealed that the substrate and product specificities of ADS are principally derived from a unique active-site geometry, whereas those of AQS are attributed to a novel CoA-binding tunnel architecture.
Results
Sequence analyses of ADS and AQS
Two full-length cDNAs encoding the novel type III PKSs, ADS and AQS, were cloned from the buds of E. rutaecarpa by the RT-PCR method. The full-length ADS and AQS cDNAs contained 1,155- and 1,200-bp open reading frames encoding Mr 42,705 and 44,065 proteins with 384 and 399 amino acids, respectively. ADS and AQS shared 76–46% amino acid sequence identities with other functionally characterized plant type III PKSs (Fig. 2). ADS shared 62% amino acid sequence identity with Medicago sativa chalcone synthase (MsCHS), which produces naringenin chalcone from the condensation of three molecules of 1 with p-coumaroyl-CoA (7) (10); 61% identity with AQS; 59% identity with Citrus microcarpa quinolone synthase (CmQNS), which produces 4-hydroxy-N-methyl-2-quinolone (8) from the decarboxylative condensation of 1 with N-methylanthraniloyl-CoA (3) (11); 59% identity with Rheum palmatum benzalacetone synthase (RpBAS), which produces benzalacetone from the decarboxylative condensation of 1 with 7 (12); 54% identity with ClDCS (9); 51% identity with Rhododendron dauricum orcinol synthase (RdORS), which generates orcinol from the condensation of three molecules of 1 with acetyl-CoA (9) (13); 50% identity with HsPKS3 (8); 49% identity with ClCURS1 (9); and 46% identity with Oriza sativa curcuminoid synthase (OsCUS), which catalyzes the one-pot formation of bisdemethoxycurcumin from the condensation of two molecules of 7 and one molecule of 1 (14). AQS shared 76% amino acid sequence identity with CmQNS, 61% identity with RpBAS, 60% identity with MsCHS, 56% identity with ClDCS, 53% identity with ClCURS1, 50% identity with OsCUS, 50% identity with HsPKS3, and 50% identity with RdORS. No additional cDNAs encoding other type III PKS isomers were obtained in this study.
Figure 2.

Comparison of the primary sequences of ADS, AQS, and other type III PKSs. The secondary structures of ADS are delineated as follows: α-helices (rectangles) and β-strands (arrows). The Cys, His, and Asn catalytic triad and the residues lining the active-site cavity of CHS are colored red and blue, respectively. Tyr-215 in AQS and the corresponding residues in other type III PKSs are highlighted in green. GenBankTM accession numbers are as follows: MsCHS, L02902; CmQNS, AB823730; ClDCS, AB535216; ClCURS1, AB495007; OsCUS, BAC79571; RpBAS, AF326911; RdORS, LC133082. The sequence of HsPKS3 has not yet been released from GenBankTM.
ADS and AQS conserved the catalytic triad of type III PKS (Cys-164, His-303, and Asn-336) and the active-site residues of CHS (Ser-133, Pro-375, and the “gatekeeper” Phe-215 (MsCHS numbering)) (Fig. 2). In contrast, Thr-132, Thr-194, Thr-197, Ser-338, and the gatekeeper Phe-265, lining the active-site cavity of CHS, were uniquely altered with Ser, Met, Cys, Trp, and Tyr, respectively, in ADS. These residues differ in numerous functionally distinct type III PKSs and are thought to control the substrate and product specificities of the enzymes (15, 16). The substitution of the conserved Thr-197 of CHS with Cys was also observed in RpBAS (12). Simultaneous small-to-large substitutions of Thr-194, Ser-338, and Phe-265 with Met, Trp, and Tyr were also found in RdORS (13). Interestingly, AQS completely retained the active-site residues of CmQNS that specifically generate 8 (11). Thus, Thr-132, Met-137, Thr-197, Gly-256, Phe-265, and Ser-338 in MsCHS were simultaneously substituted with Met, Ile, Tyr, Ala, Leu, and Gly in AQS. A phylogenetic tree analysis suggested that ADS and AQS are grouped in the non-chalcone-producing type III PKSs and are closest to CmQNS (Fig. 3).
Figure 3.

Phylogenetic tree analysis of plant type III PKSs. The bacterial β-ketoacyl carrier protein synthase IIIs (KAS III and FABH) from E. coli were employed as out groups. The scale represents 0.1 amino acid substitutions per site. ADS and AQS are highlighted with arrows. ACS, acridone synthase; ALS, aloesone synthase; BAS, benzalacetone synthase; CURS1, curcumin synthase 1; CUS, curcuminoid synthase; DCS, diketide-CoA synthase; OKS, octaketide synthase; OLS, olivetol synthase; ORS, orcinol synthase; PCS, pentaketide chromone synthase; 2PS, 2-pyrone synthase; STS, stilbene synthase; THNS, tetrahydroxynaphthalene synthase; VAS, phloroisovalerophenone synthase; VPS, valerophenone synthase. GenBankTM registration numbers are shown in parentheses.
In vitro functional analyses of ADS and AQS by co-incubation
Based on the hypothesis that the two type III PKSs from E. rutaecarpa generate 2AQ, ADS and AQS were individually expressed in Escherichia coli M15 as the N-terminally His6-tagged proteins, and both purified enzymes were subjected to enzyme reactions. The substrates and the products obtained in this study are summarized in Fig. 4 and supplemental Table S1. Because 2AQs were extracted with organic solvents from the enzyme reaction mixture, the initial functional analyses of ADS and AQS were performed for the ethyl acetate (EtOAc)-soluble portions of the enzyme reaction mixtures, using 3, 1, and C6–C16 fatty acyl-CoAs (hexanoyl-CoA (10), 2, decanoyl-CoA (11), lauroyl-CoA (12), myristoyl-CoA (13), and palmitoyl-CoA (14)) as the substrates (Fig. 5A). The enzyme reaction products were determined by comparing the retention times and the UV and MS spectra with those of authentic compounds. The co-incubation of ADS and AQS and the sole incubation of ADS produced 2-methyltriketide lactone (15) and 2-acetylmethyltetraketide lactone (16), derived from three and four molecules of 1, respectively (Fig. 5, A and B). However, the co-incubation of ADS and AQS with 3, 1, and the C10 fatty acyl-CoA 11 generated 2-nonyl-N-methyl-4-quinolone (17), derived from these three substrates as the major product, along with a 2-alkyltriketide lactone (2ATL), 2-nonyltriketide lactone (18), derived from 11 and two molecules of 1 as the minor product. The co-incubation of ADS and AQS with 3, 1, and the C8 fatty acyl-CoA 2 also yielded the corresponding 2AQ 4 as the major product, along with 2-heptyltriketide lactone (19) as the minor product. In contrast, the co-incubation with 3, 1, and the C12 fatty acyl-CoA 12 only generated 2-undecyl-N-methyl-4-quinolone (20). Among the produced 2AQs, 17 was predominantly yielded by 21.3% (Fig. 5A). However, no products were produced by the ADS and AQS co-incubation reactions using 3 and 1 with either 7 or the C6, C14 and C16 fatty acyl-CoAs 10, 13, and 14 as the substrates (Fig. 5A). Furthermore, the co-incubations of CmQNS with ADS or ADS, and the sole incubation of CmQNS in the presence of 3, 1, and either the C6–C16 fatty acyl-CoAs 10, 2, and 11–13 or 7 as the substrates only afforded 8 (Fig. 6). Interestingly, the sole incubations of ADS using 3, 1, and either the C6–C16 fatty acyl-CoAs or 7 as the substrates revealed that ADS lacks the 2AQ-forming activity; ADS only afforded the 2ATLs 19 and 18, derived from C8 and C10 fatty acyl-CoAs 2 and 11 and two molecules of 1, respectively (Fig. 5B). Furthermore, AQS no longer yielded any products from the sets of the three substrates (Fig. 5C). These observations clearly suggested that both ADS and AQS lack the one-pot 2AQ-forming activity, in contrast to HsPKS3, and the collaboration of ADS and AQS is required for the formation of 2AQ.
Figure 4.

Substrates used and products obtained in this study.
Figure 5.

Enzyme reaction products by co-incubation and sole incubations of ADS and AQS. Shown are the enzyme reaction products from N-methylanthraniloyl-CoA (3), malonyl-CoA (1), and fatty acyl-CoAs (hexanoyl (10)-, octanoyl (2)-, decanoyl (11)-, lauroyl (12)-, myristoyl (13)-, or palmitoyl (14)-CoAs) or p-coumaroyl-CoA (7) by the co-incubation of ADS and AQS (A), the sole incubation of ADS (B), and the sole incubation of AQS (C).
Figure 6.

HPLC elution profiles of EtOAc-soluble enzyme reaction products by the co-incubation of ADS, AQS, and CmQNS and by the sole incubation of CmQNS and AQS. Shown are enzyme reaction products from N-methylanthraniloyl-CoA (3), malonyl-CoA (1), and fatty acyl-CoAs (hexanoyl-CoA (10), octanoyl-CoA (2), decanoyl-CoA (11), lauroyl-CoA (12), or myristoyl-CoA (13)) or p-coumaroyl-CoA (7) by the co-incubation of ADS and CmQNS (A), the co-incubation of AQS and CmQNS (B), and the sole incubation of CmQNS (C). D, enzyme reaction products from 1 and 10, 2, 11-14, 3, or 7 by the sole incubation of AQS.
In vitro functional analysis of ADS
To further clarify the catalytic role of ADS in the formation of 2AQ, the substrate and product specificities of ADS were investigated by directly subjecting the enzyme reaction mixtures to HPLC, without extracting the products with EtOAc. The HPLC analysis revealed the presence of decanoyldiketide-CoA (21) and decanoyldiketide acid (22) in the enzyme reaction mixture, when ADS was incubated with 1 and the C10 fatty acyl-CoA 11 at pH 7.0 (Fig. 7A). The productions of 21 and 22 were not observed in a control experiment using boiled ADS (Fig. 7A). However, ADS predominantly afforded 21 at pH 6.5, whereas the production of 22 was drastically decreased at this pH. Furthermore, the production of 21 was dramatically decreased at pH 7.5, and instead, that of 22 was remarkably increased, as compared with those of the reaction at pH 7.0 (Fig. 7A). These observations suggested that either ADS lacks thioesterase activity toward 21 or this activity toward 21 was disrupted by the acidic conditions. To test the thioesterase activity of ADS, ADS was reacted with the C10 AD-CoA 21 as the substrate at pH 6.5, 7.0, and 7.5, as the possible assay system. However, the HPLC analyses did not reveal any differences in the production of 22 at these different pH values, as compared with those in the control experiment using the boiled ADS (Fig. 7B). These results strongly suggested that ADS catalyzes the decarboxylative condensation of 1 with the C10 fatty acyl-CoA 11 to produce the corresponding AD-CoA 21, whereas the corresponding AD acid 22 is a by-product generated by the non-enzymatic hydrolysis of 21, in a manner similar to the productions of feruloyldiketide-CoA and the corresponding diketide acid, as observed in the ClDCS enzyme reaction (9).
Figure 7.
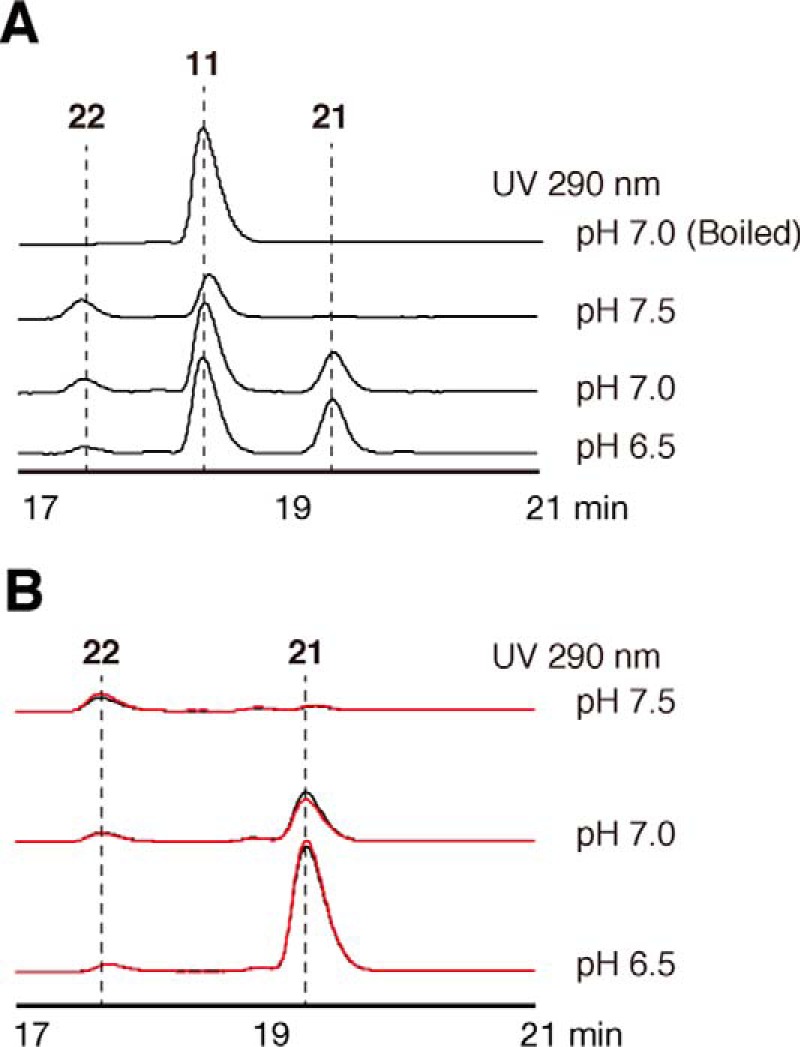
Analysis of all ADS enzyme reaction products. A, the ADS reaction products from decanoyl-CoA (11) and malonyl-CoA (1) at pH 6.5, 7.0, and 7.5. B, analysis of the thioesterase activity of ADS with decanoyldiketide-CoA (21) at pH 6.5, 7.0, and 7.5. The HPLC elution profiles obtained by ADS and boiled ADS are indicated as black and red lines, respectively.
To evaluate the substrate and product specificities of ADS, all of the ADS-producing AD-CoAs were converted to the corresponding AD acids by the alkaline hydrolysis reaction, and then the AD-CoA-forming activity of ADS was evaluated by TLC-based assays, utilizing 2-14C-labeled 1 as the extender substrate, because the UV absorption of the AD acids is quite low (Fig. 8). The TLC-based assays revealed that ADS accepts the C8 and C10 fatty acyl-CoAs 2 and 11 as starter substrates, to produce the AD-CoAs 5 and 21 (6 and 22 in the TLC-based assay) as the major products, along with the 2ATLs 19 and 18, respectively (Figs. 8 (B and C) and 9 (A and B)). Furthermore, ADS produced a trace amount of lauroyldiketide-CoA (23) (lauroyldiketide acid (24) in the TLC-based assay) as the single product, from the condensation of 1 with the C12 fatty acyl-CoA 12 (Figs. 8D and 9C). In contrast, ADS lacked the abilities to accept the C6, C14, and C16 fatty acyl-CoAs 10, 13, and 14 as the starter substrates as well as 3 and 7 as the starter substrates to produce starter substrate-derived products (Fig. 8, A and E–H). The AD-CoA- and 2ATL-forming activities observed were maximal at pH 7.5 and pH 6.0, respectively, within the range of pH 5.5–8.0 (Fig. 9).
Figure 8.

Enzyme reaction products from ADS and the ADS mutant enzymes with structurally distinct starter substrates. A–H, the TLC-based analysis of radiolabeled products from malonyl-CoA (1) and hexanoyl-CoA (10) (A), octanoyl-CoA (2) (B), decanoyl-CoA (11) (C), lauroyl-CoA (12) (D), myristyl-CoA (13) (E), palmitoyl-CoA (14) (F), N-methylanthraniloyl-CoA (3) (G), and p-coumaroyl-CoA (7) (H) by the ADS and ADS mutant enzymes. I, HPLC elution profiles of enzyme reaction products obtained by ADS and the boiled ADS reactions with acetyl-CoA (9) and 1 as the substrates.
Figure 9.

pH-dependent AD acid- and 2ATL-forming activities of ADS. Shown are the relative yields of octanoyldiketide acid (6) and 2-heptyltrikeide lactone (19) from malonyl-CoA (1) and octanoyl-CoA (2) (A), decanoyldiketide acid (22) and 2-nonyltriketide lactone (18) from 1 and decanoyl-CoA (11) (B), and lauroyldiketide acid (24) from 1 and lauroyl-CoA (12) (C). The yields of AD acids at pH 6.0–8.0, 2ATL at pH 5.5 and pH 6.0, and 2ATL at pH 6.0–8.0 are indicated as closed squares and closed and open circles, respectively.
In the production of AD-CoAs and 2ATLs, 21 (7.0% yield, according to the yield of 22 at pH 7.5) and 19 (4.6% yield at pH 6.0) were the best products, respectively (Fig. 9). The higher yield of the AD-CoA over 2ATL and the lack of reports for the isolation of the 2ATL derivatives from E. rutaecarpa might indicate that the 2ATLs are by-products limited to the in vitro enzyme reaction. Interestingly, despite the fact that ADS conserves the CHS Thr-194, Ser-338, and Phe-265 with Met, Trp, and Tyr, as in the case of RdORS, our functional analysis of ADS using 9 and 1 as the substrates did not afford the RdORS-producing orcinol; ADS only produced 15 and 16 (Fig. 8I). Thus, ADS is a novel type III PKS that generates AD-CoA in a manner similar to that of ClDCS (Fig. 1, C and E).
ClDCS was reported as an allosteric enzyme, due to the sigmoidal curve in the rate-feruloyl-CoA concentration profile (9). In contrast, the rate-fatty acyl-CoA concentration profiles with ADS generated a hyperbolic curve, which is a feature of the Michaelis-Menten model. ADS exhibited a Km value of 12.5 μm and a kcat/Km value of 3.84 min−1 mm−1 for the C10 fatty acyl-CoA 11, with respect to the 21-forming activity at the pH optimum of 7.5 (Table 1), which are 5.9 and 1.2 times higher than those of 74.4 μm and 3.23 min−1 mm−1 for the C8 fatty acyl-CoA 2, respectively. Thus, ADS preferred 11 over 2 as the starter substrate and efficiently produced 21, as observed in the functional analysis of ADS described above. The steady-state kinetic analysis revealed that ADS exhibited a kcat/Km value of 1.65 min−1 mm−1 for 1, with respect to the 21-forming activity at the pH optimum of 7.5 (Table 1).
Table 1.
Steady-state kinetic parameters of ADS and AQS
| Starter substrate | Km | kcat | kcat/Km |
|---|---|---|---|
| μm | min−1 | min−1 mm−1 | |
| ADS | |||
| 2 | 74.4 | 0.24 | 3.23 |
| 11 | 12.5 | 0.05 | 3.84 |
| 12a | NDb | ND | ND |
| 1 | 35.8 | 0.06 | 1.65 |
| AQS | |||
| 3 | 22.9 | 1.20 | 51.6 |
| 25 | 9.0 | 0.01 | 1.10 |
| 6 | 11.8 | 0.33 | 28.0 |
| 22 | 5.0 | 0.30 | 60.0 |
| 24 | 9.0 | 0.49 | 54.4 |
| 26 | 9.0 | 0.01 | 1.44 |
| 27 | 7.1 | 0.01 | 1.43 |
a The kinetic parameters were not detected, due to the trace amount of the product.
b ND, not detected.
In vitro functional analysis of AQS
Next, we investigated the substrate and product specificities of AQS, by directly subjecting the enzyme reaction mixtures to HPLC. In contrast to the other type III PKSs, AQS did not accept 3, 7, and the C6–C16 fatty acyl-CoAs 10, 2, and 11-14 as the starter substrates with 1 to generate products (Fig. 6D). However, the formations of the C8–C12 AD-CoAs 5, 21, and 23 by ADS (Fig. 8) and the C7–C11 2AQs 4, 17, and 20 by ADS and AQS from 3, 1, and the C8–C12 fatty acyl-CoAs 2, 11, and 12 (Fig. 5A) encouraged us to investigate the AQS reaction products derived from 3 as a starter substrate and the C10 AD-CoA 21 as an extender substrate. As expected, the HPLC analysis indicated that AQS accepts 21 as the extender substrate with 3, to produce the corresponding 2AQ 17 (Fig. 10A) via C–C/C–N bond formations. However, 21 was retained in the AQS and boiled AQS reaction mixtures at comparable levels. In contrast, the hydrolyzed product of 21, 22, was obviously consumed in the AQS reaction (Fig. 10A). This result suggested that AQS utilizes 22 as the extender substrate but lacks the thioesterase activity for the formation of 22 from 21. Indeed, AQS accepted 22 as the extender substrate with 3 and provided the C10 2AQ 17 with 7.2 times higher efficiency than that from 21 at the optimum pH 7.5 (Fig. 10A). Quite interestingly, AQS was capable of accepting the C6–C16 AD acids (hexanoyldiketide acid (25), 6, 22, 24, and myristoyl (26)- and palmitoyl (27)-diketide acids) with 3 to produce the corresponding 2AQs, 2-pentyl-N-methyl-4-quinolone (28), 4, 17, 20, and 2-tridecyl (29)-, and 2-pentadecyl (30)-N-methyl-4-quinolones, respectively (Fig. 10B). However, AQS lacked the ability to accept 7 as the starter substrate and did not yield any products (Fig. 10B). These observations suggested that AQS is a novel type III PKS that specifically catalyzes the decarboxylative condensation of AD acid with 3 to generate 2AQ via the C–N bond formation, in a manner similar to that of ClCURS1 (Fig. 1, D and F).
Figure 10.

Enzyme reaction products from AQS and the AQS Y215V mutant enzyme with structurally distinct substrates. A, enzyme reaction products from N-methylanthraniloyl-CoA (3) as the starter substrate and decanoyldiketide-CoA (21) or decanoyldiketide acid (22) as the extender substrate by AQS and the boiled AQS. B and C, enzyme reaction products from 3 and AD acids (hexanoyl (25)-, octanoyl (6)-, decanoyl (22)-, lauroyl- (24), myristoyl (26)-, or palmitoyl (27)-diketide acid) or p-coumaroyl-CoA (7) by AQS and its boiled enzyme (B) and the AQS Y215V mutant enzyme and its boiled enzyme (C). As the negative control data, the HPLC elution profiles of the enzyme reaction products from 3 and 22 by the boiled enzyme are shown. D, enzyme reaction products from 3 and malonyl-CoA (1) by AQS, the AQS Y215V mutant enzyme, and CmQNS.
The steady-state kinetic parameters revealed that the C10 AD acid 22 is the best extender substrate (Table 1) of AQS and efficiently generates the C10 2AQ 17, in good agreement with the best production of 17 in the ADS and AQS co-incubation reactions (Fig. 5A). AQS accepted 3 as the starter substrate with a Km value of 22.9 μm, which is 1.6 times higher than that of CmQNS (Km = 37.8 μm (11)) (Table 1).
Overall structures of ADS and AQS
To clarify the structural details of the AD-CoA- and 2AQ-forming catalytic mechanisms of ADS and AQS, the crystal structure of ADS complexed with CoASH and the apo-crystal structure of AQS were solved at 1.80 and 2.20 Å resolutions, respectively (Fig. 11). The crystallographic data and refinement statistics are summarized in supplemental Table S2. The asymmetric units of ADS and AQS contained four and three monomers, respectively. Significant backbone changes were not observed between the monomers in the structures of ADS and AQS, respectively. The structures of ADS and AQS are nearly identical, with a root-mean-square-deviation (r.m.s.d.) of 1.0 Å for the Cα-atoms. The overall structures of ADS and AQS revealed the typical type III PKS-folds. The catalytic triad (Cys-157/His-297/Asn-330 in ADS and Cys-170/His-309/Asn-342 in AQS) and the 16-Å-long CoA-binding tunnel are also conserved in the structures, in locations and orientations very similar to those in other plant type III PKSs. The overall structures of ADS and AQS are highly homologous to those of the structurally characterized plant type III PKSs (r.m.s.d. values of 0.8–1.1 and 0.7–1.1 Å for ADS and AQS, respectively) and exhibited r.m.s.d. values of 1.0 and 1.0 Å with MsCHS (PDB code 1BI5) (10), 1.1 and 0.8 Å with RpBAS (PDB code 3A5Q) (17), 0.8 and 0.7 Å with CmQNS (PDB code 3WD8) (11), and 1.1 and 1.1 Å with ClCURS1 (PDB code 3OV2) (18), respectively.
Figure 11.

Overall structures of ADS and AQS. A and B, overall structures of ADS (A) and AQS (B). ADS and AQS form homodimeric structures. The monomers are indicated as blue and gray schematic models, respectively. The CoASH molecule is shown as a green stick model. C–F, close-up views of the active-site entrances of ADS (C), RpBAS (D), MsCHS (E), and the ADS W332G mutant enzyme (F). G, stereo view of the active-site cavity of the docking model of the ADS-bound decanoyl moiety to the catalytic center Cys. The decanoyl moiety is indicated by a pink stick model.
Active-site structure and structure-based mutagenesis studies of ADS
In the absence of the structure of the AD-CoA-producing type III PKS, such as ClDCS, we compared the active-site structures of ADS and the aromatic diketide-producing RpBAS. Most of the active-site residues of ADS and RpBAS, including the catalytic triad, were superimposable in nearly identical positions (Fig. 12, A and B). However, the side chains of Trp-332 and Met-257 in ADS protruded more toward the active-site cavity, as compared with those of Ser-331 and Leu-256 in RpBAS, respectively. Thus, the side chains of Trp-332 and Met-257 blocked the access of Cys-191, corresponding to Cys-190 at the bottom wall in the active-site cavity of RpBAS, to the active-site cavity of ADS. The substitutions of Ser-332 and Leu-257 with Trp and Met in ADS, respectively, thus narrowed and shallowed the active-site cavity of ADS, as compared with that of RpBAS.
Figure 12.

Comparison of the active-site architectures of ADS, AQS, their mutant enzymes, and other type III PKSs. Shown are surface representations of ADS (A), RpBAS (B), the ADS W332Q mutant enzyme (C), the ADS W332G mutant enzyme (D), AQS (E), ClCURS1 (F), CmQNS (G), and the AQS Y215V mutant enzyme (H). The enzyme residues and the CoASH molecules are shown as blue and green stick models. The entrances of the CoA-binding tunnels are indicated with arrows. The catalytic triads and the mutated residues are highlighted with red and cyan type, respectively.
Interestingly, in contrast to the side-chain orientation of the gatekeeper Phe-258 in RpBAS, the side chain of the corresponding residue, Tyr-259, of ADS protruded toward the opposite side of the active-site cavity. This conformational change is presumably caused by the simultaneous small to large L209F and F259Y substitutions in ADS. As a result, the side chain of Phe-209, neighboring Tyr-259 in ADS, protruded more toward the active-site cavity, as compared with that of Leu-208 of RpBAS, and restricted part of the active-site cavity of ADS. Thus, the active-site cavity of ADS, including its entrance, is much narrower than that of RpBAS, suggesting that the selective starter substrate specificity for the fatty acyl-CoA of ADS arises from this characteristic active-site architecture. The total active-site cavity volume (200 Å3) and the active-site entrance area (21 Å2) of ADS were apparently smaller than those of RpBAS (350 and 34 Å2) (Figs. 11 (C and D) and 12 (A and B)). A docking study predicted that the active-site cavity of ADS is large enough to accommodate an enzyme-bound monoketide intermediate with up to a C10 length (Fig. 11G). Considering the acceptance of the C8–C12 fatty acyl-CoAs by ADS, the side chain(s) of one or more residues lining the active-site cavity might move flexibly and open the gate to a hidden space, to accept the C12 fatty acyl moiety of the substrate.
On the basis of these observations, we constructed a set of ADS site-directed mutants, in which Trp-332 was substituted with Gln, Ile, Val, Ala, and Gly. We also constructed the ADS C191G/A mutant enzymes, because the corresponding residue at this position often controls the substrate specificity and the polyketide-chain elongation reaction in numerous type III PKSs. Among them, the ADS W332S mutant enzyme formed inclusion bodies in E. coli, and thus the mutant enzyme was not analyzed in our study. The TLC-based assay revealed that the ADS W332Q mutant enzyme enhanced the abilities to form both the C8 AD-CoA 5 (6 in the TLC-based assay) and the C8 2ATL 19, as compared with those of the wild type (Fig. 8B). Furthermore, the mutant enzyme increased the C10 2ATL 18- and C12 AD-CoA 23-forming activities (24 in the TLC-based assay) (Fig. 8, C and D).
Similar results were also obtained in the ADS W332I, W332V, W332A, and W332G mutant enzyme reactions, using the C8–C14 fatty acyl-CoAs 2, 11, 12, and 13, respectively, as the starter substrates. The ADS W332I, W332V, W332A, and W332G mutant enzymes further enhanced the 19-forming activity, as compared with that of the ADS W332Q mutant enzyme, with the loss of the 5-forming activity (6 in the TLC-based assay) (Fig. 8B). The 18-forming activity was also enhanced in the ADS W332I, W332V, W332A, and W332G mutant enzymes, in this order (Fig. 8C). In contrast, the 21-forming activity (the C10 AD acid 22 in the TLC-based assay) tended to decrease, depending on the substitutions toward the less bulky residues (Fig. 8C). The 23-forming activity (the C12 AD acid 24 in the TLC-based assay) was also decreased with the ADS W332I mutant enzyme, but this mutation conferred the ability to form 2-undecyltriketide lactone (31) (Fig. 8D). In contrast, the ADS W332V, W332A, and W332G mutant enzymes lost the 23-forming activity and afforded 31 as the single product (Fig. 8D). Amazingly, the ADS W332G mutant enzymes newly accepted the C14 fatty acyl-CoA 13 as the starter substrate, and generated myristoyldiketide-CoA (32) (26 in the TLC-based assay) and 2-tridecyltriketide lactone (33) (Fig. 8E). The ADS W332A mutant enzyme also exhibited the 33-forming activity, although the activity was significantly decreased, as compared with that of the ADS W332G mutant enzyme. These observations suggested that the mutant enzymes increased the active-site cavity volume and thereby enhanced the 2ATL-forming activity. The crystal structures of the ADS W332Q and W332G mutant enzymes indeed indicated the expansion of the total active-site cavity volume, due to the loss of the planar and bulkier indole ring at this position (Fig. 12, A, C, and D). The estimated total cavity volumes of the ADS W332Q and W332G mutant enzymes were 240 and 295 Å3, which are 1.2 and 1.5 times larger, respectively, than that of the wild type (200 Å3).
Unexpectedly, the ADS W332I, W332V, W332A, and W332G mutant enzymes newly accepted 3 as the starter substrate with 1 to produce 8 (Fig. 8G). In contrast, none of the mutant enzymes accepted 7 as the starter substrate to generate products, as in the case of the wild type (Fig. 8H). We previously reported that RpBAS accepts not only 7, but also 3, as the starter substrate to produce 8, because its active-site entrance is wider than that of MsCHS (17, 19) (Fig. 11, D and E). Wild-type ADS had an active-site entrance wider than that of MsCHS but did not accept 3 as the starter substrate with 1 to yield products (Figs. 8G and 11 (C and E)). In contrast, our crystal structure analysis of the ADS W332G mutant enzyme revealed that the mutant enzyme retains the shallow active-site cavity but slightly widens the active-site entrance, as compared with that of the wild type (Figs. 11 (C and F) and 12 (A and D)). These observations suggested that the ADS W332I, W332V, W332A, and W332G mutant enzymes allowed the acceptance of the bulky N-methylanthraniloyl-CoA starter to the catalytic center, to yield 8, because of the expansion of the active-site entrance, whereas none of mutant enzymes allowed the access of 7 to the catalytic center because of the shallow active-site cavity.
Next, we investigated the effect of the C191A and C191G substitutions of ADS on the enzyme activities (Fig. 8). Both mutant enzymes did not accept 3, 7, and the C6 and C16 fatty acyl-CoAs 10 and 14 as the starter substrate (Fig. 8, A and F–H). However, the ADS C191A mutant enzyme enhanced the abilities for the formations of the C8 and C10 AD-CoAs 5 and 21 (6 and 22 in the TLC-based assay) and the corresponding 2ATLs 19 and 18, respectively, as compared with those of the wild type (Fig. 8, B and C). In addition, the ADS C191A mutant enzyme also enhanced the C12 AD-CoA 23-forming activity (24 in the TLC-based assay). Interestingly, the C191G substitution in ADS further increased the 19- and 18-forming activities, as compared with those of the ADS C191A mutant enzyme, and generated 19 and 18 as the major products. Furthermore, the ADS C191G mutant enzyme conferred the ability to form the C12 and C14 ATLs 31 and 33 (Fig. 8, D and E). These results suggested that Cys-191 controls the dynamic motion of the side chain of Trp-332, which reflects the AD-CoA-forming activity of ADS. Thus, Trp-332 and Cys-191 are the crucial residues for the AD-CoA-forming activity of ADS. Presumably, the side chain of Trp-332 not only narrows the active-site cavity of ADS, but also forms putative hydrophobic interactions between the fatty acyl moiety of the substrate and the active-site cavity. This would be the reason why ADS exhibited the selective starter substrate specificity for fatty acyl-CoA.
Active-site structure and structure-based mutagenesis studies of AQS
A comparison of the active-site structures of AQS and the functionally most similar ClCURS1 revealed that the active-site cavity of AQS is much wider and shallower than that of ClCURS1 (Fig. 12, E and F). This is mainly caused by the differences in the bulkiness of the active-site residues, such as Asp-198, Met-200, Tyr-203, and Gly-344 in AQS and Glu-192, Thr-194, Ser-197, and Gln-338 in ClCURS1. The total active-site cavity volume of AQS is 330 Å3, which is larger than that of ClCURS1 (250 Å3). Thus, the shape and size of the active-site cavity of AQS restricted the binding of the coumaroyl starter. Interestingly, the large-to-small Q344G substitution in AQS also resulted in the expansion of its active-site entrance, as compared with that of ClCURS1. Furthermore, previous studies indicated that ClCURS1 prefers a diketide acid lacking a CoA moiety as the extender substrate, due to the presence of the characteristic hydrophobic pocket formed by Gly-211, Phe-215, Phe-265, and Phe-267 in the CoA-binding tunnel, located between the protein surface and the catalytic triad (18). However, the aromatic ring of Tyr-215 in AQS, corresponding to Ala-210 in ClCURS1, occupied the space corresponding to the hydrophobic pocket in ClCURS1 and resulted in the loss of the hydrophobic cavity from the CoA-binding tunnel of AQS (Fig. 12, E and F). None of the alternative hydrophobic cavities was observed in the CoA-binding pocket of AQS.
In contrast, the active-site resides of AQS were sterically well conserved among those of CmQNS (Fig. 12, E and G). The only difference between AQS and CmQNS was the protrusion of Met-138 in AQS, corresponding to Met-132 in CmQNS, toward Ile-143 with a slight displacement of the Cα atom. Thus, the active-site cavities of AQS (330 Å3) and CmQNS (290 Å3) have almost the same size and shape. In contrast, the CoA-binding tunnel of AQS (470 Å3) was much narrower than that of CmQNS (650 Å3) (Fig. 12, E and G). Amazingly, this significant difference in AQS was caused by the smaller-to-larger substitution of Val in CmQNS with Tyr-215 in AQS. The unique extender substrate specificity of AQS is thus most likely regulated by the CoA-binding tunnel architecture. Remarkably, HPLC analyses of the reaction products generated by the AQS Y215V mutant enzyme, using the same substrates as those of the wild type, only detected the production of a trace amount of the C10 2AQ 17 (Fig. 10C). In contrast, the mutant enzyme efficiently generated 8 as the single product, when 3 and 1 were used as the substrates (Fig. 10D). Furthermore, the crystal structure of the AQS Y215V mutant enzyme clearly indicated that the Y215V substitution widens the CoA-binding tunnel at the mutated position, without any conformational changes of the other residues (Fig. 12, G and H). Thus, the mutant enzyme possessed an apparently expanded CoA-binding tunnel (575 Å3), similar to that of CmQNS (650 Å3).
These observations strongly suggest that the wide active-site entrance and the narrow CoA-binding tunnel in AQS result in the preferences of 3 and AD acid as the starter and extender substrates, respectively, and thereby specifically generate 2AQ.
Tissue distributions of ADS and AQS gene expression and 2AQ production
The tissue-specific expressions of ADS and AQS in the leaves, buds, and fruits of E. rutaecarpa were analyzed by semiquantitative RT-PCR, using gene-specific primers. The AQS gene was expressed at the highest level in leaves, and lesser amounts were detected in buds and fruits (Fig. 13A). In contrast, the ADS gene expression was only found in buds, suggesting that ADS could collaborate with AQS in the tissue to produce the C7–C11 2AQs 4, 17, and 20, as observed in the in vitro co-incubation assay of the enzymes (Fig. 5A). However, analyses of the contents of the C7–C13 2AQs 4, 17, 20, and 29 in the buds revealed that E. rutaecarpa derives 29, as well as 4, 17, and 20, and mostly accumulates 20 (Fig. 13B). This is in contrast to the results obtained by the in vitro co-incubation assay of ADS and AQS, in which 17 was primarily produced by the enzymes. Furthermore, these 2AQs accumulated in the fruits and leaves at higher and lower levels than those in buds, respectively, despite the lack of ADS gene expression in those tissues. In addition, E. rutaecarpa reportedly derives the C2, C5, and C7–C15 2AQs (1). However, ADS and AQS were only capable of producing the C8–C12 AD-CoAs and the C5–C15 2AQs, respectively. These observations suggest the presence of ADS and AQS isozymes with substrate specificities for other alkyl-length fatty acyl-CoAs in E. rutaecarpa.
Figure 13.
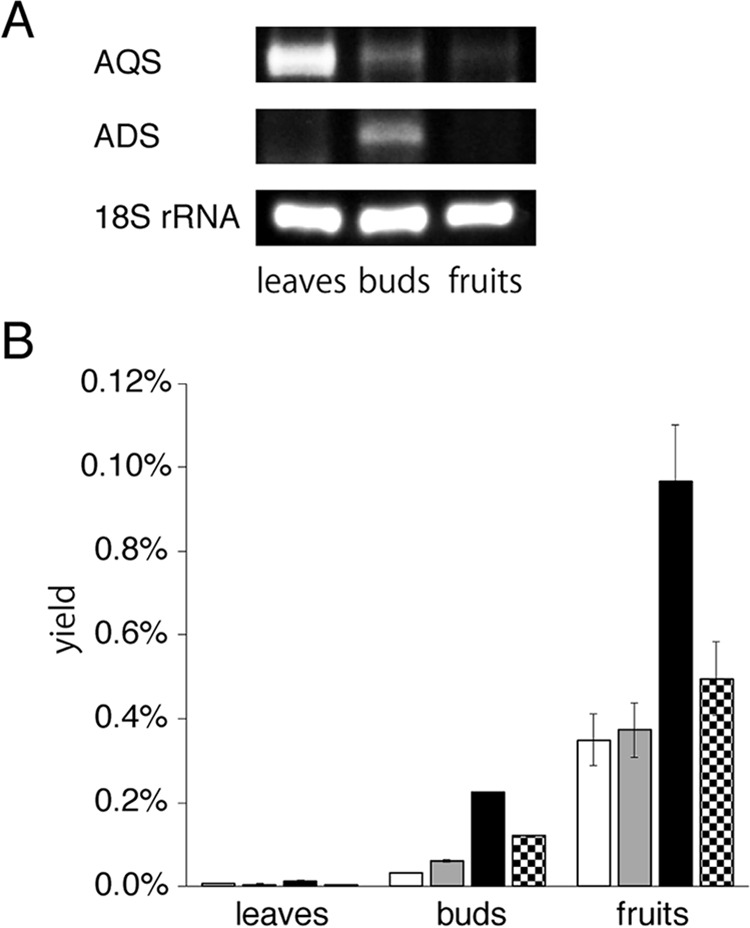
Analyses of ADS and AQS transcripts and tissue distributions of 2AQs in E. rutaecarpa. A, semiquantitative RT-PCR analyses of ADS and AQS gene expression. The 18S rRNA gene fragment was amplified as a housekeeping gene. B, 2AQs contents in leaves, buds, and fruits, analyzed by LC-ESI-MS. The data are presented as the mean ± S.D. (n = 3). The yields of the C7, C9, C11, and C13 2AQs 4, 17, 20, and 29 are represented as white, gray, black, and dotted bars, respectively. The amounts of 2AQs were estimated from the peak intensities, by comparison with those of the corresponding authentic 2AQs, and then converted to the yield percentage against the wet weights of the tissues, respectively.
Discussion
In this study, we cloned ADS and AQS and performed functional and crystal structure analyses as well as structure-based site-directed mutagenesis studies. ADS and AQS possess the unique active-site cavity and the novel CoA-binding tunnel, respectively, which have never been observed in other type III PKSs. Trp-332 and Cys-191 in ADS and Tyr-215 in AQS were indicated to control the substrate and product specificities of ADS and AQS. Notably, the active-site cavity of ADS is just large enough to accommodate up to a C10-length enzyme-bound monoketide intermediate (Fig. 11G). On the basis of these observations, we propose that ADS catalyzes the fatty acyl moiety transfer of fatty acyl-CoA to the catalytic center Cys and the condensation of one molecule of 1 with the enzyme-bound monoketide intermediate. However, due to the very tight steric restriction of the active-site cavity, especially from Trp-332, the enzyme stops the polyketide elongation reaction at the diketide stage and releases AD-CoA as the product, without transferring the diketide intermediate to the catalytic center Cys. Interestingly, the sequence comparison of ADS and ClDCS revealed that ClDCS lacks the conserved Ser-338 of the CHS, corresponding to Trp-332 in ADS, and instead has the bulky residue Gln-337 (Fig. 2), which is likely to restrict part of the active-site cavity of ClDCS, as in the case of the ADS W332Q mutant enzyme. However, Met-257, Met-188, Cys-191, and Tyr-259, lining the bottom wall of the active-site cavity of ADS, are replaced with the less bulky residues Leu-262, Thr-193, Ser-196, and Phe-265, respectively, in ClDCS (Fig. 2). Thus, the active-site cavity of ClDCS is likely to be larger and deeper than that of ADS, but due to the tight restriction of the active-site cavity by Gln-337, ClDCS seems to preferentially produce an aromatic diketide-CoA, as in the case of ADS.
Furthermore, ADS lacked the RdORS-producing orcinol-forming activity, despite the conservation of the Thr-194, Ser-338, and Phe-265 of CHS with Met, Trp, and Tyr. A sequence comparison of ADS and RdORS revealed that Met-257, at the bottom of the active-site cavity of ADS, is replaced in RdORS with the less bulky Leu-278, whereas Asn-206, neighboring the side chain of Tyr-259 in ADS, is substituted with the larger His-215 in RdORS (Fig. 2). The large-to-small M278L and small-to-large N215H substitutions in RdORS are likely to result in the protrusion of the side chain of Tyr-280, corresponding to the gatekeeper Tyr-259 in ADS, toward the active-site cavity in RdORS. Thus, the active-site cavity of RdORS might be smaller and shallower than that of ADS, which would lead to its substrate and product specificities distinct from those of RdORS.
Conversely, despite the lack of the hydrophobic cavity in the CoA-binding pocket, AQS preferred an AD acid as the extender substrate. However, as proposed for the catalytic machinery with ClCURS1 (18), the hydrophobic interaction between AQS and an AD acid would be important for the preferential acceptance of an AD acid lacking the CoA moiety. Thus, the aromatic ring of Tyr-215 could increase the hydrophobicity of the CoA binding tunnel of AQS, which controls its extender substrate specificity, in a manner similar to that of ClCURS1. In addition, Met-138, Met-200, and Tyr-203 in AQS are altered with less bulky residues, Ser, Thr, and Thr, respectively, in the 2AQ-producing HsPKS3, whereas Ile-143, Asp-198, and Gly-344 in AQS are replaced with bulkier residues Met, Glu, and Ser, respectively, in HsPKS3 (Fig. 2). Leu-271 in AQS, corresponding to the gatekeeper in other type III PKSs, is also substituted with Phe in HsPKS3. These differences suggest that HsPKS3 possesses a larger, downward expanded active-site cavity than that of AQS, and the catalytic machinery of HsPKS3 is completely different from that of AQS. OsCUS, the type III PKS from O. sativa, also catalyzes the one-pot formation of bisdemethoxycurcumin from the condensation of 7 and 1 (14) (Fig. 1C). We previously reported that OsCUS could lock both intermediates, the p-coumaroyldiketide acid generated via the decarboxylative condensation of 1 with 7 and the p-coumaroyl moiety derived from the second starter substrate 7, in the active-site cavity (20). Thus, HsPKS3 is most likely to utilize a catalytic mechanism similar to that of OsCUS.
This comprehensive assessment of the structures and functions of ADS and AQS thus demonstrates that the 2AQs derived from E. rutaecarpa are generated by two type III PKSs. However, comparisons of the kcat/Km values of ADS with those of the functionally most closely related ClDCS and other fatty-acyl-CoA-accepting type III PKSs, such as C. sativa olivetol synthase (CsOLS) and RdORS, suggested the low catalytic efficacy of ADS. Thus, the catalytic efficacy of ADS (kcat/Km = 1.65 min−1 mm−1 for 1 with respect to the C10 AD-CoA-forming activity) is 48 times lower than that of ClDCS (kcat/Km = 79.8 min−1 mm−1 for 1 with respect to the feruloyldiketide-CoA-forming activities) (9). The catalytic efficacy of ADS (kcat/Km = 3.84 min−1 mm−1 for 11 with respect to the C10 AD-CoA-forming activity) is 16 and 38 times lower than those of CsOLS (kcat/Km = 60.8 min−1 mm−1 for 10 with respect to the olivetol-forming activity) and RdORS (kcat/Km = 146.3 min−1 mm−1 for 9 with respect to the orcinol-forming activity), respectively (13, 21). In contrast, AQS exhibits catalytic efficacy (kcat/Km = 51.6 min−1 mm−1 for 3 with respect to the 17-forming activity) comparable with that of the functionally most closely related ClCURS1 (kcat/Km = 61.1 min−1 mm−1 for feruloyl-CoA with respect to the cinnamoylferuloylmethane-forming activity) (9). These observations also suggest the presence of ADS isozymes with different substrate specificities for the other alkyl-length fatty acyl-CoAs, as described above, but also the same substrate specificity as that of ADS and with higher catalytic efficacy in E. rutaecarpa.
Furthermore, our functional analyses suggest that the biosynthetic pathway of the plant 2AQ is quite different from that of the bacterial 2AQs produced by P. aeruginosa, in which three FabH-like enzymes, PqsB, -C, and -D, sharing < 19% amino acid sequence similarities with ADS and AQS, and the thioesterase PqsE complete the 2AQ biosynthesis in the order of (i) the condensation of anthraniloyl-CoA and 1 to produce 2-aminobenzoyldiketide-CoA, (ii) the hydrolysis of 2-aminobenzoyldiketide-CoA to produce 2-aminobenzoyldiketide acid, and (iii) the tail-to-tail condensation of 2-aminobenzoyldiketide acid with fatty acyl-CoA to generate 2-heptyl-4-quinolone (6, 7) (Fig. 1A). In contrast, our functional analyses of ADS and AQS also suggest that fatty acyl-CoA is initially utilized as the substrate with 1, and 3 is finally condensed with an AD acid to generate 2AQ (Fig. 1, E and F).
In addition, comparisons of the overall structures of ADS and AQS with those of the bacterial enzymes, PqsB (PDB code 5DWZ) (6), PqsC (PDB code 5DWZ) (6), and PqsD (PDB code 3H76) (22) revealed that all of the enzymes share the thiolase fold. Thus, the overall structures of PqsB, PqsC, and PqsD exhibited r.m.s.d. values of 3.2, 2.6, and 2.3 Å for ADS and 3.0, 2.7, and 2.4 Å for AQS, respectively. Furthermore, the Cys-His-Asn catalytic triad and the CoA-binding tunnel in PqsD are also well conserved in the structures of ADS and AQS, in locations and orientations similar to those in ADS and AQS. These observations suggest that ADS, AQS, and PqsD evolved from a common ancestor. However, Tyr-259 and Trp-332, within the active-site cavity of ADS, are replaced in PqsD with the less bulky Phe and Ala. Thus, the active-site cavity of PqsD is larger than that of ADS, suggesting that PqsD could catalyze the condensation of anthraniloyl-CoA with 1 to produce 2-aminobenzoyldiketide-CoA (22). In contrast, PqsC in the PqsBC heterodimer only conserves the Cys and His of the catalytic triad in ADS and AQS, whereas PqsB lacks the Cys, His, and Asn, corresponding to the catalytic triad residues. Because PqsB and PqsC share the thiolase fold, as in the case of PqsD, both enzymes are most likely to have evolved from the common ancestor of ADS, AQS, and PqsD. However, the PqsBC heterodimer possesses a unique large active-site cavity elongated from PqsC to PqsB (6). These features are in sharp contrast to the active-site architectures and catalytic machineries of ADS and AQS, suggesting that the PqsBC heterodimer specifically evolved in P. aeruginosa, and thereby could confer the ability to generate 2AQs by utilizing a different biosynthetic pathway from that of E. rutaecarpa. On the basis of these observations, we propose that the 2AQ biosynthesis evolved in a parallel but independent manner in E. rutaecarpa and P. aeruginosa.
In conclusion, our functional analyses of ADS and AQS demonstrated that two novel type III PKSs are involved in the 2AQ alkaloid biosynthesis in the medicinal plants. Furthermore, the crystal structure analyses and structure-based site-directed mutagenesis studies of ADS and AQS suggested that the unique active-site architecture principally controls the substrate and product specificities of ADS and AQS, respectively. These findings provide insights into not only the catalytic versatility of the type III PKSs and the structural bases for the catalytic machineries of ADS and AQS, but also the functional and evolutionary implications for 2AQ biosynthesis in plants and bacteria.
Experimental procedures
Materials
Malonyl-CoA (1) and acetyl-CoA (9) were purchased from Sigma-Aldrich, and N-methylanthraniloyl-CoA (3), p-coumaroyl-CoA (7), and fatty acyl-CoAs (2 and 10–14) were synthesized as described previously (23). The [2-14C]1 (52.4 mCi/mmol) was purchased from PerkinElmer Life Sciences. AD-CoA (21), AD acids (6, 22, and 24–27), and authentic samples of 2AQs (4, 17, 20, and 28–30) were synthesized according to the published reports (24–26). Authentic samples of 2ATLs (15, 18, 19, 31, and 33), 2-acetylmethyltetraketide lactone (16), and 4-hydroxy-N-methyl-2-quinolone (8) were obtained in our previous work (11, 27, 28). E. rutaecarpa (Rutaceae) buds, leaves, and fruits were harvested from the Medical Herb Garden (Faculty of Pharmaceutical Sciences, University of Toyama, Japan) and immediately frozen in liquid nitrogen.
Cloning of the ADS and AQS cDNAs
Total RNA was extracted from E. rutaecarpa buds with an RNeasy plant minikit (Qiagen) and was reverse-transcribed using Superscript II RT (Invitrogen) and the oligo(dT) primer (5′-GACTCGAGTCGACATCGATTTTTTTTTTTTTT-3′), according to the manufacturer's protocol. The single-stranded cDNA thus obtained was used as the template for the PCRs with inosine-containing degenerate oligonucleotide primers, designed on the basis of the conserved sequences of the known CHSs (112S, 174S, 368A, and 380A (the primer's number indicates the amino acid number within M. sativa CHS)), as described previously (11), and nested PCR was performed with the primer sets 112S and 380A and then with 174S and 368A, to amplify the respective 555-bp core fragments of E. rutaecarpa ADS and AQS, using the previously reported conditions (11). The 5′- and 3′-RACEs were performed with the SMARTer RACE cDNA amplification kit (Clontech), using the gene-specific primers (5′-CCATCCTCTGAATCAGGGATTGTTGTTTGATTTGC-3′ for 5′-RACE of ADS, 5′-AATGGCTTCAAAGGTGGAGAGTCGGCAGGCAGCAG-3′ for 3′-RACE of ADS, 5′-AAAACAGCTTGCCCATATAATAAATCAACGCGGTT-3′ for 5′-RACE of AQS, and 5′-TGCTGGAGCTCGAGTTCTTTTAGTGTTTTGTGATC-3′ for 3′-RACE of AQS). The full-length cDNA encoding ADS was obtained using the sense primer 5′-GCGGGATCCATGGCTTCAAAGGTGGAGAGTCGCA-3′ (the BamHI site is underlined) and the antisense primer 5′-CGCCTCGAGTCAATTGATAGCAACACTACGGAGGA-3′ (the XhoI site is underlined). The full-length cDNA encoding AQS was obtained using the sense primer 5′-GCGGGATCCATGGCTTCATCTTTTTCAATGGAAAA-3′(the BamHI site is underlined) and the antisense primer5′-CGCCTCGAGATGGTTGGAATCTAGAGGGACACTGA-3′ (the XhoI site is underlined). The amplified full-length ADS and AQS cDNA fragments were digested with BamHI/XhoI and individually cloned into the BamHI/SalI sites of pQE-80L (Novagen). Both recombinant enzymes thus contained an additional N-terminal hexahistidine tag, MRGSHHHHHHGS, before the first residue. The nucleotide sequences were determined by Eurofin Genomics.
Amino acid sequence alignment and phylogenetic tree
The amino acid sequences of 31 type III PKSs, three FabH-like enzymes, and two FabHs were aligned. A phylogenetic tree was developed with the Clustal Omega version 1.2.1 program (European Bioinformatics Institute) and was visualized by Mega version 7.0 (29).
Enzyme expression and purification
The plasmids containing the full-length cDNAs encoding ADS and AQS were individually transformed into E. coli M15. The cells harboring the plasmid were cultured to an A600 of 0.6 in LB medium containing 100 μg/ml ampicillin at 37 °C, and isopropyl 1-thio-β-d-galactopyranoside (1 mm for ADS, 0.1 mm for AQS) was then added to induce the protein expression. The culture was incubated further at 27 °C for 16 h. All of the following procedures were performed at 4 °C. The E. coli cells were harvested by centrifugation at 6,000 × g for 20 min and resuspended in buffer A (50 mm Tris-HCl buffer (pH 8.0), 200 mm NaCl, 5% (v/v) glycerol), containing 10 mm imidazole, 0.5 mg/ml lysozyme, and 0.1 units/ml benzonase nuclease (Merck). The cells were disrupted by sonication, and the lysate was centrifuged at 8,000 × g for 20 min. The supernatant was loaded onto a HisTrap HP column (GE Healthcare) equilibrated with buffer A. After the resin was washed with buffer A, the recombinant protein was subsequently eluted with buffer A containing 600 mm imidazole. The protein solution was diluted 5-fold with buffer B (50 mm Tris-HCl (pH 8.0) and 5% (v/v) glycerol) and applied to a Resource Q column (GE Healthcare). The proteins were separated with buffer B containing 50–350 mm NaCl, and the fractions containing ADS (or AQS) were collected and concentrated to 5 ml. The resultant solution was further purified to homogeneity by gel filtration chromatography on a HiLoad 16/60 Superdex 200 column (GE Healthcare), and the fractions containing ADS (or AQS) were concentrated to 10 mg/ml in buffer A with a Microcep 10k centrifugation device (PALL Life Sciences). The protein concentration was calculated by measuring the UV absorption at A280.
Co-incubation assays of ADS, AQS, and QNS
ADS and AQS, QNS and ADS, or QNS and AQS (20 μg each) were co-incubated with 55 μm 3, 60 μm fatty acyl-CoA (10, 2, 11-13, or 14), and 100 μm 1, in a final volume of 500 μl of 100 mm potassium phosphate buffer (pH 7.5), at 37 °C for 16 h. The co-incubation assay using 7 as the substrate, instead of fatty acyl-CoA, was performed under the same reaction conditions except for the substitution of fatty acyl-CoA with 7. The reactions were stopped by the addition of 20 μl of 20% HCl, and the enzyme reaction products were extracted twice with 500 μl of EtOAc. The extracts were then evaporated under reduced pressure, and the residues were dissolved in 80 μl of 100% methanol. An aliquot of the sample (20 μl) was injected into an online LC-ESI-MS system with an Agilent Technologies series 1100 HPLC connected to a Bruker Daltonics Esquire 4000 ion-trap mass spectrometer fitted with an ESI source. The enzyme reaction products were eluted with a gradient of water and CH3CN, both containing 0.1% trifluoroacetic acid: 0–5 min, 20% CH3CN; 5–45 min, 20–100% CH3CN, on a TSK-gel ODS-80Ts column (4.6-mm inner diameter × 150 mm; Tosoh) at a flow rate of 0.6 ml/min. The ESI capillary temperature and the capillary voltage were 350 °C and 4.0 V, respectively. The tube lens offset was set at 20.0 V. All spectra were obtained in the positive mode over a mass range m/z of 50–1,200 and at a range of one scan every 0.2 s. The collision gas was helium, and the relative collision energy scale was set at 30.0% (1.5 eV).
Analysis of EtOAc-soluble enzyme reaction products generated by the sole incubations of ADS, AQS, and QNS
All procedures were performed according to the same procedure used for the co-incubation assays of ADS, AQS, and QNS, except for using the sole enzymes.
Analysis of all reaction products produced by ADS and the ADS mutant enzyme
For the detection of the formations of 21 and 22, ADS (20 μg) was incubated with 300 μm 11 and 500 μm 1 in a final volume of 100 μl of 100 mm potassium phosphate buffer (pH 6.5, 7.0, or 7.5) at 37 °C for 14 h. For the assessment of the thioesterase activity of ADS, 20 μg of ADS was incubated with 60 μm 21, in a final volume of 100 μl of 100 mm potassium phosphate buffer (pH 7.5), at 37 °C for 16 h. The reactions were stopped by the addition of 4 μl of 20% HCl and a 20-μl aliquot of the enzyme reaction mixtures was directly injected into an Agilent Technologies series 1290 HPLC. The enzyme reaction products were eluted with a gradient of 25 mm potassium phosphate buffer, pH 6.5, and CH3CN: 0–5 min, 30% CH3CN; 5–21 min, 30–42% CH3CN; 21–26 min, 42–80% CH3CN: 26–40 min, 80% CH3CN, on a TSK-gel octyl-80Ts column (4.6-mm inner diameter × 150 mm; Tosoh) at a flow rate of 0.6 ml/min.
For the TLC-based assays, 4 μg of ADS was incubated with 60 μm starter substrate (10, 2, 11-14, 3, or 7) and 101.6 μm [2-14C]1 (1.1 mCi/mmol), in a final volume of 100 μl of 100 mm potassium phosphate buffer (pH 7.5), at 37 °C for 16 h. A 5-μl portion of 20% NaOH was then added, and the reaction was incubated further at 65 °C for 15 min, to hydrolyze the AD-CoA to the corresponding AD acid. The hydrolysis reaction was quenched by the addition of 10 μl of 20% HCl. The reaction products were extracted with 100 μl of EtOAc and separated with a hexane/EtOAc/acetic acid solvent system (9:1:1, v/v/v) on analytical TLC plates (Merck silica gel 60 F254). Radioactivities were quantified by autoradiography, using a Typhoon FLA-9500 biomolecular imager (GE Healthcare). The radioactive compounds on the TLC plates were analyzed with the ImageQuant TL software (GE Healthcare). The total enzyme reaction products produced by the mutant enzymes were also analyzed with the TLC assay, according to the same procedure used for the wild type ADS. The optimum pH values of ADS for the formation of 2ATL and AD-CoA were also analyzed with the TLC assays, using the same procedure as described above, except for the use of 100 mm MES-NaOH buffer (pH 5.5–6.0) and 100 mm potassium phosphate buffer (pH 6.0–8.0).
Analysis of ORS activity of ADS
ADS (20 μg) was co-incubated with 60 μm 9 and 100 μm 1, in a final volume of 500 μl of 100 mm potassium phosphate buffer (pH 7.5), at 37 °C for 16 h. The reaction was stopped, and the enzyme reaction products were extracted with EtOAc and analyzed by LC-MS, according to the same procedure used for the co-incubation assays of ADS, AQS, and QNS.
Analysis of all reaction products produced by AQS and the AQS mutant enzyme
For the determination of the type of extender substrate of AQS, 20 μg of AQS was incubated with 55 μm 3 and either 60 μm 21 or the corresponding AD acid 22, in a final volume of 100 μl of 100 mm potassium phosphate buffer (pH 7.5), at 37 °C for 16 h. The reactions were stopped, and the products were analyzed by HPLC, using the same methods as for the assessment of the thioesterase activity of ADS.
For the identification of the 2AQ-forming activity of AQS with different alkyl chain lengths of the extender substrates, AQS (20 μg) was incubated with 55 μm 3 and 60 μm AD acid (6, 22, 24–26, or 27), in a final volume of 100 μl of 100 mm potassium phosphate buffer (pH 7.5), at 37 °C for 16 h. The optimum pH value of AQS for the formation of 2AQ was evaluated using 100 mm potassium phosphate buffer (pH 6.0–8.0). The reaction products were analyzed according to the same procedure used for the determination of the type of extender substrate of AQS, except for the use of 25 mm potassium phosphate buffer (pH 6.5) and a CH3CN gradient system: 0–5 min, 15% CH3CN; 5–21 min, 15–42% CH3CN; 21–26 min, 42–80% CH3CN; 26–40 min, 80% CH3CN. The reactions of the AQS mutant enzymes and the analysis of the products generated by the mutant enzymes were performed according to the same procedure used for the wild type AQS.
Analysis of all reaction products by the sole incubations of AQS, the AQS mutant enzyme, or CmQNS
AQS, the AQS Y215 mutant enzyme, or CmQNS (20 μg) was incubated with 55 μm 3 and 100 μm 1, in a final volume of 100 μl of 100 mm potassium phosphate buffer (pH 7.5), at 37 °C for 16 h. The reactions were stopped, and the products were analyzed by HPLC, using the same methods as for the co-incubation assays of ADS, AQS, and CmQNS.
Kinetics of ADS and AQS
Steady-state kinetic parameters of ADS were determined using [2-14C]1 as the extender substrate. All experiments were performed in triplicate for each reaction condition.
For the kinetic values of ADS toward fatty acyl-CoAs (2, 11, and 12), the enzyme reaction mixture contained 4 μg of ADS, 101.6 μm [2-14C]1 (1.1 mCi/mmol), and five concentrations of fatty acyl-CoA (32, 16, 8, 4, 2 μm), in a final volume of 100 μl of 100 mm potassium phosphate buffer (pH 7.5). For the kinetic value of ADS toward 1, the reaction mixtures contained 4 μg of ADS, 60 μm 11, and five concentrations of [2-14C]1 (260, 130, 65, 32.5, and 16.25 μm containing 2.88, 1.44, 0.72, 0.36, and 0.18 mCi/mmol, respectively), in a final volume of 100 μl of 100 mm potassium phosphate buffer (pH 7.5). The mixtures were incubated at 37 °C for 20 min, and the enzyme reaction products were quantified using the same procedures as for the TLC-based assays of ADS reaction products.
For the steady-state kinetic parameters of AQS toward AD acids (6, 22, 24–27), the reaction mixture contained 4 μg of AQS, 55 μm 3, and five concentrations (32, 16, 8, 4, and 2 μm) of AD acid in a final volume of 100 μl of 100 mm potassium phosphate buffer (pH 7.5). For the steady-state kinetic parameters of AQS toward 3, the reaction mixture contained 4 μg of AQS, 180 μm 22, and five concentrations (64, 32, 16, 8, 4 μm) of 3 in a final volume of 100 μl of 100 mm potassium phosphate buffer (pH 7.5). These mixtures were incubated at 30 °C for 20 min and extracted with 100 μl of EtOAc, and the EtOAc-soluble enzyme reaction products were analyzed by using the same procedure as for the identification of the 2AQ-forming activity of AQS against the alkyl chain length of the extender substrate. Lineweaver-Burk plots of all data were generated to derive the apparent Km and kcat values, using the Excel program (Microsoft).
Site-directed mutagenesis of ADS and AQS
The plasmids expressing the ADS and AQS mutant enzymes were constructed with a QuikChange site-directed mutagenesis kit (Stratagene), according to the manufacturer's protocol, using the ADS and AQS expression plasmids as templates and primers listed in supplemental Table S3. The mutant enzymes were expressed and purified with the same procedures as described for the wild-type enzymes and used for the enzyme assays and crystallizations.
Crystallizations and structure determinations of ADS, AQS, and their mutant enzymes
All crystallization attempts were performed using the sitting-drop vapor diffusion technique at 20 °C. Diffraction quality crystals of ADS, its W332Q and W332G mutant enzymes, AQS, and its Y215V mutant enzyme were obtained in the optimized reservoir conditions listed in supplemental Table S2 after initial screenings using 96-condition crystallization screening kits (Qiagen; originally designed by Mitsubishi Chemical Corp.). All crystallization drops were prepared by mixing 0.5 μl of either the purified ADS (10 mg/ml, containing 5 mm CoASH), ADS W332Q mutant (5 mg/ml, containing 10 mm CoASH), ADS W332G mutant (5 mg/ml, containing 10 mm CoASH), AQS (22 mg/ml), or AQS Y215V mutant (10 mg/ml) protein solutions and an equal volume of reservoir solution and equilibrating the mixtures against 50 μl of reservoir solution. The crystals of the wild-type ADS were transferred into a crystallization solution containing 12% (v/v) ethylene glycol as a cryoprotectant. The crystals of the ADS W332Q and G mutant enzymes were transferred into a crystallization solution containing 10 mm CoASH and 12% (v/v) ethylene glycol as a cryoprotectant. The crystals of the wild-type AQS and AQS Y215V mutant enzymes were transferred into a crystallization solution containing 20% (v/v) glycerol as a cryoprotectant. After a few seconds, the crystals were picked up in a nylon loop and then flash-cooled to −173 °C in a nitrogen gas stream. X-ray diffraction data sets were collected on the beamlines NW-12A for the crystals of the ADS wild type complexed with CoASH and AQS wild-type apo, BL17A for the crystals of the ADS W332G mutant enzyme, and NE-3A for the crystals of the AQS Y215V mutant enzyme at the Photon Factory (Tsukuba, Japan), and BL41XU for the crystals of the ADS W332Q mutant enzyme complexed with CoASH at SPring-8 (Hyogo, Japan). Diffraction data of the crystals were processed and scaled with XDS (30). Molecular replacements were performed with the C. microcarpa acridone synthase (PDB code 3WD7) and CmQNS (PDB code 3WD8) structures as the search models, using Molrep (31) and Phaser (32), to solve the ADS and AQS wild-type structures, respectively. The ADS W332Q, ADS W332G, and AQS Y215V mutant enzyme structures were solved by the molecular replacement method with the ADS and AQS wild-type structures (PDB codes 5WX3 and 5WX4, respectively) as the search models, respectively, using Phaser (32). The structure refinements of ADS complexed with CoASH, the ADS W332Q mutant enzyme complexed with CoASH, the ADS W332G mutant enzyme complexed with CoASH, and the AQS Y215V mutant enzyme were performed using phenix.refine (33), with the twin operators (h, −k, −h−l), (h, −k, −h−l), (h, −k, −h−l), and (−h, −k, l) at 0.04, 0.04, 0.03, and 0.03, respectively. The structure refinement of AQS apo was performed using phenix.refine (33). The structures were modified manually with COOT (34) and refined with phenix.refine (33). The quality of the final models was assessed with Molprobity (35). The structure-based similarity was searched with the Dali program (36). The active-site cavity volumes and entrance areas were calculated with the CASTp program. All crystallographic figures were prepared with PyMOL.
Docking model of ADS complexed with the substrate
The three-dimensional structure of the Cys-tethered decanoylmonoketide was generated by the CHEM3D ULTRA 10 program (Cambridge Soft), and its geometry was optimized using the elbow tool in PHENIX (37). The intermediate model was manually swapped with the catalytic Cys-157 in the ADS structure with COOT (34), and then the energy minimization calculation by simulated annealing with the CNS program (38) was performed. The parameters of the intermediate for the energy minimization calculation were obtained with the PRODRG server (39).
Analyses of ADS and AQS gene expression by semiquantitative RT-PCR
Total RNA from E. rutaecarpa buds, leaves, and fruits was isolated as described above. The first-strand cDNA was then synthesized from 50 ng of each RNA sample, using SuperScript IV (Invitrogen) with a random hexamer (Invitrogen) as the primer. The ADS (678 bp) and AQS (720 bp) gene fragments were amplified with the primer pairs ADS_q and AQS_q, respectively (supplemental Table S3). The 18S rRNA gene (GenBankTM accession number AB699582, 610 bp) was amplified with the primer pair 18S (supplemental Table S3) and was loaded on the 2% agarose gel as the PCR product control. All genes were amplified with PrimeSTAR HS DNA polymerase (TaKaRa). The amplified DNA fragments were stained by ethidium bromide and visualized with the ImageQuant TL software (GE Healthcare).
Tissue distributions of C7, C9, C11, and C13 2AQs
Methanol extracts (100 mg/ml) were prepared from the buds, leaves, and fruits of E. rutaecarpa, and the C7, C9, C11, and C13 2AQs contents were analyzed by LC-ESI-MS, under the conditions described under “Co-incubation assays of ADS, AQS, and QNS.” The 2AQs in the extracts were determined by comparing the retention times and the UV and MS spectra with those of authentic compounds.
Author contributions
H. N., I. A., and H. M. designed the experiments. T. M., T. K., T. M., T. T., and H. M. performed the experiments. T. M., T. K., and H. M. analyzed the data. T. M., T. K., I. A., and H. M. wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgment
The X-ray diffraction experiments were performed at the Photon Factory, under proposals 14G504, 14G521, 14G530, 15G007, and 16G507.
This work was supported in part by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan (JSPS KAKENHI Grants JP15H01836, JP16H06443, JP25242067, JP26282210, and JP26560435); by Grants-in-Aid for the Cooperative Research Project from the Institute of Natural Medicine, University of Toyama, in 2013 (to H. N. and H. M.) and in 2015 (to T. M. and H. M.); and by the Platform Project for Supporting Drug Discovery and Life Science Research (Platform for Drug Discovery, Informatics, and Structural Life Science) from the Japan Agency for Medical Research and Development (AMED) (to T. M.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Tables S1–S3.
- 2AQ
- 2-alkylquinolone
- AD acid
- alkyldiketide acid
- AD-CoA
- alkyldiketide-CoA
- ADS
- alkyldiketide-CoA synthase
- AQS
- alkylquinolone synthase
- 2ATL
- 2-alkyltriketide lactone
- CsOLS
- C. sativa olivetol synthase
- ClDCS
- C. longa diketide-CoA synthase
- ClCURS1
- C. longa curcumin synthase 1
- QNS
- quinolone synthase
- CmQNS
- C. microcarpa quinolone synthase
- EtOAc
- ethyl acetate
- FabH
- β-ketoacyl carrier protein synthase IIIs
- HsPKS3
- H. serrata type III polyketide synthase 3
- CHS
- chalcone synthase
- MsCHS
- M. sativa chalcone synthase
- OsCUS
- O. sativa curcuminoid synthase
- PKS
- polyketide synthase
- RdORS
- R. dauricum orcinol synthase
- r.m.s.d.
- root mean square deviation
- RpBAS
- R. palmatum benzalacetone synthase
- PDB
- Protein Data Bank
- ESI
- electrospray ionization.
References
- 1. Wang X. X., Zan K., Shi S. P., Zeng K. W., Jiang Y., Guan Y., Xiao C. L., Gao H. Y., Wu L. J., and Tu P. F. (2013) Quinolone alkaloids with antibacterial and cytotoxic activities from the fruits of Evodia rutaecarpa. Fitoterapia 89, 1–7 [DOI] [PubMed] [Google Scholar]
- 2. Cardoso-Lopes E. M., Maier J. A., da Silva M. R., Regasini L. O., Simote S. Y., Lopes N. P., Pirani J. R., Bolzani Vda S., and Young M. C. (2010) Alkaloids from stems of Esenbeckia leiocarpa Engl. (Rutaceae) as potential treatment for Alzheimer disease. Molecules 15, 9205–9213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Heeb S., Fletcher M. P., Chhabra S. R., Diggle S. P., Williams P., and Cámara M. (2011) Quinolones: from antibiotics to autoinducers. FEMS Microbiol. Rev. 35, 247–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang C. F., Fan L., Tian M., Qi X. S., Liu J. X., Feng J. B., Du S. S., Su X., and Wang Y. Y. (2014) Radiosensitizing effect of schinifoline from Zanthoxylum schinifolium Sieb et Zucc on human non-small cell lung cancer A549 cells: a preliminary in vitro investigation. Molecules 19, 20128–20138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dulcey C. E., Dekimpe V., Fauvelle D. A., Milot S., Groleau M. C., Doucet N., Rahme L. G., Lépine F., and Déziel E. (2013) The end of an old hypothesis: the Pseudomonas signaling molecules 4-hydroxy-2-alkylquinolines derive from fatty acids, not 3-ketofatty acids. Chem. Biol. 20, 1481–1491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Drees S. L., Li C., Prasetya F., Saleem M., Dreveny I., Williams P., Hennecke U., Emsley J., and Fetzner S. (2016) PqsBC, a condensing enzyme in the biosynthesis of the Pseudomonas aeruginosa quinolone signal: crystal structure, inhibition, and reaction mechanism. J. Biol. Chem. 291, 6610–6624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Drees S. L., and Fetzner S. (2015) PqsE of Pseudomonas aeruginosa acts as pathway-specific thioesterase in the biosynthesis of alkylquinolone signaling molecules. Chem. Biol. 22, 611–618 [DOI] [PubMed] [Google Scholar]
- 8. Wang J., Wang X. H., Liu X., Li J., Shi X. P., Song Y. L., Zeng K. W., Zhang L., Tu P. F., and Shi S. P. (2016) Synthesis of unnatural 2-substituted quinolones and 1,3-diketones by a member of type III polyketide synthases from Huperzia serrata. Org. Lett. 18, 3550–3553 [DOI] [PubMed] [Google Scholar]
- 9. Katsuyama Y., Kita T., Funa N., and Horinouchi S. (2009) Curcuminoid biosynthesis by two type III polyketide synthases in the herb Curcuma longa. J. Biol. Chem. 284, 11160–11170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ferrer J. L., Jez J. M., Bowman M. E., Dixon R. A., and Noel J. P. (1999) Structure of chalcone synthase and the molecular basis of plant polyketide biosynthesis. Nat. Struct. Biol. 6, 775–784 [DOI] [PubMed] [Google Scholar]
- 11. Mori T., Shimokawa Y., Matsui T., Kinjo K., Kato R., Noguchi H., Sugio S., Morita H., and Abe I. (2013) Cloning and structure-function analyses of quinolone- and acridone-producing novel type III polyketide synthases from Citrus microcarpa. J. Biol. Chem. 288, 28845–28858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Abe I., Takahashi Y., Morita H., and Noguchi H. (2001) Benzalacetone synthase: a novel polyketide synthase that plays a crucial role in the biosynthesis of phenylbutanones in Rheum palmatum. Eur. J. Biochem. 268, 3354–3359 [DOI] [PubMed] [Google Scholar]
- 13. Taura F., Iijima M., Yamanaka E., Takahashi H., Kenmoku H., Saeki H., Morimoto S., Asakawa Y., Kurosaki F., and Morita H. (2016) A novel class of plant type III polyketide synthase involved in orsellinic acid biosynthesis from Rhododendron dauricum. Front. Plant Sci. 7, 1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Katsuyama Y., Matsuzawa M., Funa N., and Horinouchi S. (2007) In vitro synthesis of curcuminoids by type III polyketide synthase from Oryza sativa. J. Biol. Chem. 282, 37702–37709 [DOI] [PubMed] [Google Scholar]
- 15. Austin M. B., and Noel J. P. (2003) The chalcone synthase superfamily of type III polyketide synthases. Nat. Prod. Rep. 20, 79–110 [DOI] [PubMed] [Google Scholar]
- 16. Abe I., and Morita H. (2010) Structure and function of the chalcone synthase superfamily of plant type III polyketide syntheses. Nat. Prod. Rep. 27, 809–838 [DOI] [PubMed] [Google Scholar]
- 17. Morita H., Shimokawa Y., Tanio M., Kato R., Noguchi H., Sugio S., Kohno T., and Abe I. (2010) A structure-based mechanism for benzalacetone synthase from Rheum palmatum. Proc. Natl. Acad. Sci. U.S.A. 107, 669–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Katsuyama Y., Miyazono K., Tanokura M., Ohnishi Y., and Horinouchi S. (2011) Structural and biochemical elucidation of mechanism for decarboxylative condensation of β-keto acid by curcumin synthase. J. Biol. Chem. 286, 6659–6668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Abe I., Abe T., Wanibuchi K., and Noguchi H. (2006) Enzymatic formation of quinolone alkaloids by a plant type III polyketide synthase. Org. Lett. 8, 6063–6065 [DOI] [PubMed] [Google Scholar]
- 20. Morita H., Wanibuchi K., Nii H., Kato R., Sugio S., and Abe I. (2010b) Structural basis for the one-pot formation of the diarylheptanoid scaffold by curcuminoid synthase from Oryza sativa. Proc. Natl. Acad. Sci. U.S.A. 107, 19778–19783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Taura F., Tanaka S., Taguchi C., Fukamizu T., Tanaka H., Shoyama Y., and Morimoto S. (2009) Characterization of olivetol synthase, a polyketide synthase putatively involved in cannabinoid biosynthetic pathway. FEBS Lett. 583, 2061–2066 [DOI] [PubMed] [Google Scholar]
- 22. Bera A. K., Atanasova V., Robinson H., Eisenstein E., Coleman J. P., Pesci E. C., and Parsons J. F. (2009) Structure of PqsD, a Pseudomonas quinolone signal biosynthetic enzyme, in complex with anthranilate. Biochemistry 48, 8644–8655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nevarez D. M., Mengistu Y. A., Nawarathne I. N., and Walker K. D. (2009) An N-aroyltransferase of the BAHD superfamily has broad aroyl CoA specificity in vitro with analogues of N-dearoylpaclitaxel. J. Am. Chem. Soc. 131, 5994–6002 [DOI] [PubMed] [Google Scholar]
- 24. Tang M. C., He H. Y., Zhang F., and Tang G. L. (2013) Baeyer-Villiger oxidation of acyl carrier protein-tethered thioester to acyl carrier protein-linked thiocarbonate catalyzed by a monooxygenase domain in FR901464 biosynthesis. ACS Catal. 3, 444–447 [Google Scholar]
- 25. Giddens A. C., Nielsen L., Boshoff H. I., Tasdemir D., Perozzo R., Kaiser M., Wang F., Sacchettini J. C., and Copp B. R. (2008) Natural product inhibitors of fatty acid biosynthesis: synthesis of the marine microbial metabolites pseudopyronines A and B and evaluation of their anti-infective activities. Tetrahedron 64, 1242–1249 [Google Scholar]
- 26. Coppola G. M. (1985) The chemistry of 2h-3, 1-benzoxazine-2, 4 (1H)-dione (isatoic anhydride): 17. synthesis of 2-alkyl-4-quinolone alkaloids via a one-step reaction of N-methylisatoic anhydride with methyl ketone enolates. J. Heterocyclic Chem. 22, 491–494 [Google Scholar]
- 27. Abe I., Oguro S., Utsumi Y., Sano Y., and Noguchi H. (2005) Engineered biosynthesis of plant polyketides: chain length control in an octaketide-producing plant type III polyketide synthase. J. Am. Chem. Soc. 127, 12709–12716 [DOI] [PubMed] [Google Scholar]
- 28. Abe I., Watanabe T., and Noguchi H. (2004) Enzymatic formation of long-chain polyketide pyrones by plant type III polyketide synthases. Phytochemistry 65, 2447–2453 [DOI] [PubMed] [Google Scholar]
- 29. Kumar S., Stecher G., and Tamura K. (2016) MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vagin A., and Teplyakov A. (2010) Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 66, 22–25 [DOI] [PubMed] [Google Scholar]
- 32. McCoy A. J. (2007) Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr. D Biol. Crystallogr. 63, 32–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., and Adams P. D. (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 35. Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Holm L., and Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Echols N., Headd J. J., Hung L. W., Jain S., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R. D., Read R. J., Richardson D. C., et al. (2011) The Phenix software for automated determination of macromolecular structures. Methods 55, 94–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., and Warren G. L. (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 39. Schüttelkopf A. W., and van Aalten D. M. F. (2004) PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. 60, 1355–1363 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.