

Abstract
Ribonucleotide reductase (RR) is the rate-limiting enzyme in DNA synthesis, catalyzing the reduction of ribonucleotides to deoxyribonucleotides. During each enzymatic turnover, reduction of the active site disulfide in the catalytic large subunit is performed by a pair of shuttle cysteine residues in its C-terminal tail. Thioredoxin (Trx) and glutaredoxin (Grx) are ubiquitous redox proteins, catalyzing thiol-disulfide exchange reactions. Here, immunohistochemical examination of clinical colorectal cancer (CRC) specimens revealed that human thioredoxin1 (hTrx1), but not human glutaredoxin1 (hGrx1), was up-regulated along with human RR large subunit (RRM1) in cancer tissues, and the expression levels of both proteins were correlated with cancer malignancy stage. Ectopically expressed hTrx1 significantly increased RR activity, DNA synthesis, and cell proliferation and migration. Importantly, inhibition of both hTrx1 and RRM1 produced a synergistic anticancer effect in CRC cells and xenograft mice. Furthermore, hTrx1 rather than hGrx1 was the efficient reductase for RRM1 regeneration. We also observed a direct protein-protein interaction between RRM1 and hTrx1 in CRC cells. Interestingly, besides the known two conserved cysteines, a third cysteine (Cys779) in the RRM1 C terminus was essential for RRM1 regeneration and binding to hTrx1, whereas both Cys32 and Cys35 in hTrx1 played a counterpart role. Our findings suggest that the up-regulated RRM1 and hTrx1 in CRC directly interact with each other and promote RR activity, resulting in enhanced DNA synthesis and cancer malignancy. We propose that the RRM1-hTrx1 interaction might be a novel potential therapeutic target for cancer treatment.
Keywords: colorectal cancer, enzyme catalysis, protein-protein interaction, ribonucleotide reductase, thioredoxin
Introduction
Ribonucleotide reductase (RR)2 is a multisubunit enzyme that catalyzes the reduction of ribonucleotides to their corresponding deoxyribonucleotides, which are the building blocks required for DNA replication and repair (1). Three main classes of RR have been described based on the pathways of radical initiation and requirements of metal cofactors (2, 3). Class I RRs are further divided into three subgroups (Ia, Ib, and Ic) based on allosteric regulation and polypeptide sequence homologies (4, 5). Class Ia RRs are found in all types of eukaryotes (including human, mouse, and yeast), several viruses, a few prokaryotes (including Escherichia coli), and some bacteriophages. In Class Ia RRs, the large subunit R1 (named as RRM1 in human RR) contains the substrate-binding site as well as binding sites for allosteric regulators that control overall activity and substrate specificity, respectively. Using E. coli Class Ia RR as a prototype model, it has been proposed that substrate turnover by R1 is initiated by a thiyl radical that is present on Cys439 in the substrate-binding site. Disulfide bridges that connect Cys225 and Cys462 at the active site and Cys754 and Cys759 at the C-terminal tail also participate in the catalytic cycle. The small subunit R2 of Class Ia RRs (named as RRM2 and RRM2B, two types of R2, in human RR) contains a diiron-tyrosyl radical cofactor essential for enzyme activity (1, 2).
The expression and activity of RR are well regulated because of its unique function in DNA synthesis. The expression of RR subunit proteins is controlled by RNA transcription and protein degradation in response to cell cycle and DNA repair signals. At the same time, the enzymatic activity of RR is regulated by multiple factors that include allosteric regulation, post-translational modification, subcellular localization, and intracellular environment (6, 7). Dysregulated expression and activity of human RR are associated with genomic instability, malignant transformation, and cancer development, which make it an important target for anticancer agents (4–9). RR inhibitors, either alone or in combination with other therapies, have proven to be a useful strategy for treating solid tumors and hematological malignancies (10, 11).
Thioredoxin (Trx) is ubiquitous in the cytosol of all living cells and functions as a powerful protein-disulfide reductase to regenerate an active site dithiol after each catalytic cycle, whereas the conserved active site of Trx, Trp-Cys-Gly-Pro-Cys, becomes oxidized to a disulfide (Trx-(SH)2 + protein-S2 → Trx-S2 + protein-(SH)2). In the presence of NADPH, this disulfide is reduced back to the dithiol by thioredoxin reductase (TrxR) (12–14). Glutaredoxin (Grx) is a small redox enzyme participating in thiol-disulfide exchange reactions in the presence of glutathione (GSH), glutathione reductase (GR), and NADPH (12). It has been reported that for Class I and Class II RR enzymes the electrons are supplied by NADPH through Trx or Grx systems (15–18). Reduction of ribonucleotide in the RR catalysis involves the formation of a disulfide in the active site of R1 subunit. Structural studies with E. coli RR show that the active site cleft of the R1 subunit is not wide enough to permit the direct reduction by the external redoxin systems, so the reduction of active site disulfide (Cys225 and Cys462) is carried out by a pair of shuttle cysteine residues in the C-terminal mobile tail of R1 subunit (Cys754 and Cys759). Sequence alignment shows that the C-terminal shuttle dithiols of E. coli R1 subunit has the CXXXXC sequence, whereas mammalian R1 and yeast R1 have a CXXC sequence (19–22). In vitro mutagenesis and kinetic studies support a critical role for the C-terminal cysteine pair of R1 in regeneration of the active site (22, 23). The resulting disulfide bond in the C-terminal tail of R1 is then reduced by either the Trx or Grx system to continue the next catalytic cycle (23).
Human Trx1 (hTrx1) has been shown to play a role in a variety of human diseases including cancer (24). Cancer cells stably transfected with hTrx1 exhibit increased proliferation and decreased spontaneous and drug-induced apoptosis (25). Increased levels of hTrx1 occur in a number of cancers and are associated with increased cell proliferation, decreased apoptosis, and decreased patient survival (24). Although oxidative stress may disrupt biological functions, redox reactions in a cell are often tightly regulated and play essential physiological roles (26). The mechanism for the increase of hTrx1 in cancer cells remains unknown. There are a number of mechanisms by which hTrx1 might cause increased cell growth including an increased supply of reducing equivalents for DNA synthesis, activation of transcription factors that regulate cell growth, and an increase in the sensitivity of cells to other cytokines and growth factors (27–32). Because of its role as a stimulator of cell growth and an inhibitor of apoptosis, hTrx1 has served as a target for the development of drugs to treat and potentially prevent cancer (24). In addition, human Grx1 (hGrx1) expression in the previous studies is decreased in dysplastic tissue and even more decreased in the tumors as compared with non-neoplastic tissue. The down-regulation of hGrx1 has previously been described in lung cancer tissue where an inverse correlation of hGrx1 expression and cell proliferation of the tumors was found (33).
However, how human redox system regulates RRM1 catalysis reduction remains unclear (22, 23). Based on all the available crystal structures of RRM1, the C-terminal regions are thermally labile and structurally undefined; therefore, the location of the C terminus in the tertiary structure of RRM1 is not well resolved. Which and how the human cellular redox proteins (e.g. hTrx1 or hGrx1) regulate human RR enzymatic activity are unclear. In addition, based on our knowledge, there is still no experimental demonstration of the genetic/physical interaction between hTrx1 and RRM1 as well as the potential molecular mechanisms involved. Although the general concepts of redox signaling have been established, the identity and function of many regulatory switches remain unclear, especially in the context of cancer (24, 27). Whether and how the human cellular redox proteins affect the malignant proliferation of tumor cells via regulating human RR enzymatic activity are still unclear.
In the present study, we found that hTrx1 but not hGrx1 expression increased in parallel with RRM1 levels and malignant progression in the cancer tissues of CRC patients. Furthermore, hTrx1 significantly promoted RR activity, DNA synthesis, and cancer phenotypes in an RRM1-dependent way in cancer cells. Combined inhibition of hTrx1 and RRM1 produced a synergistic anticancer effect in cancer cell and xenograft mouse models. Subsequent biochemical evidence suggested that hTrx1 rather than hGrx1was responsible for RRM1 regeneration and RR activity. RRM1 directly bound to and was reduced by hTrx1 through the two known cysteines with a novel Cys779 residue in its C-terminal tail also required for RRM1 activity, whereas the Cys32 and Cys35 in hTrx1 played a catalytic role against RRM1. Overall, we, for the first time, have demonstrated the genetic and physical interaction of RRM1 and hTrx1 in the CRC context, providing a new potential target for drug discovery for CRC.
Results
The expression of hTrx1 but not hGrx1 was up-regulated in parallel with RRM1 increasing malignant progression in colorectal cancer
Immunohistochemical analysis was performed on 228 cases of clinical specimens of CRC (Table 1). The results showed that the expressions of RRM1, hTrx1, and hTrxR1 were up-regulated in most of the CRC tissues over their paired normal tissues (>90% specimens), whereas the hGrx1 expression level was relatively low in both normal and cancer tissues (Fig. 1, A–C, and supplemental Fig. S1). The hTrx1 expression level paralleled that of either RRM1 or hTrxR1, whereas the hGrx1 expression did not (Fig. 1D). The expressions of RRM1 and hTrx1 were significantly increased in the cancer tissues and positively correlated with cancer invasion (T1–T3) and metastasis (M0-M1) (p < 0.01) (Fig. 1E and Table 1). Consistently, The Cancer Genome Atlas data analysis showed that the hTrx1 mRNA expression level positively correlated with the mRNA levels of RRM1 in 376 CRC cases (supplemental Fig. S2 and supplemental methods). Therefore, the results showed that the expression of hTrx1 but not hGrx1 was up-regulated in parallel with RRM1, and both were correlated with the pathological staging of CRC, suggesting a potential connection among hTrx1, RRM1, and CRC malignant progression.
Table 1.
Correlation of the expression of RRM1 and hTrx1 with clinicopathological features in CRC
TNM: T, Tumor; N, regional lymph node; M, metastasis.
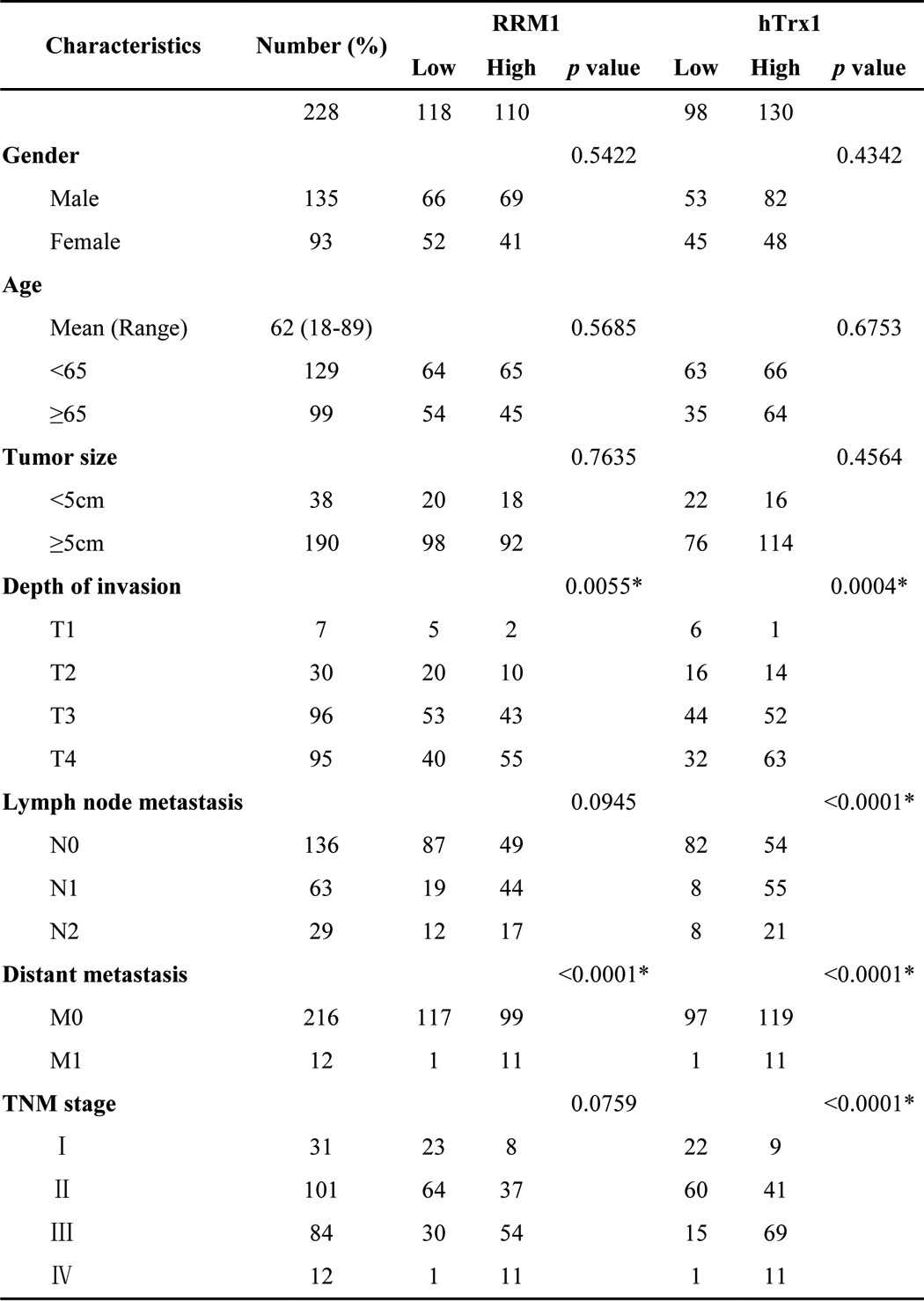
Figure 1.
The expression of hTrx1 but not hGrx1 was up-regulated in parallel with RRM1 increasing malignant progression in colorectal cancer. A, representative immunostainings of RRM1, hTrx1, hTrxR1, and hGrx1 in CRC and adjacent non-cancerous tissues. B, representative immunostainings of RRM1, hTrx1, hTrxR1, and hGrx1 in one CRC patient (consecutive sections). C, RRM1 and hTrx1 expression levels of paired cancer and adjacent normal tissues from 228 CRC patients. D, correlation of concurrent immunostaining scores of RRM1, hTrx1, hTrxR1, and hGrx1 in 228 CRC tissues. E, RRM1 and hTrx1 immunostaining scores of 228 cases of CRC patients at different progression stages. The data shown represent the mean ± S.D. *, p < 0.01. Error bars represent S.D. TNM: T, Tumor; N, regional lymph node; M, metastasis.
hTrx1 increased RR activity by RRM1 regulation, which promoted malignant phenotypes in CRC cells
To unravel the molecular detail of the relationship between hTrx1 and RRM1 in CRC, we performed a series of knockdown and overexpression experiments for hTrx1 and RRM1 in selected cancer cell lines to monitor cellular RR activity, cell viability, and DNA synthesis. Ectopic expression of hTrx1 increased RR enzymatic activity and cell proliferation in cancer cells, whereas RRM1 knockdown with siRNA completely reversed the effect (Fig. 2, A and B, and supplemental Fig. S3). EdU incorporation assays showed that hTrx1 enhanced DNA synthesis that is dependent on RRM1 in cancer cells (Fig. 2C). Moreover, wound healing assays showed that hTrx1 increased the migration of the transfected cancer cells, whereas hTrx1 knockdown with siRNA showed the opposite effect (Fig. 2D). The results indicated that hTrx1 significantly increased RR activity and DNA synthesis in cancer cells, which might then enhance CRC malignancy.
Figure 2.

hTrx1 increased RR activity by RRM1 regulation, which promoted malignant phenotypes in CRC cells. A, the RR activity of cell lysates was measured by RR activity assay (using DTT as a reductant as indicated in supplemental Table S1) in KB and CRC cells after the indicated transfection. RR activity = [3H]dCDP/([3H]CDP + [3H]dCDP) × 100%. Relative RR activity (y axis) is the RR activity of treatment group/control group × 100%. B, cell proliferation was measured by Cell Counting Kit-8 assay in KB and CRC cells after the indicated transfection. The y axis is cell viability of treatment group/control group × 100%. The data shown represent the mean ± S.D. (n = 3). *, p < 0.01, treatment group versus control group. C, the DNA synthesis was measured by EdU assay in SW480 cells after the indicated transfection. On the left is the live staining, and on the right is the quantitation. The data shown represent the mean ± S.D. (n = 3). *, p < 0.01, treatment group versus control group. D, cell migration was measured by wound healing assay in KB and CRC cells after the indicated transfection. At the top is the live staining, and on the bottom is the quantitation. Bottom panel, the y axis is wound distance of EV group/hTrx1 group × 100% or si-control (si-ctr) group/si-hTrx1 group × 100%. The data shown represent the mean ± S.D. (n = 3). *, p < 0.01; #, p < 0.05, EV group versus hTrx1 group, si-control group versus si-hTrx1. Error bars represent S.D.
Combined inhibition of hTrx1 and RRM1 produced a synergistic anticancer effect in cancer cells and xenograft mice
Gemcitabine is a nucleoside analog widely used in cancer chemotherapy. It is converted intracellularly to the active metabolites dFdCDP and dFdCTP. dFdCDP inhibits RRM1, thereby decreasing the deoxynucleotide pool available for DNA synthesis (7). PX-12 is an irreversible hTrx1 inhibitor with antitumor properties. PX-12 decreases the activity of hTrx1 by thioalkylating the critical cysteine residue (Cys73) or by increasing the dimerization of its oxidative form (34–36). As shown in Fig. 3A, gemcitabine and PX-12 showed different orders of potencies in selected cell lines. The IC50 values for gemcitabine were 25.0 ± 1.1 and 37.7 ± 1.3 nm for SW480 and SW620, respectively. The IC50 values for PX-12 were 11.9 ± 1.0 and 18.5 ± 1.1 μm for SW480 and SW620, respectively. Combined treatment with gemcitabine and PX-12 yielded a significantly greater growth inhibition than either agent used alone in both SW480 and SW620 cells (Fig. 3B). Quantitative analyses with the Chou-Talalay method (37) indicated a synergistic antiproliferation effect (combination index (CI) <1) (Fig. 3C). Plate clone formation assays also demonstrated a significantly combined action of the two drugs against CRC cells (Fig. 3D).
Figure 3.
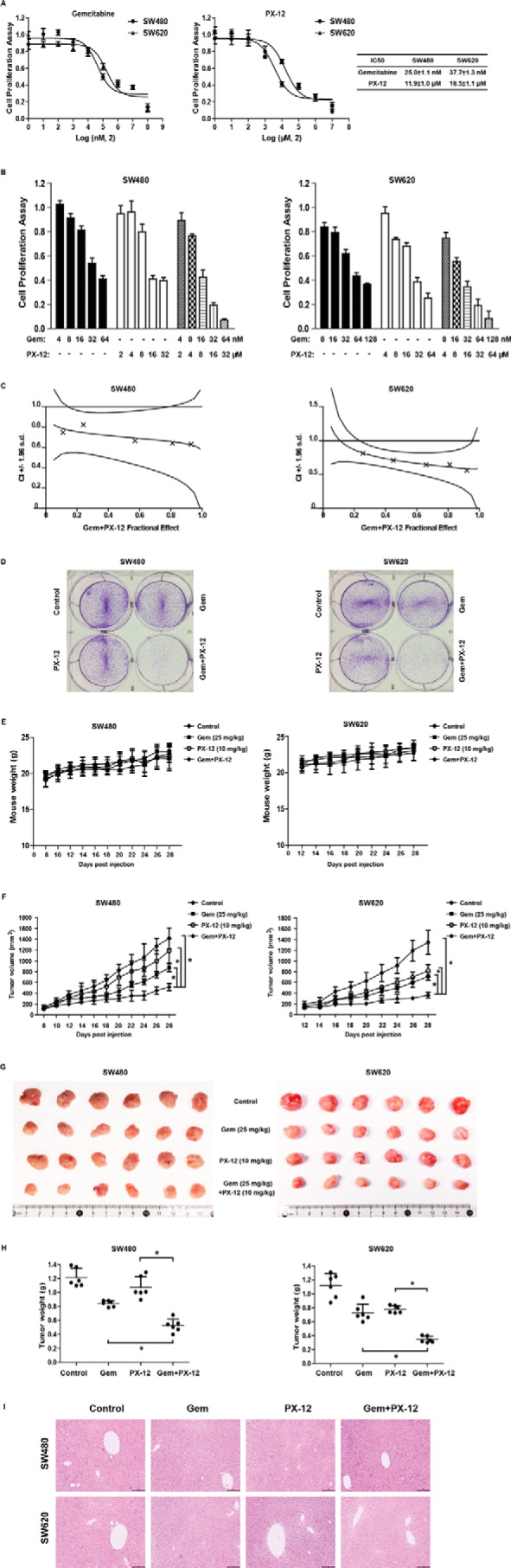
Combined inhibition of hTrx1 and RRM1 produced a synergistic anticancer effect in cancer cells and xenograft mice. A, SW480 or SW620 cells were treated with the indicated concentrations of gemcitabine or PX-12 for 72 h, and cell viability was measured with Cell Counting Kit-8 assays. B, SW480 or SW620 cells were seeded into 96-well plates and treated with gemcitabine (Gem) and/or PX-12 at the indicated concentrations. The concentrations of gemcitabine and PX-12 maintained a 2 nm:1 μm ratio for the combination group according to their IC50 values for the two cell lines. After 72 h, the cell viability was measured with Cell Counting Kit-8 assays. The data shown represent the mean ± S.D. (n = 3). C, the synergistic effect of gemcitabine combined with PX-12 was quantitatively analyzed and expressed as CI versus fractional effect. Where calculable, 95% confidence intervals are shown. D, plate clone formation of SW480 and SW620 cells with gemcitabine (8 nm in SW480 and 16 nm in SW620) and/or PX-12 (4 μm in SW480 and 8 μm in SW620). E, the body weight curves of mice with the indicated treatment. The data are presented as the mean ± S.D. (n = 6 mice per group). F, the growth curves of the tumors formed by SW480 or SW620 cells with indicated treatment. G, images of tumors formed by SW480 or SW620 cells with the indicated treatment. H, weight differences in tumors formed by SW480 or SW620 cells with the indicated treatment. The data shown represent the mean ± S.D. (n = 6 mice per group). *, p < 0.01. Error bars represent S.D. I, liver tissue sections were examined by H&E staining. Scale bar: 500 μm.
To further investigate the in vivo role, SW480 or SW620 cells were subcutaneously injected into nude mice to generate CRC xenografts followed by treatment with gemcitabine and/or PX-12. Although each group of mice showed similar body weights during the treatment (Fig. 3E), combined treatment with gemcitabine and PX-12 significantly reduced the volumes (Fig. 3, F and G) and weights (Fig. 3H) of the xenografts compared with the control and single-drug groups. No apparent histopathological changes were observed with H&E staining in liver tissue sections of the treated mice, suggesting the combined treatment did not cause liver damage in mice (Fig. 3I). These results indicated that combined use of hTrx1 and RRM1 inhibitors produced a synergistic anticancer effect in CRC cell lines and xenograft mice at the indicated dosages.
hTrx1 was the efficient reductase for RRM1 regeneration rather than hGrx1
In Class Ia RRs, Grx1 is the most efficient electron donor for the E. coli enzyme, whereas Trx1 and Grx1 show similar reduction efficiencies for the mouse enzyme (23, 38). Different reductant or redoxin systems were evaluated for their redox potentials for human RR activity (supplemental Table S1). As shown in Fig. 4A, the hTrx1/hTrxR1 system activated the RRM1 specific activity with much higher efficiency than the hGrx1 system and the generic reductant DTT. The specific activity of RRM1 was significantly up-regulated with the increase of hTrx1 concentration, which is not the case for the parallel hGrx1 group (Fig. 4, B and C). The apparent Km value for hTrx1 was about 1.45 μm, whereas Vmax was around 1.58 dCDP/s/RRM1 monomer. The results indicated that, in the external electron donors, hTrx1 is the most efficient reductase for human RR catalysis rather than hGrx1 and the small reductant DTT.
Figure 4.

hTrx1 was the efficient reductase for RRM1 regeneration rather than hGrx1. A, RRM1 specific activity was measured with generic reductant or redoxin systems as indicated in supplemental Table S1 (50 mm DTT, 25 μm hTrx1, or 25 μm hGrx1). B, RRM1 specific activity was measured with different concentration of hGrx1 (25, 50, 75, and 100 μm). C, RRM1 specific activity was measured with different concentration of hTrx1 (1, 2, 3, 4, 5, 10, 15, 20, and 25 μm). The data shown represent the mean ± S.D. (n = 3). *, p < 0.01 versus control. Error bars represent S.D. yGR, yeast glutathione reductase.
RRM1 bound with hTrx1 by direct protein-protein interaction
There was no significant difference in RRM1 and hTrx1 protein levels among different CRC cell lines (supplemental Fig. S4). Immunofluorescence revealed co-localization of RRM1 and hTrx1 in SW480 cells (supplemental Fig. S5 and supplemental methods).
SW480 cells were transfected with RRM1-FLAG expression plasmids. The cell lysates were analyzed by immunoprecipitation using anti-FLAG® M2 magnetic beads followed by SDS-PAGE and Coomassie Brilliant Blue staining. Protein bands were excised and analyzed by mass spectrometry (LC-MS/MS). We also extracted proteins from clinical CRC tissue specimens (the cancer tissues versus their paired normal tissues). Then the proteins were analyzed by immunoprecipitation using RRM1 antibody followed by SDS-PAGE and LC-MS/MS as above. The results showed that hTrx1 was one of the RRM1-interacting redox proteins in CRC cells and tissues (supplemental Tables S2 and S3 and supplemental methods).
To validate the interaction between RRM1 and hTrx1, SW480 cell lysates were co-immunoprecipitated with hTrx1 antibody and detected by immunoblotting (Fig. 5A). The result showed that RRM1 bound with hTrx1 in SW480 cells. Next, SW480 cells were co-transfected with expression plasmids for c-myc-RRM1 with FLAG-hTrx1 or FLAG-RRM1 with c-myc-hTrx1. Co-immunoprecipitation followed by Western blot analyses of the cell lysates indicated the existence of the protein-protein interaction between RRM1 and hTrx1 in cells (Fig. 5, B and C). Glutathione S-transferase (GST) pulldown combined with Western blotting confirmed the direct interaction between purified RRM1 and hTrx1 proteins in vitro (GST-hTrx1 and RRM1-His in Fig. 5D and GST-RRM1 and hTrx1-His in Fig. 5E). Real-time kinetic measurements of RRM1-hTrx1 interaction using the Octet equipment showed a high affinity (Kd = 17.5 nm) between purified RRM1 and hTrx1 proteins (supplemental Fig. S6 and supplemental methods).
Figure 5.

RRM1 bound with hTrx1 by direct protein-protein interaction. A, SW480 cell lysates were co-immunoprecipitated with hTrx1 antibody and detected by immunoblotting. IgG was used as a negative control. B, SW480 cells were co-transfected with c-myc-RRM1 and FLAG-hTrx1 expression plasmids. The cell lysates were co-immunoprecipitated with FLAG antibody and detected by immunoblotting. C, SW480 cells were co-transfected with FLAG-RRM1 and c-myc-hTrx1 expression plasmids. The cell lysates were co-immunoprecipitated with FLAG antibody and detected by immunoblotting. Ten percent of each cell lysate for the co-immunoprecipitation was used for the input control. D, GST pulldown assay was performed by incubating purified RRM1-His protein with GST-hTrx1 immobilized on GST resin and analyzed by immunoblotting. E, GST pulldown assay was performed by incubating purified hTrx1-His protein with GST-RRM1 immobilized on GST resin and analyzed by immunoblotting. F, GST pulldown assay was performed by incubating purified RRM1-His or RRM2-His protein with GST-hTrx1 immobilized on GST resin at different time points in different buffers and analyzed by immunoblotting. G, SW480 cells were co-transfected with c-myc-RRM1 and si-RRM2. GST pulldown assay was performed by incubating the cell lysates with GST-hTrx1 immobilized on GST resin and analyzed by immunoblotting. H, SW480 cells were co-transfected with FLAG-RRM2 and si-RRM1. GST pulldown assay was performed by incubating the cell lysates with GST-hTrx1 immobilized on GST resin and analyzed by immunoblotting. IP, immunoprecipitation; ctr, control.
Furthermore, GST pulldown assays in RR assay buffer and PBS buffer showed that RRM1 but not RRM2 could bind with hTrx1 (Fig. 5F). In the RRM1 or RRM2 knockdown SW480 cell lysates, GST pulldown assays showed that RRM1-hTrx1 interaction was not dependent on RRM2, but RRM2 bound to hTrx1 only in the presence of RRM1(Fig. 5, G and H). The data indicated that RRM1 but not RRM2 bound with hTrx1 by direct protein-protein interaction.
In addition to the known Cys787 and Cys790, a novel cysteine (Cys779) in the C terminus of RRM1 was essential for RRM1 regeneration and RRM1-hTrx1 binding
It is known that in most eukaryotes and prokaryotes two highly conserved cysteine residues in the C-terminal sequences of RR large subunits (Cys787 and Cys790 for RRM1) are required for the reduction of the catalytic site. Homolog alignment analysis further disclosed that a third cysteine residue is present in the C-terminal sequences of the eukaryotic Class Ia RR large subunits (Cys779 for RRM1) (Fig. 6A). A series of C-terminal mutants of RRM1 were prepared (supplemental Figs. S7 and S8) and assayed for their enzymatic activities and binding abilities with hTrx1. All the site-directed and truncated mutants were almost as active as the wild-type RRM1 with the small molecule reductant DTT, indicating that the mutations in Cys779, Cys787, and Cys790 of RRM1 did not cause any apparent RRM1 misfolding. However, as compared with the wild-type RRM1, the enzymatic activities of all the mutants significantly were reduced when assayed with the hTrx1/hTrxR1 system (Fig. 6B). The specific activity of the C779S, C787S, and C790S mutants was about 6.0, 0.3, and 1.3% of that of the wild-type RRM1, respectively, suggesting a critical role of Cys779 in RRM1 reduction by hTrx1, although it may be less important than Cys787 and Cys790. It is known that ectopic expression of RRM1 (wild type) increases RR enzymatic activity and cell proliferation in CRC cells, whereas the RR-inactive mutant RRM1-Y738F fails to provide deoxyribonucleoside diphosphates, thus blocking DNA synthesis and cell proliferation in the transfected CRC cells (9). As shown in supplemental Fig. S9, ectopic expression of RRM1 wild type but not the cysteine mutants significantly increased cell proliferation, suggesting that the three C-terminal cysteines of RRM1 were essential for RRM1 activity regeneration in CRC cells. In addition, the wild-type and all the mutants of RRM1 had very low specific activities with the hGrx1 system. It was also confirmed that hGrx1 was not the efficient reductase for human RR activity (Fig. 6B).
Figure 6.

The Cys779, Cys787, and Cys790 of RRM1 and Cys32 and Cys35 of hTrx1 played vital roles during RRM1 enzymatic activity regeneration and RRM1-hTrx1 interaction. A, sequence alignment of the C-terminal regions of the RR large subunits from different species. B, RRM1 (wild type or C-terminal cysteine mutants) specific activities with different reductants as indicated in supplemental Table S1 (50 mm DTT, 25 μm hTrx1, or 25 μm hGrx1). C, GST pulldown assay was performed by incubating purified RRM1-His (wild type or C-terminal cysteine mutants) proteins with GST-hTrx1 immobilized on GST resin and analyzed by immunoblotting. D, schematic diagram of human thioredoxin1. E, GST pulldown assay was performed by incubating purified RRM1-His (wild-type) protein with GST-hTrx1 (wild type or mutants) immobilized on GST resin and analyzed by immunoblotting. The data shown represent the mean ± S.D. (n = 3). *, p < 0.01. Error bars represent S.D. yGR, yeast glutathione reductase.
Furthermore, GST-hTrx1 pulldown followed by Western blot analyses showed that the RRM1 mutants totally lost their abilities to bind with hTrx1 (Fig. 6C). Therefore, all three C-terminal cysteines (Cys779, Cys787, and Cys790) of RRM1 are essential for RRM1 activity regeneration with hTrx1 and the RRM1-hTrx1 interaction.
The Cys32 and Cys35 of hTrx1 played a vital role during RRM1 regeneration and RRM1-hTrx1 interaction
As shown in Fig. 6D, unlike Trxs from prokaryotes such as E. coli, mammalian Trxs contain additional structural cysteines (Cys62, Cys69, and Cys73 for hTrx1) beyond the active cysteines (Cys32 and Cys35 for hTrx1) (13, 29). The cysteine mutants of hTrx1 were prepared (supplemental Fig. S10), and their activities to reduce RRM1 were measured. The results showed that the redox-active site mutants C32S, C35S, and C32S/C35S of hTrx1 almost lost their abilities for RRM1 regeneration, whereas the mutants C69S, C73S, and C69S/C73S of hTrx1 still kept the reduction activity for RRM1 (Fig. 6B), which indicated that the Cys32 and Cys35 were the key residues of hTrx1 to regenerate RRM1 activity.
GST pulldown assays further demonstrated that the hTrx1 mutants C32S and C35S significantly lost their abilities to bind with RRM1, whereas the C73S mutant bound with RRM1 similarly to the wild type (Fig. 6E), suggesting that the Cys32 and Cys35 in hTrx1 also played an important role for interaction between RRM1 and hTrx1.
Discussion
Ribonucleotide reductase, encompassing both conventional reductase and possible moonlighting (non-reductase) functions, clearly has important, subunit-specific roles in cancer biology, influencing tumor initiation, progression, and therapeutic sensitivity while also serving as a target for anticancer drug (6). Increased expression and activity of human RR are associated with malignant transformation and cancer development (4, 6). RR activity can be regulated by expression of different subunits, subcellular localization, post-translational modifications, and allosteric regulation involving both enzymatic activity and substrate specificity. In the ONCOMINE database, RRM1 mRNA is among the top 10% most overexpressed genes in 30 of the 170 studies including brain and central nervous system cancer, lung cancer, and sarcoma (6). Cellular redox proteins such as Trx and Grx systems are multifunctional thiol-dependent redoxins (39, 40). Reduction of ribonucleotide during RR catalysis involves the formation of a disulfide in the active site of R1 subunit (22, 23). However, structural studies with E. coli RR show that the active site cleft of R1 subunit is not very wide to permit the direct reduction by the external redoxin system. Instead, the reduction of active site disulfide is performed by a pair of highly conserved shuttle cysteine residues in the C-terminal mobile tail of R1 subunit. The resulting C-terminal disulfide bond is then reduced by NADPH through Trx or Grx systems to regenerate R1 for the next cycle of RR catalysis (23). hTrx1 can act as an anticancer agent by protecting cells from carcinogenic oxidants or ROS or by repairing oxidized proteins; however, it can also protect and enhance angiogenesis in different types of cancers by up-regulating HIF-1α. Increased levels of hTrx1 occur in a number of cancers, which may contribute to the resistance of cancers to therapy by scavenging ROS that are generated by various anticancer agents (18). Trx participates in signaling pathways interacting with different proteins to control their dynamic regulation of structure and function (30). hTrx1 can interact with various signaling molecules (such as AP-1, NFκB, ASK-1, etc.), and the mechanisms of hTrx1-induced growth control are multifaceted. Direct antioxidant properties, transcription regulation, and ASK-1 inhibitory antiapoptotic effects are mechanisms by which hTrx1 can exert its growth control (15).
In this study, to clarify the redox regulation of human RR, analyses of clinical specimens from CRC patients showed that RRM1, hTrx1, and hTrxR1 protein expressions were up-regulated in most of the cancer tissues over their paired normal tissues, and the hTrx1 expression levels paralleled the changes of RRM1 and hTrxR1 in most CRC cases, which were supported by The Cancer Genome Atlas data analysis. However, the hGrx1 expression level was relatively low in both the normal and cancer tissues of CRC (Fig. 1, A–D). Furthermore, the expressions of RRM1 and hTrx1 were significantly positively correlated with invasion and distant metastasis (Fig. 1E and Table 1). This clinical evidence suggested that RRM1 and hTrx1 might play an important role in CRC malignant progression, and the hTrx1-RRM1 correlation could be a potential biomarker and therapeutic target in CRC. In contrast, the immunostaining data showed that RRM1 was significantly up-regulated at the T1–T3 stages but not at the T4 stage in CRC tissues, suggesting that although the cancer-up-regulated RRM1 contributes to RR activity as the large subunit essential for cancer development the protein itself may switch to a non-RR role in a late cancer stage by enhancing the PTEN (phosphatase and tensin homolog deleted on chromosome ten) pathway (9).
Small molecule compounds targeting human RR can be divided into RRM1 inhibitors including nucleoside analogs, sulfhydryl group inhibitors (such as gemcitabine, clofarabine, cisplatin, caracemide, etc.), RRM2 inhibitors including radical scavengers (hydroxyurea) and metal chelators (Triapine), and RRM1-RRM2 polymerization inhibitors (oligopeptides) (4, 7). Clinical data demonstrate that RR inhibitors have antitumor activities when administered both as a single agent and as enhancers of other anticancer drugs. Diverse clinical application strategies and novel classes of RR inhibitors are being vigorously investigated to further improve cancer therapy (23). Herein, we demonstrated that hTrx1 can increase human RR activity by RRM1 regeneration that might further promote malignant phenotypes in CRC cells (Fig. 2). Combination treatment with gemcitabine and PX-12, used as representative inhibitory drugs of RRM1 and hTrx1, respectively, synergistically inhibited CRC cells and xenograft mice (Fig. 3). This suggests that combinatorial use of the inhibitors of RR and hTrx1 may prove to be a novel anticancer strategy by producing synergistic therapeutic effects. Moreover, interrupting the interaction between RRM1 and hTrx1 may also be a potential cancer treatment in the future.
The Class I RR introduces a complication because it has been reported to be a substrate for both Trx and Grx (23). Both Trx and Grx are dithiol electron donors of E. coli RR, whereas Grx1 is the most efficient electron donor for the enzyme activity. However, Trx1 and Grx1 systems showed similar catalytic efficiencies (kcat/Km) as recombinant mouse RR complex (23, 34). Here, we showed that, for human, hTrx1 was a much more efficient reductase for RRM1 regeneration than hGrx1 (Fig. 4). The underlying mechanism is of great interest. First, there is a mechanistic difference between E. coli and mammalian RRs in using Trx and Grx systems as electron donors (23, 41, 42). Trx is reduced directly by TrxR and NADPH, whereas in the glutaredoxin system Grx is reduced via the tripeptide GSH, and then the oxidized glutathione (GSSG) formed is reduced by GR and NADPH (42, 43). This characteristic prevents the channeling of reducing equivalents from the Trx system to the Grx system, thus keeping the two reducing systems isolated from each other and able to act on their respective substrates (22, 23, 43). Second, under normal growth conditions, both Trx and Grx are mostly in the reduced state. However, during oxidative stress where GSSG accumulates and GSSG is a poor substrate for Trx, it is possible that an “RR shunt” serves to channel reducing equivalents from the Trx to the Grx system. This would occur in a sequence where Trx reduces RR, which reduces Grx, which reduces GSSG (39, 40). It is also a possible explanation why Trx and Grx systems play different roles for RR regeneration under different conditions and in different organisms.
It is known that the C-terminal shuttle dithiols of E. coli R1 have the CXXXXC sequence, whereas yeast and mammalian R1 has a CXXC motif. It is proposed that the CXXC disulfide is rigid, and therefore high activation energy may be required to distort it to a reactive conformation. This may explain why many RRs that have the CXXC motif (e.g. T4 phage, Listeria, and Class Ib RRs) have a dedicated redoxin (T4 Trx, Listeria Trx, and NrdH, respectively) (44–47). This is also consistent with the observation that Class II and Class Ia RRs related to the E. coli enzyme, which have a CXXXXC motif, never have a dedicated redoxin in the operon (39, 47). In this study, we proved the direct protein-protein interaction between RRM1 and hTrx1 (Fig. 5). Close examination of the C-terminal tail sequences of some eukaryotic Class Ia RRs showed that there was a third partially conserved cysteine beside, which was Cys779 for human RRM1, in addition to Cys787 and Cys790 (Fig. 6A). Importantly, all three cysteine residues (Cys779, Cys787, and Cys790) were required for RRM1 reduction and the RRM1-hTrx1 interaction (Fig. 6, B and C).
As compared with prokaryotes, why a third cysteine is required in addition to the conserved CXXC sequence in eukaryotes Class Ia R1 is also an attractive question. It is possible that in addition to the formation of the disulfide of Cys787-Cys790 an isomerization might occur to form a mixture of disulfide (Cys779-Cys787 or Cys779-Cys790), which provides more flexible disulfide substrates for hTrx reduction with a longer linker between these cysteines in RRM1 C terminus. Furthermore, the redox reactions are highly active, and much more ROS are produced in higher species, especially in cancer cell metabolism. The Cys779 of RRM1 may be evolutionally favorable to the binding between RRM1 and hTrx1, keeping the reduction state of RRM1 and increasing RR activity in vivo. The unique Cys779 in RRM1 seems to be essential for selecting the endogenous reductant and important for hTrx1-RRM1 interaction. Therefore, the difference in bacterial and human Trx-R1 interaction could serve as a novel benchmark for antibiotic discovery against bacteria of interest.
The functional activity of Trx1 is critically associated with the conserved disulfide motif (Cys32-Gly-Pro-Cys35 of hTrx) in the active site. Compared with lower species such as E. coli, mammalian Trx contains additional structural cysteines (Cys62, Cys69, and Cys73 of hTrx1) (Fig. 6D). Whether these non-active site Cys residues have biologic function remains unknown. Cys73 was present as an intermolecular disulfide bond for Trx1 homodimer in X-ray crystal studies. The Trx active (Cys32 and Cys35) site is blocked by dimer formation. However, the hTrx1 mutant C73S still appeared as a homodimer in the crystal structure (13, 15). Under oxidative stress, besides the active site disulfide, a second disulfide could form between Cys62 and Cys69 with a considerable effect on hTrx1 activity. Upon nitrosative stress, a disulfide forms in the active site (Cys32-Cys35), and Cys69 and Cys73 become nitrosylated. The non-active site disulfide or nitrosothiols are not substrates for reduction by hTrxR1 and delay the reduction of the active site disulfide by hTrxR, which provides a means to transiently inhibit hTrx1 activity under different stress conditions, allowing more time for the sensing and transmission of stress signals (28). Post-translational modifications of hTrx1 via these cysteine residues, such as thiol oxidation, glutathionylation, and S-nitrosylation, have been proposed to play an essential role in regulation of functions and signal transduction pathways (16). As shown in Fig. 6B, we observed that Cys32 and Cys35 were the key sites for hTrx1 to regenerate RRM1 rather than Cys69 and Cys73. The hTrx1 mutants C32S and C35S lost their ability to bind with RRM1 (Fig. 6E), whereas the C73S mutant still appeared as a homodimer, which did not affect its binding with RRM1. These results suggested that the Cys32 and Cys35 in hTrx1 play a vital role during RRM1 regeneration through interacting with the Cys779, Cys787, and Cys790 in RRM1 (Fig. 6, B and E).
This study demonstrated that in cancer cells human RR activity can be significantly increased by the hTrx1 system through RRM1 regeneration in addition to protein expression regulation, which plays a critical role in promoting human RR activity, DNA synthesis, and cancer development. RRM1 directly binds to hTrx1 through its three C-terminal Cys residues, all of which are indispensable for the enzymatic activity. Clinical specimen analysis and animal experimental evidence suggest that hTrx1-RRM1 correlation may possess clinical significance as a potential combination or interruption target for cancer therapy. Further insight into the roles of redox proteins and how they act together with RR in cancer progression will no doubt provide important clues into more effective treatments for cancer patients.
Experimental procedures
Clinical tissues and immunohistochemistry
All patient tissue samples were obtained during surgery at the Affiliated Hospitals of Zhejiang University School of Medicine using an approved Institutional Review Board Protocol (Zhejiang University School of Medicine). Samples from patients who received preoperative radiation or chemotherapy were excluded. The clinicopathological characteristics of the 228 clinical specimens are summarized in Table 1. The primary cancer tissues were formalin-fixed and paraffin-embedded for immunohistochemistry. All the CRC patients were stochastically selected without any preference.
The immunohistochemistry was performed using an Envision detection system (Dako, Denmark) according to the manufacturer's instructions. We used commercial antibodies against RRM1 (10526-1-AP, Proteintech), hTrx1 (sc-20146, Santa Cruz Biotechnology), hTrxR1 (sc-28321, Santa Cruz Biotechnology), and hGrx1 (sc-293250, Santa Cruz Biotechnology) for immunohistochemistry. PBS was used as a negative control. To estimate the score of each slide, at least eight individual fields at 200× were selected, and 100 cancer cells were counted in each field. The score for each slide was measured as the cross-product of the value of immunostaining intensity and the value of the proportion of positive-staining cells. The immunostaining intensity was divided into five grades: 0, negative; 1, weak; 2, moderate; 3, strong; and 4, very strong (supplemental Fig. S1). The proportion of positive-staining cells was also divided into five grades: 0, <5%; 1, 6–25%; 2, 26–50%; 3, 51–75%; and 4, >75%. The score was calculated using the following formula: Total score = Intensity score × Proportion score. The judging criteria for protein expression level were as follows: low expression, total score 0 ≤ total score ≤ 8; high expression, 8 < total score ≤ 16. Consecutive sections and corresponding similar regions were used to score the immunostaining intensity. The staining results were assessed and confirmed by two independent investigators blinded to the clinical data.
Plasmid construction
pET28a-RRM1, pET28a-RRM2, pET28a-hTrxR1, pET3a-hTrx1, and pET24a-hGrx1 plasmids were obtained from Professor JoAnne Stubbe at the Massachusetts Institute of Technology. The constructs for RRM1 and hTrx1 with different tags were prepared as reported previously (9). The mutants for RRM1 and hTrx1 were generated using the QuikChange site-directed mutagenesis kit (Stratagene) according to the manufacturer's instructions. The primers for the above constructs are listed in supplemental Tables S4 and S5. All constructs were confirmed by DNA sequencing.
Protein expression and purification
RRM1, RRM2, hTrx1, and hTrxR1 proteins with a His tag were expressed from BL21(DE3) cells with pET28a-RRM1, pET28a-RRM2, pET28a-hTrx1, and pET28a-hTrxR1 plasmids and purified using TALONTM affinity chromatography (Clontech) (48). hTrx1 protein without a His tag was expressed from BL21(DE3)pLysS competent cells with pET3a-hTrx1 plasmid and purified using a Q Sepharose Fast Flow column and Sephacryl 26/60 S-200 column (GE Healthcare) (48). hGrx1 protein was expressed from BL21(DE3)pLysS competent cells with pET24a-hGrx1 plasmid and purified using a DEAE Fast Flow column (GE Healthcare) column (49).
Cell culture and viability assays
KB, SW480, and SW620 (American Type Culture Collection) were cultured in RPMI 1640 medium (Gibco) supplemented with 10% fetal bovine serum (Gibco) at 37 °C in a humidified 5% CO2 atmosphere.
To determine cell viability, cells in logarithmic growth were plated into 96-well plates and incubated overnight. The cells were then treated with the designated expression plasmids, siRNA, or drugs. After 72 h of culture, Cell Counting Kit-8 (Dojindo, Japan) was added, and the A450 was measured using an automatic plate reader. Data were obtained from at least three separate experiments done in triplicate.
Transfection and siRNA interference
The c-myc-RRM1 and FLAG-hTrx1 constructs for cell transfection were prepared (9) and transfected using X-treme GENE HP DNA transfection reagent (Roche Applied Science) according to the manufacturer's protocol. RRM1 siRNA (sc-37640), hTrx1 siRNA (sc-106984), and scrambled siRNA (sc-37007, all from Santa Cruz Biotechnology) were transfected with LipofectamineTM RNAiMAX (Invitrogen) according to the manufacturer's instructions.
Western blot analysis
The whole-cell lysate was analyzed with the antibodies goat anti-human RRM1, rabbit anti-human Trx1, mouse anti-human GST tag, mouse anti-human His tag, mouse anti-human FLAG tag, mouse anti-human c-myc tag, and anti-GAPDH (Santa Cruz Biotechnology). IRDye® 800CW- or IRDye 680-conjugated secondary antibodies (LI-COR Biosciences, Lincoln, NE) were used for staining and then detected by an Odyssey® infrared imaging system (9).
GST pulldown
GST and GST fusion proteins were expressed and purified from E. coli BL21(DE3) cells. For the pulldown assays, GST-EV and GST-hTrx1 (or GST-RRM1) proteins were immobilized onto glutathione-Sepharose resin (GE Healthcare) at 4 °C for 1 h. Following incubation, resins were washed with PBST buffer (1% TritonX-100 in PBS) three times. Then purified RRM1-His (or hTrx1-His) protein (10 μg) was incubated with GST-EV and GST-hTrx1 (or GST-RRM1) proteins-resins in RR activity assay buffer at room temperature for 30 min. Resins were washed with PBST buffer three times, and bound proteins were eluted from the beads using GST elution buffer (50 mm Tris-HCl, pH 8.0, containing 10 mm reduced glutathione). Interactions were analyzed by Western blot analysis (50).
Co-immunoprecipitation
The cells were co-transfected with expression vectors for c-myc-RRM1 and hTrx1-FLAG. The cell lysates (2 mg) were then analyzed by immunoprecipitation using anti-FLAG M2 magnetic beads (Sigma-Aldrich) followed by Western blotting with c-myc or FLAG antibody. Ten percent of the cell lysate for each co-immunoprecipitation was used for the input control.
RR activity assay
The method was modified for assaying the enzymatic activity of recombinant RR proteins as described previously (48). Briefly, the specific activity of RRM1 was determined by measuring the reduction of [5-3H]CDP in the presence of a 5-fold excess of RRM2 subunit. The system for the RRM1 specific activity with different reductants or reductases (DTT, hTrx1/hTrxR1 system, or hGrx1/yeast glutathione reductase/GSH system) are listed in supplemental Table S1. The RR activity in the cultured cells was assayed with a further optimized and standardized procedure as described previously (9). Briefly, an appropriate amount of proteins, prepared by streptomycin sulfate-(NH4)2SO4 double precipitation, was added to 100 μl of reaction mixture containing 0.125 mm [3H]CDP (24 Ci/mmol), 50 mm Hepes, pH 7.2, 6 mm DTT, 4 mm MgOAc, 2 mm ATP, 0.05 mm CDP, and 100 mm KCl. The assay mixture was incubated at 37 °C for 30 min, then dephosphorylated, and analyzed by HPLC and liquid scintillation counting. The enzyme activity of RR in the cultured cells was calculated as follows: RR activity = [3H]dCDP/([3H]CDP + [3H]dCDP) × 100%. Relative RR activity was the RR activity of treatment group/control group × 100%.
EdU incorporation assay
DNA synthesis was assessed using the Cell-Light EdU DNA cell proliferation kit (RiboBio Co., China). The cancer cells (2.5 × 104 cells/well) were seeded in 96-well plates in triplicate and treated with 50 μm EdU for 2 h at 37 °C. After being fixed with 4% paraformaldehyde for 30 min, the cells were treated with 0.5% Triton X-100 for 20 min and washed with PBS three times. The cells were then exposed to 100 μl of 1× Apollo® reaction mixture for 30 min and incubated with 1× Hoechst 33342 to stain the cell nuclei for 30 min. Images of the cells were captured with a fluorescence microscope (Nikon, Japan). ImageJ software (National Institutes of Health) was used to count the fluorescent points.
Wound healing assay
Cells were seeded in 6-well plates. After treatment for 24 h, the monolayer was gently and slowly scratched with a pipette tip across the center of the well. While scratching across the surface, the long axis of the tip was always perpendicular to the bottom of the wells. After scratching, the wells were gently washed several times with PBS to remove detached cells. The wells were replenished with fresh medium without fetal bovine serum, which prohibited cell growth. Then the cells were allowed to grow for an additional 48 h when images of the stained monolayer were captured on a microscope. The wound was evaluated using ImageJ.
Drug combination effect analysis
To assess whether gemcitabine (2,2-difluorodeoxycytidine; MedChem Express) and PX-12 (1-methylpropyl 2-imidazolyl disulfide; MedChem Express) have a synergistic effect, SW480 and SW620 cells were treated with serial dilutions of the two drugs either alone or in combination. The concentrations of gemcitabine and PX-12 were maintained at a 2 nm:1 μm ratio for the combination group according to their IC50 values for the two cell lines. After 72 h, cell viability was measured with Cell Counting Kit-8 assays. The data shown represent the mean ± S.D. (n = 3). The combination effects were determined using the median effect Chou-Talalay method (37) with the CalcuSyn program (Biosoft, Cambridge, UK). Interaction between the drugs was quantified by determining a CI. Using this method, CI < 1 indicates synergy, CI = 1 indicates an additive effect, and CI > 1 indicates antagonism.
Plate colony formation assay
Cells were plated in 6-well culture plates at 200 cells/well. After treatment and incubation for 12 days at 37 °C, cells were washed twice with PBS and stained with a crystal violet staining solution. The colony formation efficiency was calculated as (Number of colonies/Number of cells inoculated) × 100%.
Nude mouse tumor xenograft model
Animal experiments were approved by Zhejiang Chinese Medical University Laboratory Animals Research Center. The BALB/C nude mice (5 weeks old) were randomly divided into eight groups (n = 6 per group), and SW480 or SW620 cells (2 × 106) were subcutaneously injected into the left armpit of nude mice. Then the mice were intraperitoneally injected with gemcitabine or PX-12 every other day at 25 and 10 mg/kg, respectively, for 21 days. For the combination group, mice were intraperitoneally injected with a mixture of gemcitabine and PX-12. 1% DMSO in physiological saline was used as a control. The long diameter (a) and short diameter (b) of the tumors were measured, and then the volume (V) was calculated using the formula V = 1/2a × b2. Mice were sacrificed after 28 days, and tumors were harvested, weighted, and examined histologically.
Statistics
Statistical data analysis was performed using the two-tailed Student's t test and one-way analysis of variance, and the results are presented as the mean ± S.D. of three separate experiments.
Author contributions
M. L. and Q. L. conducted most of the experiments and analyzed the results. M. L., H. L., and J. S. designed the experiments and wrote the paper. G. R., J. Z., and X. X. performed experiments in Fig. 1 and Table 1. Y. D. and T. Z. analyzed The Cancer Genome Atlas data. Q. L. purified RRM2-His protein. X. L., L. Z., H. Q., and J. S. performed experiments in Fig. 3. All authors read and approved the final manuscript.
Supplementary Material
This work was supported by National Natural Science Foundation of China Grants 81372138, 81572384, and 81472543; National Key Research and Development Project of China Grant 2016YFC1303401; and the Foundation of Zhejiang University for International Cooperation and Exchanges. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental methods, Figs. S1–S10, and Tables S1–S5.
- RR
- ribonucleotide reductase
- CI
- combination index
- IP
- immunoprecipitation
- CRC
- colorectal cancer
- GR
- glutathione reductase
- Grx
- glutaredoxin1
- hGrx1
- human glutaredoxin1
- Trx
- thioredoxin
- TrxR
- thioredoxin reductase
- hTrx1
- human thioredoxin1
- hTrxR1
- human thioredoxin reductase1
- RRM1
- human ribonucleotide reductase M1
- RRM2
- human ribonucleotide reductase M2
- EdU
- 5-ethynyl-2′-deoxyuridine
- dFdCDP
- difluorodeoxycytidine diphosphate
- dFdCTP
- difluorodeoxycytidine triphosphate
- ROS
- reactive oxygen species
- EV
- empty vector.
References
- 1. Kolberg M., Strand K. R., Graff P., and Andersson K. K. (2004) Structure, function, and mechanism of ribonucleotide reductases. Biochim. Biophys. Acta 1699, 1–34 [DOI] [PubMed] [Google Scholar]
- 2. Nordlund P., and Reichard P. (2006) Ribonucleotide reductases. Annu. Rev. Biochem. 75, 681–706 [DOI] [PubMed] [Google Scholar]
- 3. Mathews C. K. (2016) The most interesting enzyme in the world. Structure 24, 843–844 [DOI] [PubMed] [Google Scholar]
- 4. Shao J., Liu X., Zhu L., and Yen Y. (2013) Targeting ribonucleotide reductase for cancer therapy. Expert Opin. Ther. Targets 17, 1423–1437 [DOI] [PubMed] [Google Scholar]
- 5. Hofer A., Crona M., Logan D. T., and Sjöberg B. M. (2012) DNA building blocks: keeping control of manufacture. Crit. Rev. Biochem. Mol. Biol. 47, 50–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Aye Y., Li M., Long M. J., and Weiss R. S. (2015) Ribonucleotide reductase and cancer: biological mechanisms and targeted therapies. Oncogene 34, 2011–2021 [DOI] [PubMed] [Google Scholar]
- 7. Jordheim L. P., Sève P., Trédan O., and Dumontet C. (2011) The ribonucleotide reductase large subunit (RRM1) as a predictive factor in patients with cancer. Lancet Oncol. 12, 693–702 [DOI] [PubMed] [Google Scholar]
- 8. Bepler G., Sharma S., Cantor A., Gautam A., Haura E., Simon G., Sharma A., Sommers E., and Robinson L. (2004) RRM1 and PTEN as prognostic parameters for overall and disease-free survival in patients with non-small-cell lung cancer. J. Clin. Oncol. 22, 1878–1885 [DOI] [PubMed] [Google Scholar]
- 9. Qi H., Lou M., Chen Y., Liu X., Chen N., Shan J., Ling Z., Shen J., Zhu L., Yen Y., Zheng S., and Shao J. (2015) Non-enzymatic action of RRM1 protein upregulates PTEN leading to inhibition of colorectal cancer metastasis. Tumour Biol. 36, 4833–4842 [DOI] [PubMed] [Google Scholar]
- 10. Lee J. J., Maeng C. H., Baek S. K., Kim G. Y., Yoo J. H., Choi C. W., Kim Y. H., Kwak Y. T., Kim D. H., Lee Y. K., Kim J. B., and Kim S. Y. (2010) The immunohistochemical overexpression of ribonucleotide reductase regulatory subunit M1 (RRM1) protein is a predictor of shorter survival to gemcitabine-based chemotherapy in advanced non-small cell lung cancer (NSCLC). Lung Cancer 70, 205–210 [DOI] [PubMed] [Google Scholar]
- 11. Shao J., Zhou B., Chu B., and Yen Y. (2006) Ribonucleotide reductase inhibitors and future drug design. Curr. Cancer Drug Targets 6, 409–431 [DOI] [PubMed] [Google Scholar]
- 12. Lu J., and Holmgren A. (2014) The thioredoxin antioxidant system. Free Radic. Biol. Med. 66, 75–87 [DOI] [PubMed] [Google Scholar]
- 13. Powis G., and Montfort W. R. (2001) Properties and biological activities of thioredoxins. Annu. Rev. Biophys. Biomol. Struct. 30, 421–455 [DOI] [PubMed] [Google Scholar]
- 14. Collet J. F., and Messens J. (2010) Structure, function, and mechanism of thioredoxin proteins. Antioxid. Redox Signal. 13, 1205–1216 [DOI] [PubMed] [Google Scholar]
- 15. Yoshioka J., Schreiter E. R., and Lee R. T. (2006) Role of thioredoxin in cell growth through interactions with signaling molecules. Antioxid. Redox Signal. 8, 2143–2151 [DOI] [PubMed] [Google Scholar]
- 16. Hashemy S. I., and Holmgren A. (2008) Regulation of the catalytic activity and structure of human thioredoxin 1 via oxidation and S-nitrosylation of cysteine residues. J. Biol. Chem. 283, 21890–21898 [DOI] [PubMed] [Google Scholar]
- 17. Husbeck B., Stringer D. E., Gerner E. W., and Powis G. (2003) Increased thioredoxin-1 inhibits SSAT expression in MCF-7 human breast cancer cells. Biochem. Biophys. Res. Commun. 306, 469–475 [DOI] [PubMed] [Google Scholar]
- 18. Marks P. A. (2006) Thioredoxin in cancer—role of histone deacetylase inhibitors. Semin. Cancer Biol. 16, 436–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang Z., Yang K., Chen C. C., Feser J., and Huang M. (2007) Role of the C terminus of the ribonucleotide reductase large subunit in enzyme regeneration and its inhibition by Sml1. Proc. Natl. Acad. Sci. U.S.A. 104, 2217–2222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lin A. N., Ashley G. W., and Stubbe J. (1987) Location of the redox-active thiols of ribonucleotide reductase: sequence similarity between the Escherichia coli and Lactobacillus leichmannii enzymes. Biochemistry 26, 6905–6909 [DOI] [PubMed] [Google Scholar]
- 21. Aberg A., Hahne S., Karlsson M., Larsson A., Ormö M., Ahgren A., and Sjöberg B. M. (1989) Evidence for two different classes of redox-active cysteines in ribonucleotide reductase of Escherichia coli. J. Biol. Chem. 264, 12249–12252 [PubMed] [Google Scholar]
- 22. Holmgren A., and Sengupta R. (2010) The use of thiols by ribonucleotide reductase. Free Radic. Biol. Med. 49, 1617–1628 [DOI] [PubMed] [Google Scholar]
- 23. Sengupta R., and Holmgren A. (2014) Thioredoxin and glutaredoxin-mediated redox regulation of ribonucleotide reductase. World J. Biol. Chem. 5, 68–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Arnér E. S., and Holmgren A. (2006) The thioredoxin system in cancer. Semin. Cancer Biol. 16, 420–426 [DOI] [PubMed] [Google Scholar]
- 25. Mahmood D. F., Abderrazak A., El Hadri K., Simmet T., and Rouis M. (2013) The thioredoxin system as a therapeutic target in human health and disease. Antioxid. Redox Signal. 19, 1266–1303 [DOI] [PubMed] [Google Scholar]
- 26. Trachootham D., Lu W., Ogasawara M. A., Nilsa R. D., and Huang P. (2008) Redox regulation of cell survival. Antioxid. Redox Signal. 10, 1343–1374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Harris I. S., Treloar A. E., Inoue S., Sasaki M., Gorrini C., Lee K. C., Yung K. Y., Brenner D., Knobbe-Thomsen C. B., Cox M. A., Elia A., Berger T., Cescon D. W., Adeoye A., Brüstle A., et al. (2015) Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 27, 211–222 [DOI] [PubMed] [Google Scholar]
- 28. Watson W. H., Pohl J., Montfort W. R., Stuchlik O., Reed M. S., Powis G., and Jones D. P. (2003) Redox potential of human thioredoxin 1 and identification of a second dithiol/disulfide motif. J. Biol. Chem. 278, 33408–33415 [DOI] [PubMed] [Google Scholar]
- 29. Nakamura H. (2005) Thioredoxin and its related molecules: update 2005. Antioxid. Redox Signal. 7, 823–828 [DOI] [PubMed] [Google Scholar]
- 30. Lee S., Kim S. M., and Lee R. T. (2013) Thioredoxin and thioredoxin target proteins: from molecular mechanisms to functional significance. Antioxid. Redox Signal. 18, 1165–1207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Spindel O. N., World C., and Berk B. C. (2012) Thioredoxin interacting protein: redox dependent and independent regulatory mechanisms. Antioxid. Redox Signal. 16, 587–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ago T., and Sadoshima J. (2007) Thioredoxin1 as a negative regulator of cardiac hypertrophy. Antioxid. Redox Signal. 9, 679–687 [DOI] [PubMed] [Google Scholar]
- 33. Said K., Glaumann H., Björnstedt M., and Bergquist A. (2012) The value of thioredoxin family proteins and proliferation markers in dysplastic and malignant gallbladders in patients with primary sclerosing cholangitis. Dig. Dis. Sci. 57, 1163–1170 [DOI] [PubMed] [Google Scholar]
- 34. Powis G., Mustacich D., and Coon A. (2000) The role of the redox protein thioredoxin in cell growth and cancer. Free Radic. Biol. Med. 29, 312–322 [DOI] [PubMed] [Google Scholar]
- 35. Baker A. F., Dragovich T., Tate W. R., Ramanathan R. K., Roe D., Hsu C. H., Kirkpatrick D. L., and Powis G. (2006) The antitumor thioredoxin-1 inhibitor PX-12 (1-methylpropyl 2-imidazolyl disulfide) decreases thioredoxin-1 and VEGF levels in cancer patient plasma. J. Lab. Clin. Med. 147, 83–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ramanathan R. K., Kirkpatrick D. L., Belani C. P., Friedland D., Green S. B., Chow H. H., Cordova C. A., Stratton S. P., Sharlow E. R., Baker A., and Dragovich T. (2007) A Phase I pharmacokinetic and pharmacodynamic study of PX-12, a novel inhibitor of thioredoxin-1, in patients with advanced solid tumors. Clin. Cancer Res. 13, 2109–2114 [DOI] [PubMed] [Google Scholar]
- 37. Ashton J. C. (2015) Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 75, 2400. [DOI] [PubMed] [Google Scholar]
- 38. Aslund F., Ehn B., Miranda-Vizuete A., Pueyo C., and Holmgren A. (1994) Two additional glutaredoxins exist in Escherichia coli: glutaredoxin 3 is a hydrogen donor for ribonucleotide reductase in a thioredoxin/glutaredoxin 1 double mutant. Proc. Natl. Acad. Sci. U.S.A. 91, 9813–9817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zahedi Avval F., and Holmgren A. (2009) Molecular mechanisms of thioredoxin and glutaredoxin as hydrogen donors for mammalian S phase ribonucleotide reductase. J. Biol. Chem. 284, 8233–8240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ortenberg R., Gon S., Porat A., and Beckwith J. (2004) Interactions of glutaredoxins, ribonucleotide reductase, and components of the DNA replication system of Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 101, 7439–7444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Meyer Y., Buchanan B. B., Vignols F., and Reichheld J. P. (2009) Thioredoxins and glutaredoxins: unifying elements in redox biology. Annu. Rev. Genet. 43, 335–367 [DOI] [PubMed] [Google Scholar]
- 42. Gon S., Faulkner M. J., and Beckwith J. (2006) In vivo requirement for glutaredoxins and thioredoxins in the reduction of the ribonucleotide reductases of Escherichia coli. Antioxid. Redox Signal. 8, 735–742 [DOI] [PubMed] [Google Scholar]
- 43. Arnér E. S., Nakamura H., Sasada T., Yodoi J., Holmgren A., and Spyrou G. (2001) Analysis of the inhibition of mammalian thioredoxin, thioredoxin reductase, and glutaredoxin by cis-diamminedichloroplatinum (II) and its major metabolite, the glutathione-platinum complex. Free Radic. Biol. Med. 31, 1170–1178 [DOI] [PubMed] [Google Scholar]
- 44. Jordan A., Aslund F., Pontis E., Reichard P., and Holmgren A. (1997) Characterization of Escherichia coli NrdH. A glutaredoxin-like protein with a thioredoxin-like activity profile. J. Biol. Chem. 272, 18044–18050 [DOI] [PubMed] [Google Scholar]
- 45. Boal A. K., Cotruvo J. A. Jr., Stubbe J., and Rosenzweig A. C. (2010) Structural basis for activation of class Ib ribonucleotide reductase. Science 329, 1526–1530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Makhlynets O., Boal A. K., Rhodes D. V., Kitten T., Rosenzweig A. C., and Stubbe J. (2014) Streptococcus sanguinis class Ib ribonucleotide reductase: high activity with both iron and manganese cofactors and structural insights. J. Biol. Chem. 289, 6259–6272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wei Y., Mathies G., Yokoyama K., Chen J., Griffin R. G., and Stubbe J. (2014) A chemically competent thiosulfuranyl radical on the Escherichia coli class III ribonucleotide reductase. J. Am. Chem. Soc. 136, 9001–9013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ando N., Li H., Brignole E. J., Thompson S., McLaughlin M. I., Page J. E., Asturias F. J., Stubbe J., and Drennan C. L. (2016) Allosteric inhibition of human ribonucleotide reductase by dATP entails the stabilization of a hexamer. Biochemistry 55, 373–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bouldin S. D., Darch M. A., Hart P. J., and Outten C. E. (2012) Redox properties of the disulfide bond of human Cu, Zn superoxide dismutase and the effects of human glutaredoxin 1. Biochem. J. 446, 59–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Arnaoutov A., and Dasso M. (2014) Enzyme regulation. IRBIT is a novel regulator of ribonucleotide reductase in higher eukaryotes. Science 345, 1512–1515 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.