

Abstract
O-GlcNAcylation is the covalent addition of an O-linked β-N-acetylglucosamine (O-GlcNAc) sugar moiety to hydroxyl groups of serine/threonine residues of cytosolic and nuclear proteins. O-GlcNAcylation, analogous to phosphorylation, plays critical roles in gene expression through direct modification of transcription factors, such as NF-κB. Aberrantly increased NF-κB O-GlcNAcylation has been linked to NF-κB constitutive activation and cancer development. Therefore, it is of a great biological and clinical significance to dissect the molecular mechanisms that tune NF-κB activity. Recently, we and others have shown that O-GlcNAcylation affects the phosphorylation and acetylation of NF-κB subunit p65/RelA. However, the mechanism of how O-GlcNAcylation activates NF-κB signaling through phosphorylation and acetylation is not fully understood. In this study, we mapped O-GlcNAcylation sites of p65 at Thr-305, Ser-319, Ser-337, Thr-352, and Ser-374. O-GlcNAcylation of p65 at Thr-305 and Ser-319 increased CREB-binding protein (CBP)/p300-dependent activating acetylation of p65 at Lys-310, contributing to NF-κB transcriptional activation. Moreover, elevation of O-GlcNAcylation by overexpression of OGT increased the expression of p300, IKKα, and IKKβ and promoted IKK-mediated activating phosphorylation of p65 at Ser-536, contributing to NF-κB activation. In addition, we also identified phosphorylation of p65 at Thr-308, which might impair the O-GlcNAcylation of p65 at Thr-305. These results indicate mechanisms through which both non-pathological and oncogenic O-GlcNAcylation regulate NF-κB signaling through interplay with phosphorylation and acetylation.
Keywords: acetylation, E1A-binding protein p300 (p300), NF-kappaB, O-GlcNAcylation, phosphorylation
Introduction
O-GlcNAcylation is the attachment of the carbohydrate moiety O-linked β-N-acetylglucosamine (O-GlcNAc)3 to serine/threonine residues of cytoplasmic and nuclear proteins (1, 2). This post-translational modification is reversible and dynamic, its addition catalyzed by O-GlcNAc transferase (OGT) using UDP-GlcNAc as the substrate and its removal catalyzed by O-GlcNAcase (OGA). UDP-GlcNAc is a high-energy end product of the hexosamine biosynthesis pathway that branches from glycolysis, linking cellular metabolism to O-GlcNAcylation (3, 4). O-GlcNAcylation, analogous to phosphorylation, regulates a variety of cellular processes, including protein trafficking and turnover, cell cycle, gene expression, cellular stress response, and signal transduction (1, 4, 5). For example, O-GlcNAcylation regulates gene expression through direct modification of transcription factors and co-activators, such as FoxO1 (6, 7), PGC-1α (7, 8), ChREBP (9), and NF-κB (10, 11). One mechanism through which O-GlcNAcylation may regulate transcription factors is through competition with phosphorylation at the same site or a nearby site, as in the case of Thr-58 on c-Myc (12); Ser-1177 on endothelial nitric oxide synthase (13); and Ser-16 on ER-β (14), p53 (15), and synapsin I (16).
NF-κB is a family of transcription factors including RelA/p65, RelB, c-Rel/Rel, p105/p50, and p100/p52. They are characterized by an N-terminal Rel-homology domain, which is responsible for DNA binding and dimerization (17). NF-κB controls many biological processes, such as innate immunity, inflammation, cell survival and proliferation, and tumorigenesis (18, 19). The prototypical NF-κB form is the heterodimer p65/p50, which is mainly sequestered in the cytoplasm by inhibitor of NF-κBα (IκBα) in an inactive state. Upon stimulation by pro-inflammatory cytokines, lipopolysaccharide, or genotoxic agents, signal transduction activates the IKK complex consisting of kinases IKKα and IKKβ and a regulatory subunit, IKKγ. Activated IKK complex then phosphorylates IκBα at Ser-32 and Ser-36, leading to its polyubiquitination and subsequent proteasomal degradation. Once liberated, the p65/p50 heterodimer translocates to the nucleus and binds to NF-κB promoter/enhancer sites, initiating transcription of NF-κB target genes through recruitment of the transcriptional co-activator CBP/p300 mediated by interaction with the transcriptional activation domain of p65 (17).
The transcription of NF-κB-regulated genes is further regulated by posttranslational modifications of p65, such as phosphorylation and acetylation. For example, p65 phosphorylation at Ser-276 weakens the intramolecular interaction of p65, thereby facilitating the binding of p65 to co-activator CBP/p300, which up-regulates NF-κB gene transcription at least in part through CBP/p300 acetyltransferase activity (20). Phosphorylation of p65 Ser-536 also promotes recruitment of CBP/p300 to p65 (21, 22). Furthermore, acetylation at Lys-310 of p65 by CBP/p300 is required for full activation of p65 transcription in a manner independent of altered p65 DNA binding or IκBα association (23). We and others have shown that O-GlcNAcylation enhances the activity of NF-κB in many cell types, including pancreatic cancer cells (10), vascular smooth muscle cells (24), and lymphocytes (25). O-GlcNAc modifies the p65 subunit of NF-κB and upstream kinases IKKα and IKKβ. Lowering hyper-O-GlcNAcylation in pancreatic cancer cells decreases IKKβ expression and attenuates p65-activating phosphorylation (Ser-536), nuclear translocation, and NF-κB transcriptional activity. Conversely, elevating O-GlcNAc increases IKKα and p65 O-GlcNAcylation accompanied by increased IKK- and p65-activating phosphorylation, p65 nuclear localization, and NF-κB transcriptional activity (10). Moreover, O-GlcNAcylation has also been recently shown to facilitate the acetylation of p65 at Lys-310, a known positive regulatory event (26). However, the mechanism of how O-GlcNAcylation activates NF-κB signaling through phosphorylation and acetylation has not been fully elucidated.
Aberrant regulation of NF-κB activity has been linked to many human diseases, such as cancer and autoimmune disorders. Therefore, fully elucidating the molecular mechanisms through which O-GlcNAc tunes NF-κB activity may have great biological and clinical significance. Here, through strategies of O-GlcNAc site mapping and mutagenesis, we test how NF-κB O-GlcNAcylation interplays with phosphorylation and acetylation to regulate the activity of NF-κB.
Results
Mapping novel O-GlcNAc modification sites on the NF-κB subunit p65
We have shown that cancer cell hyper-O-GlcNAcylation contributes to oncogenic up-regulation of NF-κB signaling (10). Several p65 O-GlcNAc modification sites (including Thr-305, Thr-322, and Thr-352) have been reported (24, 26). However, little is known about mechanisms through which O-GlcNAcylation up-regulates NF-κB activity. To begin to understand possible direct roles for p65 O-GlcNAcylation, we set out to comprehensively map p65 O-GlcNAc sites. We developed two strategies to identify potential additional p65 O-GlcNAc sites. First, either 3xFLAG-tagged human WT p65, serine/threonine to alanine mutants of potential O-GlcNAc sites, or truncation mutants were transiently expressed in HEK 293T cells. To maximize cellular O-GlcNAcylation, we co-expressed OGT and pharmacologically inhibited OGA with NButGT (27). p65 proteins were immunoprecipitated and subjected to Western blot analysis for O-GlcNAc levels. Additionally, human recombinant full-length p65 or fragments (Fig. 1) prepared from bacteria or immunoprecipitated from HEK 293T cell lysates were in vitro O-GlcNAcylated by recombinant human OGT, and O-GlcNAc levels were detected by immunoblotting. As shown in Fig. 1A, mutating Thr-305 to alanine decreased O-GlcNAc levels on a p65 T322A/T352A double mutant, suggesting that Thr-305 is a p65 O-GlcNAc site. O-GlcNAc levels on p65(2–518) T322A/T352A were not reduced by further truncation to residues 2–450, suggesting an absence of O-GlcNAc sites between residues 450 and 518 (Fig. 1B). O-GlcNAcylation was not observed on an N-terminal p65 fragment (residues 2–280) (Fig. 1C) or a very C-terminal p65 fragment (residues 431–551) (Fig. 1D), suggesting an absence of O-GlcNAc sites in these regions. A p65 fragment (residues 2–450) containing mutations of all known O-GlcNAc sites in this region (T305A/T308A/T322A/T352A) was expressed in 293T cells, and O-GlcNAc signal was detected on this quadruple mutant when co-expressed with OGT in 293T cells (Fig. 1E). These results suggested that additional O-GlcNAc sites occur in the region from amino acid 280 to 430. Therefore, a series of truncation mutants between amino acids 321 and 450 were made to help localize additional novel O-GlcNAc sites. Deletion of amino acids from 360 to 421 significantly reduced O-GlcNAcylation (Fig. 1F), indicating potential O-GlcNAc sites in this region. We next individually mutated each serine or threonine residue in the region spanning 360–421 to alanine on the background of the quadruple mutant p65(2–450) T305A/T308A/T322A/T352A. Replacing Ser-374 or Ser-377 with alanine strongly reduced O-GlcNAc levels (Fig. 1G). Moreover, O-GlcNAc was detected on p65(2–321) T305A/T308A, but not on p65(2–313) T305A/T308A (Fig. 1, F and H), suggesting that a potential O-GlcNAc site(s) existed between residues 313 and 321. Further mutational analysis revealed that S319A mutation completely abolished O-GlcNAcylation on p65(2–321) T305A/T308A (Fig. 1I). These results indicate that Ser-319, Ser-374, and Ser-377 are probably novel p65 O-GlcNAc sites in addition to previously reported p65 O-GlcNAc sites (Thr-305, Thr-322, and Thr-352).
Figure 1.

Identification of O-GlcNAcylation sites on the NF-κB subunit p65 by site-directed mutagenesis and truncation analysis. A, HEK 293T cells were transfected with plasmids expressing 3xFLAG-tagged p65 T322A/T352A or 3xFLAG-tagged p65 T305A/T322A/T352A together with the HA-OGT expression plasmid and then treated with the OGA inhibitor NButGT (50 μm) overnight. FLAG-p65 was immunoprecipitated with the anti-FLAG antibody and blotted for O-GlcNAc. G250 was used to stain for total p65. B, HEK 293T cells were cotransfected with the indicated FLAG-tagged p65 plasmids (2–450, 2–450 T322A/T352A, or 2–518 T322A/T352A) and the HA-OGT plasmid and then incubated overnight with NButGT. FLAG immunoprecipitates were blotted for O-GlcNAc. G250 was used to stain for total p65. C, HEK 293T cells were transfected with the indicated FLAG-p65 constructs (2–153, 2–213, 2–280, or 2–313). FLAG-tagged p65 was immunoprecipitated and O-GlcNAcylated with Rec OGT in vitro. O-GlcNAc was detected by Western blotting. D, recombinant p65 fragments (2–313 and 431–551) prepared from bacteria were O-GlcNAcylated with Rec OGT in vitro. O-GlcNAc was detected by Western blotting. E, HEK 293T cells were transfected with the indicated FLAG-tagged p65(2–450) plasmids containing either T322A/T352A, T305A/T322A/T352A, or T305A/T308A/T322A/T352A mutations together with the HA-OGT plasmid and then incubated overnight with NButGT. FLAG immunoprecipitates were blotted for O-GlcNAc. G250 was used to stain for total p65. F, HEK 293T cells were cotransfected with the various indicated plasmids expressing FLAG-tagged p65 mutants and the HA-OGT plasmid. O-GlcNAc was detected as above (left) and quantified by normalization to total p65 (right). G, HEK 293T cells were transfected with plasmids expressing either FLAG-tagged p65(2–450) T305A/T308A/T322A/T352A or p65(2–450) T305A/T308A/T322A/T352A with additional mutations of T365A, S370A, S374A, S377A, or S401A together with the HA-OGT plasmid and then incubated overnight with NButGT. O-GlcNAc was detected as in A. H, HEK 293T cells were cotransfected with the various indicated plasmids expressing FLAG-tagged p65 mutants and the HA-OGT plasmid. O-GlcNAc was detected as in A. I, HEK 293T cells were transfected with plasmids expressing either FLAG-tagged p65(2–321) T305A/T308A or p65(2–321) with additional mutations of S311A, S316A, or S319A together with the HA-OGT plasmid and then incubated overnight with NButGT. O-GlcNAc was detected as in A. Short, short exposure; Long, long exposure; #, nonspecific bands; §, O-GlcNAc-modified p65; *, recombinant p65 fragments.
O-GlcNAc site mapping on p65 by mass spectrometry
To confirm O-GlcNAc modification sites indicated from mutagenesis and truncation studies, we employed mass spectrometry (MS) to attempt direct experimental observation of p65 site-specific O-GlcNAcylation on proteolyzed peptides of p65. Our initial attempts using trypsin proteolysis of in vitro O-GlcNAcylated full-length recombinant p65 failed to identify any O-GlcNAc sites. O-GlcNAc sites indicated by truncation and mutagenesis analysis reside in three distinct regions (residues 301–323, 334–357, and 361–381). In silico digestion of the protein using MS-Digest (28) showed that trypsin would produce either very short or very long peptides in these regions, but chymotrypsin and chemical cleavage using CNBr should generate peptides of a size amenable to MS analysis of the three O-GlcNAc-modified regions of interest. For MS analysis, in vivo O-GlcNAcylated 3xFLAG-tagged p65(2–450) was immunoprecipitated and in-gel-digested with chymotrypsin or CNBr and then using higher-energy collision dissociation (HCD) and electron transfer dissociation (ETD) mass spectrometry fragmentation approaches to target O-GlcNAc site mapping (29). We were able to unambiguously map O-GlcNAc on Ser-337 (Fig. 2A) and Ser-374 (Fig. 2B). O-GlcNAc was also present between residues 352 and 357 (Fig. 2C). Based on our site mapping results and previously reported results, we reasoned that p65 could be O-GlcNAc-modified on Thr-305, Ser-319, Ser-337, Thr-352, and Ser-374. To further verify these sites, we individually mutated Thr-305, Thr-308, Ser-319, Thr-322, Ser-337, Thr-352, or Ser-374 to alanine on full-length p65 and tested O-GlcNAcylation levels on these mutants. Consistent with MS data, mutants S319A, S337A, T352A, and S374A showed reduced levels of O-GlcNAcylation compared with WT (Fig. 2D). Unexpectedly, mutants T305A, T308A, and T322A did not display a decrease in O-GlcNAcylation (Fig. 2D). Given that Thr-305 is well conserved across species (Fig. 2E) and mutating Thr-305 into alanine markedly decreased the O-GlcNAc level on p65 T322A/T352A double mutant (Fig. 1A), we reasoned that loss of O-GlcNAc on Thr-305 on mutant p65 T305A could be compensated by increased O-GlcNAcylation on Thr-352 in our system. Consistent with this hypothesis, mutating Thr-352 into alanine on mutant p65 T305A dramatically reduced the level of O-GlcNAcylation compared with parent mutant p65 T305A (Fig. 3C), suggesting that O-GlcNAc on Thr-352 contributes to the increased O-GlcNAcylation on p65 T305A compared with WT p65. Mutant p65 T305A/T352A also displayed a lower level of O-GlcNAcylation compared with mutant p65 T352A (Fig. 3C), further supporting the conclusion that Thr-305 can be O-GlcNAcylated. Taken together, p65 could be O-GlcNAc-modified at least on Thr-305, Ser-319, Ser-337, Thr-352, and Ser-374.
Figure 2.
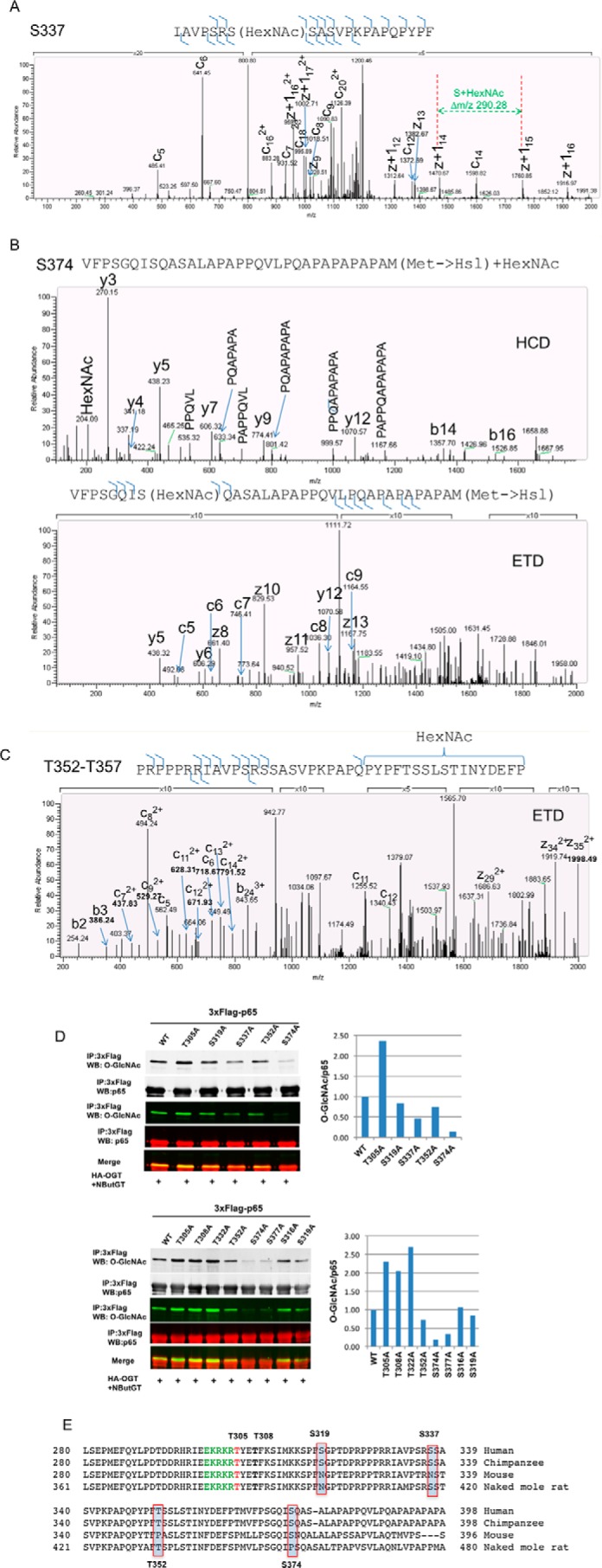
Mass spectrometry analysis of O-GlcNAcylation sites on the NF-κB subunit p65. A–C, HEK 293T cells were transfected with plasmids expressing 3xFLAG-tagged p65(2–450) together with the HA-OGT plasmid and then incubated overnight with NButGT. FLAG immunoprecipitates were digested with chymotrypsin in A and cyanogen bromide in B and C and subjected to HCD and ETD analysis. Representative mass spectra are shown. D, HEK 293T cells were transfected with plasmids expressing 3xFLAG-tagged WT p65, T305A, T308A, S316A, S319A, T322A, S337A, T352A, S374A, or S377A together with the HA-OGT plasmid and then incubated overnight with NButGT. FLAG-tagged p65 was immunoprecipitated, and O-GlcNAc was detected by Western blotting and normalized to total p65. The result represents two independent experiments. E, multiple-sequence alignment of p65 from human, chimpanzee, mouse, and naked mole rat shows the conservation of Thr-305 and Thr-308 numbered in human p65.
Figure 3.

O-GlcNAcylation of p65 at T305 enhances NF-κB transcriptional activity. A, NF-κB p65-deficient MEF cells were transfected with plasmids expressing IκBα and 3xFLAG-tagged WT p65, T305A, S319A, S337A, T352A, S374A, or vector and plasmids expressing HA-OGT or lacZ together with NF-κB luciferase and Renilla luciferase reporter vectors. The luciferase activity relative to WT p65 was measured 24 h later and normalized with the Renilla activity. B, HEK 293T cells were transfected with plasmids expressing 3xFLAG-tagged WT p65, T305A, S319A, T305A/S319A, or vector and plasmids expressing HA-OGT or lacZ together with NF-κB luciferase and Renilla luciferase reporter vectors. The luciferase activity relative to WT p65 was measured 24 h later and normalized with the Renilla activity. C, plasmids expressing 3xFLAG-tagged WT p65, T305A, T352A, or T305A/T352A and the HA-OGT plasmid were transfected into HEK 293T cells. FLAG-tagged p65 was immunoprecipitated, and O-GlcNAc and p65 were detected by Western blotting. D, NF-κB p65-deficient MEF cells were transfected with plasmids expressing IκBα and 3xFLAG-tagged WT p65, T305A, T352A, T305A/T352A, or vector and plasmids expressing HA-OGT or lacZ together with NF-κB luciferase and Renilla luciferase reporter vectors. The luciferase activity relative to WT p65 was measured 24 h later and normalized with the Renilla activity. Data represent the means ± S.D. (error bars) from at least three independent experiments. * and #, p < 0.05; ** and ##, p < 0.01; *** and ###, p < 0.001.
O-GlcNAc modification of p65 Thr-305 contributes to NF-κB transcriptional activity
To investigate p65 O-GlcNAc site-specific roles, p65 constructs with O-GlcNAc sites individually mutated to alanine were co-expressed with IκBα in p65-deficient MEF cells, and NF-κB transcriptional activity was accessed using a κB luciferase reporter assay. Mutant p65 T305A displayed reduced transcriptional activity basally and in response to OGT overexpression-induced elevated O-GlcNAcylation, whereas O-GlcNAc mutants S319A, S337A, T352A, and S374A displayed no change in NF-κB activity (Fig. 3, A and B). Moreover, the T305A mutation on a background of the T352A mutation reduced O-GlcNAc levels and led to reduced NF-κB transcriptional activity compared with WT p65 or mutant p65 T352A (Fig. 3D), further supporting the conclusion that Thr-305 O-GlcNAcylation on p65 contributes to NF-κB transcriptional activity.
O-GlcNAcylation of Thr-305 on p65 does not prevent its reassembly with IκBα
A possible regulatory mechanism for how p65 Thr-305 O-GlcNAcylation positively regulates NF-κB activity is inhibition of NF-κB p65/p50 heterodimer binding to its known inhibitor protein IκBα. The crystal structure of the p65/p50 heterodimer bound to IκBα reveals that p65 Thr-305 forms a hydrogen bond with His-84 of IκBα (30), probably representing a key contact that contributes to formation of the complex (Fig. 4A). This led us to hypothesize that p65 Thr-305 O-GlcNAcylation might interfere through steric hindrance with this hydrogen bonding as a mechanism inhibiting p65 binding to IκBα, thereby increasing NF-κB transcriptional activity. To test this hypothesis, WT p65(2–450) or p65(2–450) T305A mutant were transfected into 293T cells along with HA-OGT, and levels of IκBα associated with immunoprecipitated p65 were determined by Western blotting. The p65 T305A mutation did not alter levels of IκBα association (Fig. 4B). Similar results were obtained using full-length WT versus T305A p65 expressed in 293T cells (Fig. 4C). We further tested this hypothesis in vitro. Recombinant p65(2–313) prepared from bacteria was in vitro O-GlcNAcylated (Fig. 4D). Non-O-GlcNAcylated or in vitro O-GlcNAcylated p65(2–313) was incubated with recombinant IκBα, and levels of IκBα association were determined. In vitro p65 O-GlcNAcylation did not alter levels of IκBα association (Fig. 4E). These results suggest that inhibition of p65 association with IκBα is not a mechanism through which p65 Thr-305 O-GlcNAcylation increases NF-κB activity.
Figure 4.

O-GlcNAcylation of p65 at Thr-305 does not affect the binding of p65 to IκBα. A, crystal structure of p65/p50 heterodimer binding to IκBα (Protein Data Bank code 1NFI). Green, p65; cyan, p50; magenta, IκBα. Blue, p65 nuclear localization signal (NLS). The structure in the box shows the interactions between NLS of p65 and ankyrin repeat 1 of IκBα. Thr-305 sits right after the nuclear localization signal of p65 between helix 3 and helix 4 and forms a hydrogen bond with His-84 on IκBα. B, HEK 293T cells were cotransfected with plasmids expressing 3xFLAG-tagged p65(2–450) or T305A and the HA-OGT plasmid and then incubated overnight with NButGT. FLAG-tagged p65 was immunoprecipitated, and O-GlcNAc and p65-associated IκBα were detected by immunoblotting and quantified. Whole-cell lysates were also subjected to Western blot analysis using the indicated antibodies. C, HEK 293T cells were cotransfected with plasmids expressing 3xFLAG-tagged p65 or T305A and the HA-OGT plasmid and then incubated overnight with NButGT. FLAG-tagged p65 was immunoprecipitated, and O-GlcNAc and p65-associated IκBα were detected by immunoblotting. D, recombinant p65 fragment 2–313 prepared from bacteria was O-GlcNAcylated with Rec OGT in vitro. O-GlcNAcylated p65 was detected by Western blot. E, modified or unmodified p65 from D was incubated with recombinant IκBα for 2 h at 4 °C. p65 was immunoprecipitated, and associated IκBα was detected by immunoblotting and quantified. The experiments were repeated twice with similar results.
Thr-305 and Ser-319 O-GlcNAcylation contributes to activating acetylation of p65 at Lys-310 by p300
It has been reported that O-GlcNAcylation promotes p300-mediated activating acetylation of Lys-310 on p65, but mechanisms underlying this are not fully understood (26). Therefore, we examined potential roles of p65 Thr-305 O-GlcNAcylation specifically in p65 Lys-310 acetylation. WT p65 or O-GlcNAc site mutants and p300 were co-transfected into HEK 293T cells along with HA-OGT. Consistent with previous results, we found that acetylation of Lys-310 induced by p300 was enhanced by elevation of O-GlcNAc through OGT overexpression and/or NButGT inhibition of OGA (Fig. 5A). Mutation of p65 Thr-305 to alanine attenuated Lys-310 acetylation by p300/CBP (Fig. 5, B and E), indicating an important role for p65 Thr-305 O-GlcNAcylation in Lys-310 acetylation. Lys-310 acetylation was also found to be modestly impaired on the p65 S319A mutant, but other p65 O-GlcNAc site mutants (S337A, T352A, and S374A) did not affect Lys-310 acetylation (Fig. 5C). Thr-305 has been shown to be phosphorylated by MS (31), but the functional significance of this is not known. To rule out the possibility that the effects of p65 T305A mutant were due to loss of phosphorylation, mutants that mimic constitutive phosphorylation (T305E and T305D) were generated and tested for Lys-310 acetylation and NF-κB luciferase activity. If loss of phosphorylation resulted in T305A phenotypes, then T305E/D phosphorylation mimics may be expected to increase Lys-310 acetylation and NF-κB transcriptional activity, provided that T305E and T305D effectively mimic phosphorylation. However, the p65 mutants T305E and T305D displayed impaired acetylation of p65 at Lys-310 (Fig. 5B) and reduced transcriptional activity (Fig. 5D) similar to p65 T305A, supporting the conclusion that loss of O-GlcNAc specifically at Thr-305 accounts for reduced transcriptional activity of NF-κB. In addition, we found that overexpression of OGT led to increased expression of p300 in transiently transfected HEK 293T cells (Fig. 5F), which to some extent may explain how elevation of O-GlcNAcylation by overexpression of OGT increases activating acetylation of p65 at Lys-310 by CBP/p300.
Figure 5.

O-GlcNAcylation of p65 at Thr-305 promotes the acetylation of p65 at Lys-310. A, HEK 293T cells were cotransfected with 3xFLAG-tagged p65, p300, and HA-OGT and treated with NButGT overnight. FLAG immunoprecipitates were immunoblotted for acetylation of p65 at Lys-310 (AcK310) and O-GlcNAc. B, HEK 293T cells were cotransfected with plasmids expressing 3xFLAG-tagged WT p65, T305A, T305D, or T305E and the p300 plasmid. FLAG-tagged p65 was immunoprecipitated and immunoblotted with acetylated Lys-310. Whole-cell lysates were also subjected to Western blot analysis using the indicated antibodies. C, HEK 293T cells were transfected with plasmids expressing 3xFLAG-tagged WT p65, S374A, T352A, S337A, or S319A together with plasmids expressing p300 and HA-OGT and then incubated overnight with NButGT. FLAG-tagged p65 was immunoprecipitated and immunoblotted with acetylated Lys-310. D, NF-κB p65-deficient MEF cells were transfected with plasmids expressing IκBα and 3xFLAG-tagged WT p65, T305A, T305D, T305E, or vector together with NF-κB luciferase and Renilla luciferase reporter vectors. The luciferase activity relative to WT p65 was measured 24 h later and normalized with the Renilla activity. Data represent mean and S.D. (error bars) of at least three independent experiments. *, p < 0.05; **, p < 0.01. E, HEK 293T cells were cotransfected with plasmids expressing 3xFLAG-tagged WT p65 or T305A and the FLAG-CBP-HA plasmid. Whole-cell lysates were subjected to Western blot analysis using the indicated antibodies. F, HEK 293T cells were transfected with plasmids expressing 3xFLAG-tagged p65 together with plasmids expressing HA-OGT and p300 (pcDNA-p300) or p300-HA (pCMVβ-p300-HA) and immunoblotted with acetylated Lys-310. Whole-cell lysates were subjected to Western blot analysis using indicated antibodies. The experiments were repeated twice with similar results.
Thr-308 is phosphorylated, and phosphorylation of Thr-308 may impair the O-GlcNAcylation of Thr-305
Phosphorylation of p65 on Thr-305 and Thr-308 has previously been detected by MS (31), but there is no biochemical or functional data on these phosphorylation events. To validate whether these two sites are phosphorylated in vivo, WT p65 or mutants T305A, T308A, T322A, T352A, and T308A/T322A/T352A were transfected into HEK 293T cells, and phosphorylation was induced by treatment with the phosphatase inhibitor calyculin A. FLAG-tagged p65 was immunoprecipitated, and phosphorylation of p65 was then detected by an anti-phosphothreonine antibody. Compared with WT p65, phosphorylation on mutants T308A and T308A/T322A/T352A was reduced about 20%, but not on mutants T305A, T322A, and T352A (Fig. 6A), suggesting that Thr-308 is a potential phosphorylation site. To further examine phosphorylation of this site, SDS-PAGE containing phos-tagTM, a molecule that specifically captures phosphomonoester dianions (ROPO32−) and thus slows the migration of phosphorylated proteins in polyacrylamide gels (32), was used to detect phosphorylated p65. Compared with WT p65, the high-mobility shift of mutant T308A was not detected (Fig. 6B), and the ratio of phosphorylated p65 to non-phosphorylated p65 in mutant T308A was reduced, suggesting that p65 Thr-308 is phosphorylated in response to calyculin A treatment. Given the proximity of Thr-308 to Thr-305, we considered that Thr-308 phosphorylation may affect Thr-305 O-GlcNAcylation. To test this, phosphomimic mutant p65(2–450) T308E was generated and transfected into 293T cells along with HA-OGT. Compared with WT p65(2–450), T308E mutant, similar to T305A mutant, exhibited a lower level of O-GlcNAcylation (Fig. 6C). Moreover, mutating Thr-305 into alanine on the background of T308E (T305A/T308E) did not further reduce the O-GlcNAc level compared with phosphomimic mutant T308E (Fig. 6C), indicating that phosphorylation of Thr-308 on p65 may interfere with the O-GlcNAcylation of Thr-305. However, gain or loss of phosphorylation on Thr-308 (mutants T308E and T308A) did not affect the acetylation of p65 at Lys-310 by p300 (Fig. 6D).
Figure 6.

NF-κB subunit p65 is phosphorylated at Thr-308, and phosphorylation on Thr-308 interferes with Thr-305 O-GlcNAcylation. A, HEK 293T cells were transfected with plasmids expressing 3xFLAG-tagged WT p65, T305A, T308A, T322A, T352A, or T308A/T322A/T352A. After 48 h, cells were treated with calyculin A (100 nm) for 30 min. FLAG-tagged p65 was immunoprecipitated, immunoblotted with an anti-threonine antibody, and quantified by normalization to WT p65. B, HEK 293T cells were transfected with plasmids expressing 3xFLAG-tagged WT p65, T305A, or T308A. After 48 h, cells were treated with calyculin A (100 nm) for 30 min. FLAG-tagged p65 was immunoprecipitated and subjected to SDS-PAGE containing 50 μm phos-tagTM. -Fold changes of phosphorylated p65 to non-phosphorylated p65 were quantified and normalized to WT p65. C, plasmids expressing 3xFLAG-tagged p65(2–450), T305A, T308E, T308A, T305A/T308E, or T305A/T308A together with the HA-OGT plasmid were transfected into HEK 293T cells. FLAG-tagged p65 were immunoprecipitated, and O-GlcNAc and p65 were detected by Western blotting. Quantification was normalized to total p65. D, HEK 293T cells were cotransfected with plasmids expressing 3xFLAG-tagged p65(2–450), T308A, T308E, and the p300 plasmid. Whole-cell lysates were also subjected to Western blot analysis using the indicated antibodies. The experiments were repeated twice, and representative results are shown.
O-GlcNAcylation enhances IKK-mediated phosphorylation of p65 at Ser-536
We have shown that O-GlcNAcylation up-regulates Ser-536, activating phosphorylation on p65 in pancreatic cancer BxPC-3 cells (10). The IKK kinase complex is known to phosphorylate p65 Ser-536. To further dissect which kinases are responsible for increased p65 Ser-536 phosphorylation in response to increased O-GlcNAcylation, IKKα or IKKβ together with FLAG-tagged p65 were co-transfected into HEK 293T cells along with HA-OGT. Both IKKα and IKKβ increased the phosphorylation of p65 at Ser-536 compared with control vector (Fig. 7A). Moreover, elevation of O-GlcNAcylation by overexpression of OGT could further drive the phosphorylation of Ser-536, suggesting that both IKK kinases could mediate phosphorylation in response to increased O-GlcNAcylation (Fig. 7A). Interestingly, we found that overexpression of OGT increased the expression of IKKα and IKKβ, which indicates a mechanism probably contributing to how elevation of O-GlcNAcylation increases activating phosphorylation of p65 on Ser-536. To examine whether direct O-GlcNAcylation of p65 contributes to IKK-mediated phosphorylation of Ser-536, FLAG-tagged WT p65, T305A, S319A, S337A, T352A, or S374A was transfected into HEK 293T cells along with HA-OGT and IKKα. We found that loss of O-GlcNAc on Thr-305, Ser-319, Ser-337, Thr-352, and Ser-374 did not affect the IKK-mediated phosphorylation of p65 at Ser-536 (Fig. 7, B–D).
Figure 7.

O-GlcNAcylation facilitates IKK-dependent phosphorylation of p65 at Ser-536. A, HEK 293T cells were transfected with the plasmid expressing 3xFLAG-tagged p65 together with plasmids encoding HA-OGT and 3xHA-IKKα or 3xHA-IKKβ. After 48 h, FLAG-tagged p65 was immunoprecipitated and immunoblotted with phospho-Ser-536 of p65. Whole-cell lysates were also subjected to Western blot analysis using the indicated antibodies. B–D, HEK 293T cells were transfected with plasmids expressing 3xFLAG-tagged WT p65, S374A, T352A, S337A, S319A, or T305A together with plasmids expressing 3xHA-IKKα and HA-OGT. Whole-cell lysates were subjected to Western blot analysis using the indicated antibodies. The result represents two independent experiments.
Discussion
We and others have shown that O-GlcNAcylation promotes NF-κB activity in a variety of cell lines and that O-GlcNAcylation increases the activating phosphorylation of Ser-536 and acetylation of Lys-310 on p65 (10, 24–26). However, the underlying mechanisms for NF-κB activation by O-GlcNAcylation through interplay with phosphorylation and acetylation are not fully understood. In this study, we found that p65 was O-GlcNAcylated at Thr-305, Ser-319, Ser-337, Thr-352, and Ser-374 and phosphorylated at Thr-308; that O-GlcNAcylation of p65 at Thr-305 and Ser-319 increased CBP/p300-mediated activating acetylation of p65 at Lys-310; and that O-GlcNAcylation promoted IKK-mediated activating phosphorylation of p65 at Ser-536.
It is well known that post-translational modifications including phosphorylation, acetylation, and methylation can regulate NF-κB activity and that the interplay between phosphorylation, acetylation, and methylation can fine-tune NF-κB signaling (33). This led us to reason that O-GlcNAcylation may regulate NF-κB activity through direct modification of p65. Thus, identification of O-GlcNAcylation sites on p65 is the first step to understand the mechanisms of how O-GlcNAcylation regulates NF-κB activity. To comprehensively map O-GlcNAcylation sites on p65, we employed truncation, site-directed mutagenesis, and MS and identified new O-GlcNAc sites at Ser-337, Thr-352, and Ser-374. We also for the first time-mapped O-GlcNAc modification of Ser-319 and Thr-305 on full-length p65 using site-directed mutagenesis. In addition, we have shown that Thr-308 and Ser-377 are potential O-GlcNAc sites (Figs. 1G, 2D, and 6C) according to the mutagenesis. However, we do not have MS or functional data to support O-GlcNAcylation of these sites. Replacing Thr-305 with alanine decreased the O-GlcNAc level on p65(2–450) and p65 T322A/T352A mutants. Interestingly, the level of O-GlcNAcylation on p65 T305A single mutant was not reduced compared with WT p65. To resolve this paradox, we found that elimination of O-GlcNAcylation at Thr-305 could be compensated by increased O-GlcNAcylation at Thr-352. Likewise, increased O-GlcNAcylation on Thr-308 and Thr-322 is probably due to overcompensation for loss of other O-GlcNAc sites (Fig. 2D). This result raises a caveat that O-GlcNAcylation site mapping only by site-directed mutagenesis could be inadequate and may need to be complemented by other means, such as MS. It is also worth noting that truncation mutants p65(2–313) and p65(2-351) T305A/T308A/T322A were not observed to be O-GlcNAcylated (Fig. 1H) in our experimental setting, demonstrating that truncation and mutational mapping can miss modifications, possibly due to altered cellular localization or protein folding.
To elucidate which O-GlcNAcylation sites are functional, we screened the transcriptional activity of WT p65 and O-GlcNAc site mutants using an IκBα promoter luciferase reporter assay. Mutant p65 T305A displayed reduced transcriptional activity at both basal and elevated O-GlcNAcylation levels, whereas other O-GlcNAc mutants S319A, S337A, T352A, and S374A did not display altered activity. It has been reported that mutant p65 T352A attenuates the binding of p65 to the promoters of IL-6, TNF-1α, and MCP1 and lowers target gene mRNA expression of IL-6 and TNF-1α in MEF cells compared with WT p65 (34). Results of that study are not consistent with our observation that mutant T352A did not reduce NF-κB transcriptional activity. However, our study examines mutant effects on a distinct promoter (IκBα promoter), perhaps explaining the potential discrepancy. Moreover, mutant p65 T305A displayed attenuated p300-dependent activating acetylation of p65 at Lys-310. This result is consistent with a previous report that Thr-305 is important for acetylation of Lys-310 by p300, NF-κB-dependent gene expression, and cell survival in MEF cells (26). In addition to Thr-305, we found that the O-GlcNAcylation site Ser-319 contributed to p300-mediated acetylation of p65 at Lys-310. It is worth noting that Lys-314 and Lys-315 have also been shown to be acetylated by p300/CBP to regulate a specific set of NF-κB target genes after TNFα stimulation (35, 36). It is therefore possible that O-GlcNAcylation on Thr-305 and/or Ser-319 contributes to the acetylation of p65 at Lys-314 and Lys-315 by p300. Finally, known p300 non-histone substrates, including p53, p73, c-Myb, androgen receptor, and p300, have potential Ser/Thr O-GlcNAc modification sites five residues away from p300 acetylation sites (37), suggesting potential interplay between O-GlcNAcylation and acetylation on these proteins.
Thr-305 and Thr-308 have been identified as phosphorylated by MS (31), but they have not been validated, and the function of phosphorylation was unknown. Here, we provided evidence that Thr-308 was phosphorylated in response to phosphatase inhibitor calyculin A, whereas Thr-305, Thr-322, and Thr-352 were not. Considering the proximity of Thr-308 to Thr-305, we reasoned that there could be interplay between phosphorylation of Thr-308 and O-GlcNAcylation of Thr-305. Indeed, we found that phosphorylation of Thr-308 led to decreased O-GlcNAcylation on p65, which might be due to interference with Thr-305 O-GlcNAcylation. In support of this, it has been reported that O-GlcNAcylation of p53 at Ser-149 inhibits the phosphorylation of p53 at Thr-155, thereby stabilizing p53 by blocking the proteasomal degradation induced by COP9 signalosome that recognizes the phosphorylation at Thr-155 (15). To identify the kinase responsible for the phosphorylation of Thr-308, we searched the protein motif database Scansite with the p65 sequence. PKC was shown to be the only putative kinase targeting Thr-308 for phosphorylation at low stringency. Future studies will investigate whether any PKC isoforms could phosphorylate Thr-308 of p65. In addition, we found that phosphorylation mimic mutant T308E did not affect the acetylation of p65 at Lys-310 by p300 (Fig. 6D). Given that mutant p65 T308E reduced O-GlcNAc on p65 Thr-305 (Fig. 6C), this suggests that a role of Thr-308 phosphorylation could be suppressed by loss of O-GlcNAc on p65. Synthesized peptides with distinct phosphorylation/O-GlcNAc modifications could be used in future studies to further help dissect effects on acetylation. Likewise, Ser-316 of p65 has been reported to be phosphorylated in response to IL-1β (38). It remains to be determined whether Ser-319 O-GlcNAcylation interplays with Ser-316 phosphorylation.
In the crystal structure of p65/p50 heterodimer complexed with IκBα (30), Thr-305 is proximal to a nuclear localization signal of p65 between helix 3 and helix 4 and forms a hydrogen bond with His-84 on IκBα (Fig. 4A), probably contributing to the affinity of p65 for IκBα. Because Thr-305 was found to be O-GlcNAcylated, this led us to hypothesize that O-GlcNAcylation of Thr-305 could prevent the association of p65 with IκBα resulting from the steric hindrance of the O-GlcNAc moiety, thereby increasing the transcriptional activity of p65. Besides this hydrogen bond, the interaction between p65/p50 heterodimer and IκBα is also stabilized by the salt bridges between Lys-301, Arg-302, and Arg-304 in p65 and Asp-75, Glu-85, and Asp-73 in IκBα and a set of hydrophobic interactions among Phe-309, Ile-312, Met-313, and Phe-318 in p65 and Phe-77, Leu-80, Ala-81, Leu-89, and Val-93 in IκBα (30). Consistent with this, mutating Arg-304 into alanine in p65 reduced IκBα-NF-κB binding affinity by 2.2-fold measured by surface plasmon resonance (39) and at least by 30-fold for F309A measured by isothermal titration calorimetry (40). In this present study, we tested whether O-GlcNAcylation on Thr-305 could reduce the affinity of p65 to IκBα. Although mutating Thr-305 into alanine did not affect the association of p65 with IκBα using immunoprecipitation, future studies will further ascertain the effect of O-GlcNAcylation of p65 at Thr-305 on the association of p65 to IκBα using surface plasmon resonance and isothermal titration calorimetry in the form of synthesized O-GlcNAc-modified peptides.
We have shown that elevated O-GlcNAcylation increases the phosphorylation of p65 at Ser-536 (10). In line with the notion that IKKα and IKKβ are the major kinases to phosphorylate Ser-536 in response to different stimuli, we found that both IKKα and IKKβ could mediate the phosphorylation of Ser-536 on p65 in response to increased O-GlcNAcylation. Furthermore, elevation of O-GlcNAcylation increased the expression of IKKα and IKKβ. Because we and others have shown that both IKKα and IKKβ are O-GlcNAc-modified (10, 41), future studies will investigate the mechanism of how O-GlcNAcylation regulates the expression of IKKα and IKKβ.
It has been shown that O-GlcNAc modifies p65 and that O-GlcNAcylation is required for the activation of T and B lymphocytes (25). However, the mechanisms of how O-GlcNAcylation promotes the activation of T and B lymphocytes are unknown. Recently, another study has reported that c-Rel complexed with p65 is the major O-GlcNAcylated protein in lymphocytes and that O-GlcNAcylation of c-Rel (Q04864-2) at Ser-350 increases c-Rel-mediated gene expression of the cytokines IL-2, IFNG, and CSF2 in response to T cell receptor activation (42). Sequence alignment revealed that O-GlcNAcylation site Ser-374 in p65 is conserved with the site Ser-350 in c-Rel. Given the functional role of Ser-350 in c-Rel, it is very likely that O-GlcNAcylation of Ser-374 in p65 plays a role in the activation of lymphocytes, although we did not find that O-GlcNAcylation of Ser-374 affected NF-κB luciferase activity in MEF cells without stimuli. Future studies will investigate the effect of O-GlcNAcylation on p65 Ser-374 for lymphocyte activation.
In summary, we have identified that p65 is O-GlcNAc-modified at Thr-305, Ser-319, Ser-337, Thr-352, and Ser-374 and phosphorylated at Thr-308. O-GlcNAcylation of Thr-305 and Ser-319 contributes to the p300-dependent activating acetylation of p65 at Lys-310. In addition, elevation of O-GlcNAcylation increases the IKK-mediated phosphorylation of Ser-536. Thus, interplay between elevation of O-GlcNAcylation and increased activating p65 phosphorylation and acetylation positively regulates the activity of NF-κB.
Experimental procedures
Antibodies and reagents
Anti-O-GlcNAc antibody (CTD 110.6) has been described previously (19). The OGA inhibitor NButGT was prepared as described (27). Anti-FLAG and anti-actin were obtained from Sigma. Anti-OGT (F-12), anti-p65, anti-IKKβ, and anti-p300 were purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX). Anti-IKKα, anti-IKKβ, anti-p65, anti-phospho-p65 (Ser-536), anti-phosphothreonine, anti-acetyl-p65 (Lys-310), and anti-IκBα were obtained from Cell Signaling (Danvers, MA). Human p65 cDNA, a gift from Dr. Jianhua Yang (Baylor College of Medicine) (43), was cloned into p3xFLAG-CMV vector (Sigma-Aldrich). Plasmid pcDNA3.1-p300 was a gift from Warner Greene (Addgene number 23252), and pcDNA3β-FLAG-CBP-HA was a gift from Tso-Pang Yao (Addgene number 32908). Human recombinant IκBα protein was obtained from Sino Biological Inc. (Beijing, China). Mutagenesis on p65 was performed using the QuikChange II site-directed mutagenesis kit (Agilent Technologies).
Cell culture
Human HEK 293T cells and p65-deficient MEF cells (a gift from Dr. Jianhua Yang, Baylor College of Medicine) were cultured in DMEM containing 4.5 mg/ml d-glucose and 4 mm l-glutamine supplemented with 10% fetal bovine serum (FBS), 100 units/ml penicillin, and 100 μg/ml streptomycin. All cells were maintained at 37 °C with 5% CO2-humidified atmosphere.
Immunoblotting
Cells were lysed on ice for 10 min in radioimmune precipitation assay buffer (50 mm Tris-HCl, pH 7.4, 1% Igepal CA-630, 0.5% sodium deoxycholate, 150 mm NaCl, 1 mm EDTA, 0.1% SDS) supplemented with protease inhibitor mixture tablet (Roche Applied Science). Cell lysates were cleared by centrifuging at 16,000 × g for 20 min at 4 °C. The protein concentrations were determined using the BCA protein assay reagent kit (Pierce). Total cellular proteins were separated on SDS-PAGE and transferred to a polyvinylidene difluoride (PVDF) or nitrocellulose membrane. The membranes were incubated with HRP or IRDye®-conjugated secondary antibodies and then developed using chemiluminescence or with an Odyssey infrared imager (LI-COR).
To detect phosphorylated proteins through SDS-PAGE, 7.5% polyacrylamide gels containing 50 μm diMn(II) (1,3-bis(bis(pyridin-2-ylmethyl)amino)propan-2-olato) complex (commercially available as phos-tagTM acrylamide, Wako Chemicals) and 100 μm MnCl2 were used according to the manufacturer's protocol.
Immunoprecipitation
Cells were lysed on ice for 10 min in FLAG lysis buffer (50 mm Tris-HCl, pH 7.4, 1% Triton X-100, 150 mm NaCl, 1 mm EDTA) supplemented with protease inhibitor mixture tablet (Roche Applied Science). Cell lysates were incubated with anti-FLAG M2 affinity gel (Sigma), and immunoprecipitated 3xFLAG-p65 was eluted with 3xFLAG peptides (Sigma).
Luciferase assay
MEF p65−/− cells were seeded into 24-well plates at a concentration of 1.0 × 105/well and then transfected with vector, WT p65, or O-GlcNAc site mutants together with luciferase reporter plasmids (NF-κB luciferase reporter pNF-κB-luc and Renilla luciferase plasmid phRL-TK), pBabe-IκBα, and Plenti4-HA-OGT or Plenti4-LacZ. Cell lysates were prepared 24 h after transfection. Reporter activities were analyzed using the Promega Dual-Luciferase assay kit (Promega) according to the manufacturer's protocol. The luciferase activity was normalized to the Renilla luciferase activity, and results were reported as -fold relative to the activity of scramble-infected, vehicle-treated, or WT p65-expressing plasmids.
Protein expression and purification
Human p65(2–313) and p65(431–551) cDNAs were subcloned into pETHSUL using ligation-independent cloning as described previously (44). Expression vectors were transformed in RosettaTM 2(DE3) competent cells (EMD Millipore Chemicals). Human p65 proteins were expressed at 30 °C using an autoinduction system as described previously (44) and purified by immobilized metal-affinity chromatography. Human ncOGT constructs were kindly provided by Dr. Suzanne Walker (Harvard University), expressed and purified as described (45).
In vitro O-GlcNAcylation assay
In vitro O-GlcNAcylation assay was performed as described previously (46). Recombinant p65(2–313) and p65(431–551) were incubated with 3 μg of recombinantly expressed and immobilized metal-affinity chromatography-purified ncOGT (Rec OGT) and 1 mm UDP-GlcNAc (Sigma) in 60 μl of O-GlcNAcylation buffer (50 mm Tris-HCl, 12.5 mm MgCl2, 1 mm DTT, pH 7.5) overnight at room temperature. The reactions were stopped by 2× SDS sample-loading buffer, and proteins were separated by SDS-PAGE and subjected to immunoblotting for O-GlcNAc.
Mass spectrometry
Proteins were resolved by SDS-PAGE and stained with Coomassie Blue G-250. Gel bands were excised from the SDS-PAGE. Protein was reduced in 10 mm DTT for 30 min at 50 °C and then alkylated with 20 mm iodoacetamide for 45 min in the dark. Protein cleavage was achieved by overnight incubation with CNBr dissolved in 0.1 m HCl or chymotrypsin. Chromatography was performed using a Nanoacquity HPLC (Waters) at a flow rate of 300 nl/min. The column was a BEH130 C18, 75-μm inner diameter × 150 mm (Waters), and a 60-min gradient was employed. Solvent A was water, 0.1% formic acid, and solvent B was acetonitrile, 0.1% formic acid, and peptides were eluted by a gradient from 2 to 28% solvent B over 34 min followed by a short wash at 50% solvent B before returning to starting conditions. Peptide components eluted over a period of about 30 min during these runs.
MS was performed using an LTQ-orbitrap Velos with the ability to perform ETD analysis (Thermo). After a precursor scan of intact peptides was measured in the orbitrap by scanning from m/z 350 to 1400, the three most intense multiply charged precursors were selected for both HCD and ETD analysis, with HCD fragments measured in the orbitrap, whereas ETD fragments were measured in the ion trap. Activation times were set as 100 ms for both HCD and ETD fragmentation, but charge state-dependent activation was employed for ETD data. Supplemental activation of the charge-reduced species was employed in the ETD analysis to improve fragmentation. Dynamic exclusion for 60 s was employed to prevent repeated analysis of the same components.
Statistical analysis
All of the quantitative data are presented as means ± S.D. The statistical significance of differences was determined using Student's two-tailed t test in two groups and one-way analysis of variance in multiple groups. A p value ≤ 0.05 was considered statistically significant.
Author contributions
Z. M. and K. V. designed the study and wrote the manuscript. Z. M. performed and analyzed the experiments. K. V. supervised the experiments. R. J. C. performed mass spectrometry experiments and analyzed the data in Fig. 2. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Dr. Suzanne Walker for providing human ncOGT bacterial expression construct, Dr. Lance Wells for plenti4-HA-OGT and plenti4-lacZ plasmids, and Dr. Jianhua Yang for human p65 cDNA and p65-deficient MEF cells.
The authors declare that they have no conflicts of interest with the contents of this article.
- O-GlcNAc
- O-linked β-N-acetylglucosamine
- OGT
- O-GlcNAc transferase
- ncOGT
- nucleocytoplasmic OGT
- Rec OGT
- recombinantly expressed and immobilized metal-affinity chromatography-purified ncOGT
- OGA
- O-GlcNAcase
- CBP
- CREB-binding protein
- CREB
- cAMP-response element-binding protein
- HCD
- higher-energy collision dissociation
- ETD
- electron transfer dissociation
- MEF
- mouse embryo fibroblast
- IP
- immunoprecipitation
- WB
- Western blotting
- NButGT
- 1,2-dideoxy-2′-propyl-α-d-glucopyranoso-[2,1-d]-Δ2′-thiazoline.
References
- 1. Hart G. W., Housley M. P., and Slawson C. (2007) Cycling of O-linked β-N-acetylglucosamine on nucleocytoplasmic proteins. Nature 446, 1017–1022 [DOI] [PubMed] [Google Scholar]
- 2. Wells L., Vosseller K., and Hart G. W. (2001) Glycosylation of nucleocytoplasmic proteins: signal transduction and O-GlcNAc. Science 291, 2376–2378 [DOI] [PubMed] [Google Scholar]
- 3. Hart G. W., Slawson C., Ramirez-Correa G., and Lagerlof O. (2011) Cross talk between O-GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease. Annu. Rev. Biochem. 80, 825–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ma Z., and Vosseller K. (2013) O-GlcNAc in cancer biology. Amino Acids 45, 719–733 [DOI] [PubMed] [Google Scholar]
- 5. Ma Z., and Vosseller K. (2014) Cancer metabolism and elevated O-GlcNAc in oncogenic signaling. J. Biol. Chem. 289, 34457–34465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Housley M. P., Rodgers J. T., Udeshi N. D., Kelly T. J., Shabanowitz J., Hunt D. F., Puigserver P., and Hart G. W. (2008) O-GlcNAc regulates FoxO activation in response to glucose. J. Biol. Chem. 283, 16283–16292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Housley M. P., Udeshi N. D., Rodgers J. T., Shabanowitz J., Puigserver P., Hunt D. F., and Hart G. W. (2009) A PGC-1α-O-GlcNAc transferase complex regulates FoxO transcription factor activity in response to glucose. J. Biol. Chem. 284, 5148–5157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ruan H. B., Han X., Li M. D., Singh J. P., Qian K., Azarhoush S., Zhao L., Bennett A. M., Samuel V. T., Wu J., Yates J. R. 3rd, and Yang X. (2012) O-GlcNAc transferase/host cell factor C1 complex regulates gluconeogenesis by modulating PGC-1α stability. Cell Metab. 16, 226–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Guinez C., Filhoulaud G., Rayah-Benhamed F., Marmier S., Dubuquoy C., Dentin R., Moldes M., Burnol A. F., Yang X., Lefebvre T., Girard J., and Postic C. (2011) O-GlcNAcylation increases ChREBP protein content and transcriptional activity in the liver. Diabetes 60, 1399–1413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ma Z., Vocadlo D. J., and Vosseller K. (2013) Hyper-O-GlcNAcylation is anti-apoptotic and maintains constitutive NF-κB activity in pancreatic cancer cells. J. Biol. Chem. 288, 15121–15130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. James L. R., Tang D., Ingram A., Ly H., Thai K., Cai L., and Scholey J. W. (2002) Flux through the hexosamine pathway is a determinant of nuclear factor κB- dependent promoter activation. Diabetes 51, 1146–1156 [DOI] [PubMed] [Google Scholar]
- 12. Kamemura K., Hayes B. K., Comer F. I., and Hart G. W. (2002) Dynamic interplay between O-glycosylation and O-phosphorylation of nucleocytoplasmic proteins: alternative glycosylation/phosphorylation of THR-58, a known mutational hot spot of c-Myc in lymphomas, is regulated by mitogens. J. Biol. Chem. 277, 19229–19235 [DOI] [PubMed] [Google Scholar]
- 13. Musicki B., Kramer M. F., Becker R. E., and Burnett A. L. (2005) Inactivation of phosphorylated endothelial nitric oxide synthase (Ser-1177) by O-GlcNAc in diabetes-associated erectile dysfunction. Proc. Natl. Acad. Sci. U.S.A. 102, 11870–11875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen Y. X., Du J. T., Zhou L. X., Liu X. H., Zhao Y. F., Nakanishi H., and Li Y. M. (2006) Alternative O-GlcNAcylation/O-phosphorylation of Ser16 induce different conformational disturbances to the N terminus of murine estrogen receptor β. Chem. Biol. 13, 937–944 [DOI] [PubMed] [Google Scholar]
- 15. Yang W. H., Kim J. E., Nam H. W., Ju J. W., Kim H. S., Kim Y. S., and Cho J. W. (2006) Modification of p53 with O-linked N-acetylglucosamine regulates p53 activity and stability. Nat. Cell Biol. 8, 1074–1083 [DOI] [PubMed] [Google Scholar]
- 16. Cole R. N., and Hart G. W. (1999) Glycosylation sites flank phosphorylation sites on synapsin I: O-linked N-acetylglucosamine residues are localized within domains mediating synapsin I interactions. J. Neurochem. 73, 418–428 [DOI] [PubMed] [Google Scholar]
- 17. Hayden M. S., and Ghosh S. (2008) Shared principles in NF-κB signaling. Cell 132, 344–362 [DOI] [PubMed] [Google Scholar]
- 18. Karin M., and Greten F. R. (2005) NF-κB: linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 5, 749–759 [DOI] [PubMed] [Google Scholar]
- 19. Karin M. (2006) Nuclear factor-κB in cancer development and progression. Nature 441, 431–436 [DOI] [PubMed] [Google Scholar]
- 20. Zhong H., Voll R. E., and Ghosh S. (1998) Phosphorylation of NF-κB p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol. Cell 1, 661–671 [DOI] [PubMed] [Google Scholar]
- 21. Chen L. F., Williams S. A., Mu Y., Nakano H., Duerr J. M., Buckbinder L., and Greene W. C. (2005) NF-κB RelA phosphorylation regulates RelA acetylation. Mol. Cell Biol. 25, 7966–7975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hoberg J. E., Popko A. E., Ramsey C. S., and Mayo M. W. (2006) IκB kinase α-mediated derepression of SMRT potentiates acetylation of RelA/p65 by p300. Mol. Cell Biol. 26, 457–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen L. F., Mu Y., and Greene W. C. (2002) Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-κB. EMBO J. 21, 6539–6548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yang W. H., Park S. Y., Nam H. W., Kim D. H., Kang J. G., Kang E. S., Kim Y. S., Lee H. C., Kim K. S., and Cho J. W. (2008) NFκB activation is associated with its O-GlcNAcylation state under hyperglycemic conditions. Proc. Natl. Acad. Sci. U.S.A. 105, 17345–17350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Golks A., Tran T. T., Goetschy J. F., and Guerini D. (2007) Requirement for O-linked N-acetylglucosaminyltransferase in lymphocytes activation. EMBO J. 26, 4368–4379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Allison D. F., Wamsley J. J., Kumar M., Li D., Gray L. G., Hart G. W., Jones D. R., and Mayo M. W. (2012) Modification of RelA by O-linked N-acetylglucosamine links glucose metabolism to NF-κB acetylation and transcription. Proc. Natl. Acad. Sci. U.S.A. 109, 16888–16893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Macauley M. S., Whitworth G. E., Debowski A. W., Chin D., and Vocadlo D. J. (2005) O-GlcNAcase uses substrate-assisted catalysis: kinetic analysis and development of highly selective mechanism-inspired inhibitors. J. Biol. Chem. 280, 25313–25322 [DOI] [PubMed] [Google Scholar]
- 28. Chalkley R. J., Hansen K. C., and Baldwin M. A. (2005) Bioinformatic methods to exploit mass spectrometric data for proteomic applications. Methods Enzymol. 402, 289–312 [DOI] [PubMed] [Google Scholar]
- 29. Chalkley R. J., Thalhammer A., Schoepfer R., and Burlingame A. L. (2009) Identification of protein O-GlcNAcylation sites using electron transfer dissociation mass spectrometry on native peptides. Proc. Natl. Acad. Sci. U.S.A. 106, 8894–8899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jacobs M. D., and Harrison S. C. (1998) Structure of an IκBα/NF-κB complex. Cell 95, 749–758 [DOI] [PubMed] [Google Scholar]
- 31. Hornbeck P. V., Kornhauser J. M., Tkachev S., Zhang B., Skrzypek E., Murray B., Latham V., and Sullivan M. (2012) PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 40, D261–D270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kinoshita E., Kinoshita-Kikuta E., Takiyama K., and Koike T. (2006) Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol. Cell Proteomics 5, 749–757 [DOI] [PubMed] [Google Scholar]
- 33. Huang B., Yang X. D., Lamb A., and Chen L. F. (2010) Posttranslational modifications of NF-κB: another layer of regulation for NF-κB signaling pathway. Cell. Signal. 22, 1282–1290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yang Y. R., Kim D. H., Seo Y. K., Park D., Jang H. J., Choi S. Y., Lee Y. H., Lee G. H., Nakajima K., Taniguchi N., Kim J. M., Choi E. J., Moon H. Y., Kim I. S., Choi J. H., et al. (2015) Elevated O-GlcNAcylation promotes colonic inflammation and tumorigenesis by modulating NF-κB signaling. Oncotarget 6, 12529–12542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Buerki C., Rothgiesser K. M., Valovka T., Owen H. R., Rehrauer H., Fey M., Lane W. S., and Hottiger M. O. (2008) Functional relevance of novel p300-mediated lysine 314 and 315 acetylation of RelA/p65. Nucleic Acids Res. 36, 1665–1680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rothgiesser K. M., Fey M., and Hottiger M. O. (2010) Acetylation of p65 at lysine 314 is important for late NF-κB-dependent gene expression. BMC Genomics 11, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liu X., Wang L., Zhao K., Thompson P. R., Hwang Y., Marmorstein R., and Cole P. A. (2008) The structural basis of protein acetylation by the p300/CBP transcriptional coactivator. Nature 451, 846–850 [DOI] [PubMed] [Google Scholar]
- 38. Wang B., Wei H., Prabhu L., Zhao W., Martin M., Hartley A. V., and Lu T. (2015) Role of novel serine 316 phosphorylation of the p65 subunit of NF-κB in differential gene regulation. J. Biol. Chem. 290, 20336–20347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bergqvist S., Alverdi V., Mengel B., Hoffmann A., Ghosh G., and Komives E. A. (2009) Kinetic enhancement of NF-κBxDNA dissociation by IκBα. Proc. Natl. Acad. Sci. U.S.A. 106, 19328–19333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cervantes C. F., Bergqvist S., Kjaergaard M., Kroon G., Sue S. C., Dyson H. J., and Komives E. A. (2011) The RelA nuclear localization signal folds upon binding to IκBα. J. Mol. Biol. 405, 754–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kawauchi K., Araki K., Tobiume K., and Tanaka N. (2009) Loss of p53 enhances catalytic activity of IKKβ through O-linked β-N-acetyl glucosamine modification. Proc. Natl. Acad. Sci. U.S.A. 106, 3431–3436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ramakrishnan P., Clark P. M., Mason D. E., Peters E. C., Hsieh-Wilson L. C., and Baltimore D. (2013) Activation of the transcriptional function of the NF-κB protein c-Rel by O-GlcNAc glycosylation. Sci. Signal. 6, ra75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fan Y., Mao R., Zhao Y., Yu Y., Sun W., Song P., Shi Z., Zhang D., Yvon E., Zhang H., Fu S., and Yang J. (2009) Tumor necrosis factor-α induces RelA degradation via ubiquitination at lysine 195 to prevent excessive nuclear factor-κB activation. J. Biol. Chem. 284, 29290–29297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Weeks S. D., Drinker M., and Loll P. J. (2007) Ligation independent cloning vectors for expression of SUMO fusions. Protein Expr. Purif. 53, 40–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gross B. J., Kraybill B. C., and Walker S. (2005) Discovery of O-GlcNAc transferase inhibitors. J. Am. Chem. Soc. 127, 14588–14589 [DOI] [PubMed] [Google Scholar]
- 46. Yuzwa S. A., Yadav A. K., Skorobogatko Y., Clark T., Vosseller K., and Vocadlo D. J. (2011) Mapping O-GlcNAc modification sites on tau and generation of a site-specific O-GlcNAc tau antibody. Amino Acids 40, 857–868 [DOI] [PubMed] [Google Scholar]