

Abstract
Calcium-activated chloride channels (CaCCs) are key players in transepithelial ion transport and fluid secretion, smooth muscle constriction, neuronal excitability, and cell proliferation. The CaCC regulator 1 (CLCA1) modulates the activity of the CaCC TMEM16A/Anoctamin 1 (ANO1) by directly engaging the channel at the cell surface, but the exact mechanism is unknown. Here we demonstrate that the von Willebrand factor type A (VWA) domain within the cleaved CLCA1 N-terminal fragment is necessary and sufficient for this interaction. TMEM16A protein levels on the cell surface were increased in HEK293T cells transfected with CLCA1 constructs containing the VWA domain, and TMEM16A-like currents were activated. Similar currents were evoked in cells exposed to secreted VWA domain alone, and these currents were significantly knocked down by TMEM16A siRNA. VWA-dependent TMEM16A modulation was not modified by the S357N mutation, a VWA domain polymorphism associated with more severe meconium ileus in cystic fibrosis patients. VWA-activated currents were significantly reduced in the absence of extracellular Mg2+, and mutation of residues within the conserved metal ion-dependent adhesion site motif impaired the ability of VWA to potentiate TMEM16A activity, suggesting that CLCA1-TMEM16A interactions are Mg2+- and metal ion-dependent adhesion site-dependent. Increase in TMEM16A activity occurred within minutes of exposure to CLCA1 or after a short treatment with nocodazole, consistent with the hypothesis that CLCA1 stabilizes TMEM16A at the cell surface by preventing its internalization. Our study hints at the therapeutic potential of the selective activation of TMEM16A by the CLCA1 VWA domain in loss-of-function chloride channelopathies such as cystic fibrosis.
Keywords: chloride channel, cystic fibrosis, drug development, magnesium, protein-protein interaction, TMEM16A, calcium-activated chloride channel, calcium-activated chloride channel regulator 1 (CLCA1), metal ion-dependent adhesion site (MIDAS)
Introduction
Calcium-activated chloride channels (CaCCs)4 are key players in transepithelial ion transport and fluid secretion, smooth muscle constriction, neuronal excitability, and cell proliferation (1, 2). The first member of the family to be identified, TMEM16A/Anoctamin 1 (ANO1) (3–5), is found in airway epithelia, submucosal glands, and smooth muscle, and increased TMEM16A activity has been associated with chronic obstructive pulmonary disease and asthma manifestations such as mucus cell metaplasia and mucus hypersecretion (3, 6, 7). Conversely, targeted potentiation of TMEM16A has been suggested as a mechanism to treat certain channelopathies, such as cystic fibrosis (CF) (8). Calcium-activated chloride channel regulator (CLCA) proteins are self-cleaving metalloproteases that activate calcium-dependent chloride currents (ICaCC) in mammalian cells (9). Initially annotated as CaCCs (10), further studies have shown that CLCAs are not channels but secreted, soluble proteins that modulate CaCCs endogenous to cells (11–14). CLCA1 is expressed in lung epithelia, where it plays a role in mucus homeostasis (15), and it has been linked to some of the same airway disease traits associated with TMEM16A (16). Overexpression of CLCA1 alleviates the intestinal disease meconium ileus in Cftr-deficient mice (17), and the CLCA1 variant S357N is found with high frequency in CF patients with aggravated intestinal disease (18), all of which suggests that CLCA1 might act as a modifier in the context of CF.
Recently, we uncovered a functional link between CLCA1 and TMEM16A (13, 14). Specifically, we demonstrated that the N-terminal fragment resulting from the self-proteolysis of CLCA1 (N-CLCA1) can activate ICaCC in mammalian cells (14) and that these currents are carried by TMEM16A, which interacts directly with secreted N-CLCA1 at the cell surface (13). In this study, we apply electrophysiological and imaging methods to further investigate the mechanisms underlying CLCA1-TMEM16A interactions. We report that the von Willebrand factor type A (VWA) domain within N-CLCA1 is sufficient to increase TMEM16A at the cell surface and TMEM16A-mediated current density and that the mechanism is dependent upon the metal ion-dependent adhesion site (MIDAS) motif within N-CLCA1 VWA. Up-regulation of TMEM16A activity by CLCA1 occurs in minutes and can be mimicked by treatment with the endocytosis inhibitor nocodazole, in support of our hypothesis that CLCA1 stabilizes TMEM16A at the cell surface by preventing its internalization (13). Our findings provide novel insights into CLCA1 and TMEM16A as a cooperative pair and have direct implications for the targeting of CLCA1-TMEM16A interactions in airway disease.
Results
The VWA domain of CLCA1 is necessary and sufficient to up-regulate TMEM16A activity
We demonstrated previously that N-CLCA1 binds to TMEM16A and that this interaction can increase the surface density of TMEM16A, thereby increasing TMEM16A currents (13, 14). Because N-CLCA1 contains several discrete protein domains (Fig. 1A), we set out to identify the minimal region required for interaction with TMEM16A. First, we transfected HEK293T cells with expression constructs composed of one or more N-CLCA1 structural domains (Fig. 1A) and tested them for TMEM16A activity by means of patch clamp electrophysiology and confocal microscopy-based immunofluorescence (Fig. 1, B–E). Robust, modestly outward-rectifying currents were activated in cells expressing full-length (CLCA1) or N-CLCA1 (Fig. 1, C and D), and we have demonstrated that these currents are carried by TMEM16A (13). Substantially smaller currents were measured in cells transfected with the empty pHLsec vector, consistent with the low endogenous levels of TMEM16A at the membrane of these cells (19, 20). In cells transfected with constructs containing the VWA domain, currents with the same biophysical characteristics and comparable density as those evoked in CLCA1- or N-CLCA1-transfected cells were activated (Fig. 1, C and D), and this was not affected by the absence of the β sheet-rich domain in the CAT + VWA construct, suggesting that this domain is not required for CLCA1-dependent TMEM16A regulation. In cells transfected with a construct containing only the metalloprotease domain (CAT), the currents were indistinguishable from those measured in mock-transfected cells (Fig. 1, C and D), suggesting that proteolytic cleavage of a downstream target is not part of the TMEM16A modulation mechanism, in agreement with our previous results (14). In immunofluorescence experiments, fixed and non-permeabilized cells transfected with constructs that included the VWA domain (CLCA1, N-CLCA1, CAT + VWA, or VWA alone) stained strongly for TMEM16A surface expression, whereas cells transfected with CAT or pHLsec displayed little or no TMEM16A staining (Fig. 1E), further suggesting that N-CLCA1 interactions with TMEM16A are mediated by the VWA domain. Thus, we conclude that the VWA domain contained within N-CLCA1 is required and sufficient to stabilize TMEM16A at the cell surface.
Figure 1.

The VWA domain of CLCA1 is necessary and sufficient to activate TMEM16A-like currents and to increase TMEM16A protein at the cell surface. A, top panel, CLCA1 is a self-cleaving zincin metalloprotease. The dashed line indicates the proteolytic cleavage site. Cat, metalloprotease catalytic domain; Cys, cysteine-rich domain; VWA, von Willebrand factor type A domain; BSR, β sheet-rich domain; FnIII, fibronectin type III domain. Bottom panel, schematic of constructs used to experimentally determine the minimal domain within CLCA1 required to modulate TMEM16A. B, HEK293T cells were transfected with empty pHLsec vector or with CLCA1 constructs as in A and assayed for TMEM16A functional expression by patch clamp electrophysiology and confocal microscopy imaging. C and D, whole-cell currents measured in cells superfused with standard extracellular solution and in the presence of 10 μm free Ca2+ in the pipette. C, representative current traces. The pulse protocol is shown at the top left. Outward currents are represented by upward deflections, and dotted lines indicate zero current. Membrane capacitance was similar in all cases at ∼25 pF. D, current density at +100 mV, measured at the end of the 600-ms voltage step. Symbols represent data from individual cells (n = 19–45); error bars indicate the means ± S.E. of all experiments. Statistical differences are indicated by different lowercase letters; i.e. a group labeled with a given letter is statistically similar to any other group labeled with the same letter but significantly different from any other group labeled differently (p < 0.05, one-way ANOVA, F = 16 and p = 4 × 10−13, followed by the Tukey test). E, immunofluorescence staining of TMEM16A (green) and the cell surface marker WGA (blue).
Secreted CLCA1 VWA can activate TMEM16A-mediated currents and increase TMEM16A surface levels
In our previous studies, we showed that N-CLCA1 can be applied exogenously to potentiate the functional expression of TMEM16A in HEK293T cells (13). We therefore investigated whether the isolated VWA domain can elicit the same responses, and we also examined the cellular phenotype of S357N, a common human polymorphism within the CLCA1 VWA domain that has been associated with more severe meconium ileus in CF patients (18) (Fig. 2). We cultured untransfected cells or cells transfected with negative control scrambled RNAi (siControl) or TMEM16A siRNA overnight in medium obtained from cells transfected with either empty pHLsec vector or wild-type or S357N VWA and then assayed them for TMEM16A protein and activity as above (Fig. 2A). Typical CLCA1-dependent currents were activated in untransfected cells exposed to secreted WT or S357N VWA (Fig. 2B); the current density was similar in both cases (Fig. 2C) and comparable with that observed in VWA-transfected cells (Fig. 1D). Conversely, only small background currents were measured in mock-conditioned untransfected cells (Fig. 2, B and C). TMEM16A staining was noticeably increased in untransfected cells conditioned with either WT or S357N VWA (Fig. 2D), and currents were knocked down to near background levels in WT or mutant VWA-conditioned cells transfected with TMEM16A siRNA but remained unchanged in cells transfected with siControl (Fig. 2, B and C). These results further support the hypothesis that the VWA domain of CLCA1 is critical for CLCA1-TMEM16A interactions and suggest that external application of the CLCA1 VWA domain may be an effective means of potentiating TMEM16A activity. Finally, VWA activity was not modified by the S357N mutation, suggesting that the association of CLCA1 S357N with the etiology of meconium ileus, if any, is unrelated to TMEM16A channel regulation.
Figure 2.

Secreted CLCA1 VWA domain up-regulates TMEM16A. A, untransfected cells or cells transfected with negative control RNAi (siControl) or TMEM16A siRNA were cultured in medium from pHLsec-transfected cells or from cells expressing CLCA1 VWA, either WT or cystic fibrosis-associated variant S357N, and subsequently assayed for TMEM16A currents and surface staining. B and C, whole-cell currents measured under standard extracellular and pipette conditions. B, representative current traces, displayed as in Fig. 1C. The pulse protocol is shown at the bottom right. Membrane capacitance was similar in all cases at ∼25 pF. C, current density at +100 mV. Symbols represent data from individual cells (n = 9–31); error bars indicate the means ± S.E. of all experiments. The results of the statistical analysis are indicated by lowercase letters; i.e. groups sharing letters are statistically similar (for example, groups labeled a and ab), whereas those not sharing any letters are significantly different (for example, groups labeled a and bc) (p < 0.05, one-way ANOVA, F = 11 and p = 2 × 10−9, followed by the Tukey test). D, immunofluorescence staining of TMEM16A (green), CLCA1 (red, anti-human CLCA1 antibody 2F4), and the cell surface marker WGA (blue) in untransfected cells incubated in pHLsec-, WT, or S357N VWA-conditioned medium.
CLCA1 VWA-mediated current activation is dependent on extracellular Mg2+ and is impacted by mutations within the MIDAS motif
VWA domains, commonly found in extracellular adhesion proteins, are implicated in extracellular protein-protein interactions, largely via a MIDAS motif consisting of up to five polar or negatively charged residues that coordinate a divalent cation, usually Mg2+ (21). Sequence alignments and structure prediction (Fig. 3A) indicate that the CLCA1 VWA domain contains a five-residue (“perfect”) MIDAS motif (21), corresponding to Asp-312, Ser-314, Ser-316, Thr-383, and Asp-412 (Fig. 3A), and we investigated whether CLCA1-TMEM16A interactions are mediated by this motif (Fig. 3, B–F). We observed that CLCA1 VWA-dependent current activation requires extracellular Mg2+; thus, the current density in cells incubated overnight with CLCA1 VWA-conditioned medium dropped to background levels when experiments were performed in Mg2+-free extracellular solution (Fig. 3, B and C). More conclusively, disruption of the MIDAS motif, in particular mutation of Ser-316 or Thr-383 to Ala, reduced the ability of CLCA1 VWA to modulate surface protein levels and activity of TMEM16A (Fig. 3, D–F). The current density in cells treated with D312A/S314A or D312A/S314A/D412 VWA-conditioned medium was comparable with that measured in cells exposed to wild-type VWA but decreased ∼30% in cells conditioned with D312A/S314A/S316A or D312A/S314A/S316A/D412A VWA and to background levels in cells conditioned with D312A/S314A/S316A/T383A VWA (Fig. 3, D and E). Compared with cells conditioned with WT, D312A/S314A, or D312A/S314A/D412 VWA, the increase in TMEM16A surface staining was also attenuated in cells exposed to D312A/S314A/S316A, D312A/S314A/S316A/D412A, and D312A/S314A/S316A/T383A (Fig. 3F). These findings suggest that CLCA1 VWA interacts with TMEM16A in a Mg2+- and MIDAS-dependent manner and that residues Ser-316 and Thr-383 within the MIDAS motif are critical for these interactions.
Figure 3.

A MIDAS motif within the CLCA1 VWA domain is implicated in the CLCA1-TMEM16A interactions. A, homology model of the CLCA1 VWA domain based on the VWA domain of C. acidiphila (PDB code 4FX5). Inset, the MIDAS pocket with an Mg2+ ion within. The critical residues Ser-316 and Thr-383 are highlighted in red. B and C, whole-cell currents measured in wild-type CLCA1 VWA- (WT VWA) or mock-conditioned (pHLsec) cells superfused with standard ([1 mm Mg2+]out) or Mg2+-free ([0 mm Mg2+]out) extracellular solution. D and E, whole-cell currents measured in mock-conditioned cells and in cells conditioned with WT or MIDAS motif mutant VWA superfused with standard extracellular solution. B and D, representative current traces displayed as in Fig. 1C. The pulse protocol is the same as in Figs. 1 and 2. The membrane capacitance was similar in all cases at ∼25 pF. C and E, current density at +100 mV. Symbols are data from individual cells (C, n = 6–25; E, n = 18–30); error bars indicate the means ± S.E. of all experiments. The results of the statistical analysis are indicated by lowercase letters; i.e. groups sharing letters are statistically similar (for example, groups labeled a and ab or groups labeled ab and bc), whereas those not sharing any letters are significantly different (for example, groups labeled a and b or groups labeled ab and c) (p < 0.05, one-way ANOVA; C, F = 13 and p = 3 × 10−9; E, F = 12 and p = 1 × 10−10; followed by Tukey test). F, immunofluorescence staining of TMEM16A (red) in pHLsec-, WT, or MIDAS motif mutant VWA-conditioned medium.
CLCA1-dependent modulation of TMEM16A activity occurs within minutes and is mimicked by treatment with the internalization inhibitor nocodazole
We demonstrated previously that application of exogenous CLCA1 to HEK293T cells increases TMEM16A-mediated current density and TMEM16A surface staining without changing total TMEM16A protein levels. Because we found that CLCA1 directly binds TMEM16A at the cell surface, we hypothesize that this interaction may prevent recycling of the channel (13). The effects of CLCA1 on TMEM16A surface levels and function are relatively rapid (Fig. 4), consistent with a mechanism that is independent of protein synthesis. Strong TMEM16A staining was detected and maximized after just 30 min of incubation in CLCA1-conditioned medium (Fig. 4A). In patch clamp assays (Fig. 4, B–E), robust, slightly outward-rectifying currents were activated ∼1–3 min after application of purified CLCA1 VWA protein to the extracellular solution and approached steady state ∼5–7 min later (Fig. 4, C and D); the maximum current density in VWA-treated cells was up to 7-fold higher than in cells exposed to protein-free buffer (Fig. 4E). The microtubule destabilizer nocodazole disrupts microtubule-dependent internalization of channels and other membrane proteins (22–24), thus preventing the recycling of these proteins from the cell surface. In cells treated with nocodazole, but not in those treated with vehicle (0.5% DMSO), currents with properties similar to those observed in cells transfected or conditioned with CLCA1 (see, for example, Refs. 13, 14 and Figs. 1–3) were activated (Fig. 5). The current density was significantly knocked down in nocodazole-treated cells transfected with TMEM16A siRNA in contrast to those transfected with siControl (Fig. 5). Together, these data support the hypothesis that CLCA1 stabilizes TMEM16A at the cell surface by impeding its reinternalization.
Figure 4.

CLCA1-dependent modulation of TMEM16A activity occurs within minutes. A, immunofluorescence staining of TMEM16A (red), CLCA1 (green, anti-human CLCA1 antibody 1228), and the cell surface marker WGA (blue) in untransfected cells incubated in CLCA1-conditioned medium for a few seconds (0 min) or 30 or 120 min. B–E, time course of whole-cell current activation by purified CLCA1 VWA protein. B, experiments were performed under standard extracellular and pipette conditions. A descending voltage ramp was applied every 10 s for the duration of the experiment. C, current density at +80 mV measured over time in two example cells (membrane capacitance ∼20 pF). After stable conditions were achieved, 5 μl of purified CLCA1 VWA protein (final concentration, 5 μm) or dialysis buffer B was added to the bath (arrow); currents immediately before addition of protein or buffer (a) and at the post-application steady state (b) are shown in color. D, current/voltage relationships at a and at b for the examples shown in C. E, current density at +80 mV in three individual buffer-treated cells and in three individual VWA-treated cells measured at selected time points, immediately before (0 min) or up to 9 min after addition of protein or buffer. Each cell is represented with unique symbols; the colored symbols are the same as in C.
Figure 5.

Nocodazole increases TMEM16A current density. HEK293T cells, untransfected or transfected with negative control RNAi (siControl) or TMEM16A siRNA, were treated for 30 min with the microtubule polymerization inhibitor nocodazole (20 μg/ml) or with vehicle (DMSO). Whole-cell currents were measured under standard extracellular and pipette conditions and using the same pulse protocol as for Figs. 1–3. A, representative current traces. B, current density at +100 mV. Symbols are data from individual cells (n = 10–20); error bars are the means ± S.E. of all experiments. Statistical differences are indicated by different lowercase letters, i.e. a group labeled with a given letter is statistically similar to any other group labeled with the same letter but significantly different from any other group labeled differently (p < 0.05, one-way ANOVA, F = 11 and p = 2 × 10−5, followed by the Tukey test).
Discussion
The VWA domain in N-CLCA1 is the minimal requirement for interaction with TMEM16A
Here we demonstrate that the CLCA1 VWA domain is responsible for mediating the interaction with TMEM16A, resulting in increased TMEM16A at the cell surface and increased ICaCC density (Figs. 1–4). VWA domains mediate protein-protein interactions important for cell adhesion and signaling in extracellular matrix proteins, such as integrins and collagens, but are also found in auxiliary subunits of voltage-gated Ca2+ (CaV) channels (21). A common mechanism of VWA domain-dependent protein-protein interactions involves the coordination of a divalent cation, usually Mg2+, by a MIDAS motif at the binding interface (21). However, there are examples of VWA-mediated interactions in which surfaces other than the MIDAS are implicated (25–27). Our results indicate that the CLCA1 VWA-TMEM16A interaction is, at least in part, dependent on both Mg2+ and the perfect MIDAS motif within the VWA domain of CLCA1 (Fig. 3). These observations draw intriguing comparisons with the α2δ subunits of CaV channels, in particular CaV1 and CaV2 (28). Like CLCAs, α2δ proteins are posttranslationally cleaved into two fragments, α2 and δ (29), and modulate Ca2+ currents through functional and structural association with α1 pore-forming subunits (30, 31). Both α2δ-1 and α2δ-2 contain VWA domains with a perfect MIDAS motif that is required for increasing Ca2+ current density and CaV channel complex surface expression (30, 32, 33). However, unlike N- and C-CLCA1, the α2 and the δ fragments remain linked by a disulfide bond after cleavage (34) and are likely associated with the plasma membrane though a predicted glycosylphosphatidylinositol (GPI) anchor site at the C terminus of δ (35, 36). CaVα1-α2δ interactions seem to occur during the intracellular protein maturation process (32), whereas CLCA1 can interact with TMEM16A after it has been secreted, either by the same cell or by neighboring cells (13). Similar to CLCA1, a major mechanism for α2δ subunit-dependent modulation of Ca2+ currents is the increase of α1 subunit and CaV channel complex surface expression (28), but, unlike CLCA1, there is substantial evidence that α2δ subunits also inform the biophysical and pharmacological properties of CaV channels (37).
Mechanism of TMEM16A stabilization by CLCA1
Our data indicate that exogenously applied CLCA1 increases TMEM16A surface levels and, thus, ICaCC through the channel. This up-regulation occurs within minutes (Fig. 4), suggesting that it is not dependent on protein synthesis but, rather, draws from the existing pool of TMEM16A protein in the cell. TMEM16A channels are functional oligomeric homodimers, i.e. each of the two identical subunits defines its own conduction pore (38–40), but oligomerization is required for channel assembly and surface expression (41, 42); TMEM16A likely forms high-affinity dimers, as the purified protein elutes as a dimer on gel filtration columns (43), and the first crystal structure of a fungal orthologue revealed a dimer (44). Thus, it is unlikely that CLCA1 engages monomeric TMEM16A and drives dimerization, but it may stabilize the dimer at the cell surface, perhaps through a conformational change that masks a reinternalization signal. Treatment with nocodazole, an inhibitor of microtubule-dependent internalization, recapitulates the effect of exogenous application of CLCA1 (Fig. 5), suggesting that CLCA1 may prevent the recycling of TMEM16A channels. Interestingly, although the α2δ subunits of CaV channels are thought to act mostly by enhancing forward trafficking of the channel complexes (45, 46), there is evidence that they may also delay their turnover (47), but the mechanisms involved remain unclear.
A model for CLCA1-TMEM16A interactions
Based on our previous findings (13, 14) and the results reported here, we propose the following model for the up-regulation of TMEM16A-mediated ICaCC by secreted CLCA1 (Fig. 6). CLCA1 is synthesized as a full-length protein, and, in this form, the VWA domain of CLCA1 is prevented from interacting with TMEM16A by the C-terminal region of the protein (Fig. 6A); accordingly, disruption of the catalytic or cleavage sites impairs the ability of CLCA1 to activate ICaCC (14). This masking of the VWA domain might be achieved through interaction with the fibronectin III (FnIII) domain found in the C-terminal fragment (Fig. 1A), as FnIII domains are known ligands for VWA domains (21). Upon proteolytic self-cleavage, the CLCA1 VWA domain is exposed and free to engage TMEM16A, likely via the last extracellular (α9-α10) loop of the channel (Fig. 6A); thus, an antibody raised against this region of TMEM16A blocks the binding of N-CLCA1 to TMEM16A-expressing cells (13). The VWA domain of secreted N-CLCA1 binds and stabilizes TMEM16A at the cell surface, likely by preventing its reinternalization (Fig. 6B), thereby leading to increased TMEM16A surface expression and TMEM16A-mediated ICaCC density.
Figure 6.
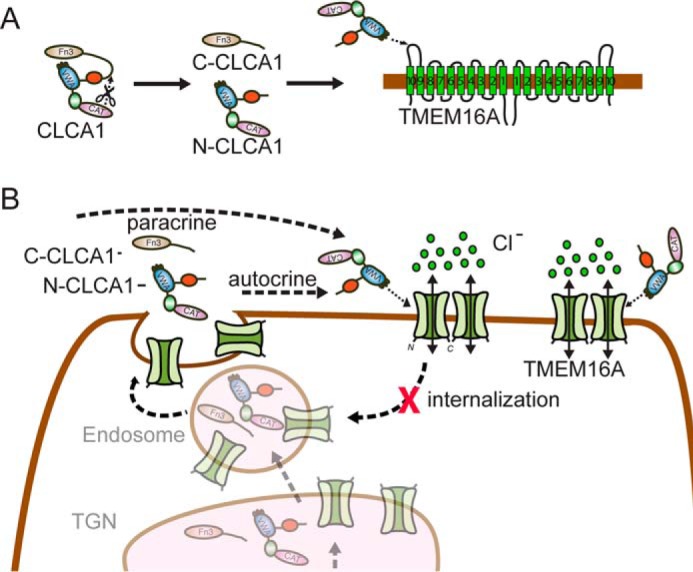
Model for CLCA1-mediated activation of TMEM16A. A, in full-length CLCA1, the VWA domain within the N-terminal region of the protein (N-CLCA1) is unable to engage TMEM16A, possibly because of a self-interaction with the FnIII domain in the C-terminal portion (C-CLCA1) that masks the VWA. Following self-cleavage, N-CLCA1 and C-CLCA1 likely dissociate, allowing the N-CLCA1 VWA domain to directly engage TMEM16A, likely via the α9-α10 loop of the channel. B, N-CLCA1 secreted by the same cell (autocrine) or by adjacent cells (paracrine) binds and stabilizes TMEM16A at the cell surface, likely by preventing its recycling, thereby increasing TMEM16A surface expression and ICaCC density.
Implications for airway disease
CLCA1 has been proposed as a modifier gene in the context of CF. Cftr−/− mice, which mostly die from severe meconium ileus, show decreased expression of Clca1 in the intestine, but up-regulation of Clca1 in these mice ameliorates the intestinal phenotype and improves survival (17). One report linked the S357N variant within the CLCA1 VWA domain to increased risk of developing meconium ileus in a subset of CF patients (18). However, this variant had no effect on VWA; neither TMEM16A current activation (Fig. 2, B and C) nor TMEM16A surface staining (Fig. 2D) were altered by S357N, suggesting that any potential contribution of this mutation to the development of meconium ileus is not linked to decreased ability of the VWA domain to engage TMEM16A.
A significant gap in our knowledge still exists regarding the cooperative roles of CLCA1 and TMEM16A in physiology and disease (48). For example, both proteins are up-regulated in mucus cell metaplasia (7, 49), a hallmark of asthma and chronic obstructive pulmonary disease, but no mechanistic link has yet been established. Increased TMEM16A activity is associated with pulmonary hypertension in a rat model of the disease (50), but whether CLCA1 is involved remains to be addressed. Both CLCA1 and TMEM16A have been proposed as therapeutic targets for airway disease (9), and our results suggest that they may be used in tandem to selectively activate anion currents. This may be advantageous in the case of certain channelopathies, such as CF, which arises from impaired Cl− and HCO3− conductance in mucosal epithelia because of partial or total loss of function of the cystic fibrosis transmembrane conductance regulator (CFTR). Therapeutic approaches have mostly focused on the symptoms, but significant efforts are being made to target the defective CFTR itself. Approximately 2000 mutations in the CFTR gene have been found that disrupt CFTR function by one of six general mechanisms, including defective trafficking, folding, and gating of the channel protein (51). Although a few mutation-specific therapies are available (52), the activation of an alternate anion channel, such as TMEM16A, would be agnostic to the CFTR mutation and, thus, a potentially universal approach. We show that exogenously applied CLCA1, specifically the VWA domain including a classic protein-protein interaction MIDAS motif, directly binds and stabilizes TMEM16A at the cell surface, thereby increasing Cl− conductance. This raises the intriguing possibility that CLCA1 VWA, or a peptide derived from it, might be developed as a drug to compensate for decreased or absent CFTR conductance in CF patients.
Experimental procedures
Reagents
Commercial antibodies were used according to the specifications of the manufacturer. Primary antibodies were as follows: goat-anti-human-TMEM16A polyclonal antibody S-20 raised against a 15- to 20-amino acid peptide within residues 820-870, corresponding to the last (α9-α10) extracellular loop (Santa Cruz Biotechnology, Santa Cruz, CA); rabbit anti-human CLCA1 polyclonal antibody 1228 (Biosystems, Rockford, IL); and wheat germ agglutinin (WGA)-Alexa Fluor 633 conjugate (Life Technologies). Secondary antibodies were as follows: donkey anti-goat IgG-Alexa Fluor 488, donkey anti-rabbit IgG-Alexa Fluor 488, donkey anti-mouse IgG-Alexa Fluor 555, and donkey anti-goat IgG-Alexa Fluor 594 conjugates (all from Life Technologies). Mouse anti-human CLCA1 monoclonal antibody 2F4 was produced in-house and used as described previously (14, 16). Nocodazole (Sigma-Aldrich, St. Louis, MO) was prepared as a 2 mg/ml stock solution in DMSO and was used at a working concentration of 20 μg/ml.
Cloning and mutagenesis
All human CLCA1 cDNAs were cloned into the pHLsec vector, which contains an optimized secretion signal and a C-terminal hexahistidine tag (53). Constructs with the cDNA sequences for full-length CLCA1 (22–914), the CLCA1 N-terminal fragment (N-CLCA1, 22–694), or the following CLCA1 domains were prepared (Fig. 1A): CAT (22–293), including the metalloprotease catalytic domain; VWA (294–478), containing von Willebrand factor type A; and CAT + VWA (22–478). Domain boundaries were predicted by means of the Phyre2 Protein Fold Recognition Server (54). Mutations were introduced by means of QuikChange Lightning (Agilent, Santa Clara, CA). Constructs were verified by sequencing. Expression and secretion of each of the different CLCA1 proteins were confirmed by Western blotting (supplemental Fig. S1).
Heterologous expression of CLCA1
HEK293T cells were cultured in 6-well dishes in Dulbecco's modified Eagle's medium (Life Technologies) supplemented with 10% fetal bovine serum, 105 units/liter penicillin, 100 mg/liter streptomycin, 1% glutamine, and 1% non-essential amino acids (Corning Inc., Corning, NY) at 37 °C and 5% CO2. Cells were transfected at 80% confluency with 293fectin transfection reagent (Life Technologies) at a 1:2 ratio (micrograms of DNA:microliters of 293fectin) using 1–2 μg of plasmid DNA per 1 million cells. Experiments were conducted in cells that were transiently transfected with the appropriate constructs or in cells that were exposed to exogenous CLCA1 protein by treatment with medium conditioned with CLCA1. For conditioned medium experiments, cells were transfected with either CLCA1 constructs or empty pHLsec vector (pHLsec). After 6 h, the transfection medium was removed, cells were washed with sterile PBS, and fresh medium was applied. Following 24–48 h of incubation, medium from these cells was harvested and centrifuged gently (1500 rpm, 5 min) to remove non-adherent cells. Untransfected cells were plated at low density onto UV-sterilized, 8-mm round German glass coverslips (Electron Microscopy Sciences, Hatfield, PA) and incubated for up to 24 h in 2 ml of cleared CLCA1- or pHLsec-conditioned medium supernatants.
siRNA knockdown of TMEM16A
Cells plated in 48-well plates were transfected with either 200 nm TMEM16A siRNA (HSS123904, 5′-AAG UUA GUG AGG UAG GCU GGG AAC C-3′, Life Technologies) or 200 nm medium GC content Stealth RNAi negative control (12935300, 5′-GGU UCC CAG CCU ACC UCA CUA ACU U-3′, Life Technologies) using Lipofectamine 2000 (Life Technologies) at a 20:2 ratio (picomoles of siRNA:microliters of Lipofectamine 2000). 24 h later, cells were plated onto round coverslips and incubated for an additional 24 h in CLCA1- or pHLsec-conditioned medium as described above. TMEM16A knockdown was estimated at 60–70% as assayed by quantitative PCR.
Recombinant expression of CLCA1 VWA
The VWA domain of CLCA1 was expressed in 293F cells via transient transfection with Hype-5 (OZ Biosciences, San Diego, CA) at a 1:1.5 ratio (micrograms of DNA:microliters of Hype-5) using 1 μg of plasmid per 1 million cells. Media from supernatants were harvested after 72 h. Protein was purified from medium supernatant using nickel-nitrilotriacetic acid superflow resin (Qiagen, Hilden, Germany) and eluted in 5 ml of buffer A containing 50 mm K2HPO4 (pH 8), 300 mm NaCl, and 250 mm imidazole. Purified CLCA1 VWA was dialyzed into buffer B containing 20 mm HEPES and 150 mm NaCl (pH 7.4) and concentrated in a centrifuge concentrator to 1 mm, calculated from absorbance at 280 nm.
Whole-cell patch clamp recordings
Experiments were performed at 25 °C up to 24 h after transfection or incubation in conditioned medium. Micropipettes were prepared from non-heparinized hematocrit glass (Kimble-Chase, Vineland, NJ) on a horizontal puller (Sutter Instruments, Novato, CA) and filled to a typical electrode resistance of 3 megohms with a pipette solution containing 150 mm N-methyl-d-glucamine chloride, 10 mm Hepes, 2 mm MgCl2, 8 mm HEDTA, and 5.8 mm CaCl2 to attain 10 μm free Ca2+, as calculated by means of the CaBuf program (available through Katholieke Universiteit Leuven). The pH of the pipette solution was adjusted to 7.1 with Tris. The standard bath solution was 150 mm NaCl, 10 mm Hepes, 1 mm CaCl2, and 1 mm MgCl2 and adjusted to pH 7.4 with Tris. Selected experiments were performed in the absence of extracellular Mg2+ with a bath solution composed of 150 mm NaCl, 10 mm Hepes, 1 mm Na-EDTA, and 2 mm CaCl2 to maintain the extracellular free Ca2+ at 1 mm (Fig. 3, B and C). After formation of a gigaohm seal and establishment of the whole-cell configuration, cells were voltage-clamped at 0 mV. For most experiments, a pulse protocol was applied in which the membrane potential was held at 0 mV for 50 ms and stepped to a test value (−100 to +100 mV in 20-mV increments) for 600 ms before returning to the holding potential for an additional 400 ms. For time course experiments (Fig. 4), a 400-ms descending voltage ramp protocol (+80 to −80 mV in 400 ms) was applied every 10 s, and were currents continuously recorded. Following a brief period of stabilization (∼1 min after break-in), a 5 μm bolus of purified CLCA1 VWA protein or the equivalent volume of buffer B (5 μl) was added to the bath, and the response was monitored until a steady state was reached. Membrane capacitance was calculated from the integral of the current transient in response to 10-mV depolarizing pulses and monitored for stability throughout the experiments. Patch clamp data were filtered at 2 kHz, and signals were digitized at 5 kHz with a Digidata 1322A (Molecular Devices, Sunnyvale, CA). MultiClamp 700B Commander and pClamp software (Molecular Devices) were used for pulse or ramp protocol application and data acquisition. Data were analyzed using Clampfit 10.1 (Molecular Devices). Results are presented as mean ± S.E. Statistical differences between groups were assessed by one-way ANOVA and post hoc all-pairwise Tukey test (Prism 5.0c, GraphPad Software, San Diego, CA). The results of the statistical analysis are indicated in the figures by a lowercase letter system, whereby groups that share letters are statistically similar (for example, “a” and “ab” or “ab” and “bc”), whereas those not sharing any letters are significantly different (for example, “a” and “b” or “a” and “bc”).
Immunofluorescence
For staining experiments, cells were either transfected or exposed to conditioned medium as described above. Cells were fixed for 5 min on glass slides with 4% paraformaldehyde and blocked for 1 h at room temperature with 1% blocking solution (Life Technologies). For the experiments shown in Figs. 1E and 2D, cells were incubated with primary antibodies overnight at 4 °C, i.e. goat anti-human-TMEM16A polyclonal antibody S-20 at 1:50 dilution (Figs. 1E and 2D) and mouse anti-human CLCA1 monoclonal antibody 2F4 at 1:100 dilution (Fig. 2D). Slides were washed and incubated with WGA-Alexa Fluor 633 conjugate (5 μg/ml) for 30 min at room temperature, followed by secondary antibodies for 2 h at room temperature; i.e. donkey anti-goat IgG-Alexa Fluor 488 conjugate at 1:200 dilution (Figs. 1E and 2D) and donkey anti-mouse IgG-Alexa Fluor 555 conjugate at 1:250 dilution (Fig. 2D). For the experiments shown in Fig. 3F, cells were incubated in goat anti-human-TMEM16A antibody S-20 as above, followed by donkey anti-goat IgG-Alexa Fluor 594 (1:200). For the experiments shown in Fig. 4A, cells were incubated with rabbit anti-human 1228 antibody at 1:100 dilution and goat anti-human-TMEM16A antibody S-20 and WGA-Alexa Fluor 633 conjugate as described above, followed by donkey anti-goat IgG-Alexa Fluor 594 conjugate (1:200) and donkey anti-rabbit IgG-Alexa Fluor 488 conjugate (1:250). Washed slides were then mounted in Vectashield H-1200 mounting medium with DAPI (Vector Laboratories, Burlingame, CA). Confocal microscopy was carried out using a Zeiss LSM 880 confocal laser-scanning microscope with Airyscan (Carl Zeiss Microscopy, Thornwood, NY). The images were acquired and batch-processed with Zen software (Carl Zeiss Microscopy). For each experiment, all cells were treated on the same day and fixed, stained and imaged in parallel using the same acquisition and display settings.
Homology modeling
The tertiary structure of the CLCA1 VWA domain was predicted using Phyre2 (54). The best template identified was the VWA domain from Catenulispora acidiphila (PDB code 4FX5), and the resulting model based on this template was built with 99.9% confidence.
Author contributions
M. S. R., Z. Y., K. N. B., C. G. N., and T. J. B. designed the experiments. M. S. R., Z. Y., and K. N. B. performed the experiments and analyzed the data. M. S. R., Z. Y., and T. J. B. wrote the manuscript. M. S. R., Z. Y., K. N. B., C. G. N., and T. J. B. revised and edited the manuscript.
Supplementary Material
This work was supported by National Institutes of Health Grants R01-HL119813 (to T. J. B.), R01-HL54171 (to C. G. N.), and T32-GM7200 and T32-HL007317 (to K. N. B.); American Lung Association Grant RG-196051 (to T. J. B.); a Center for the Investigation of Membrane Excitability Diseases (CIMED) pilot and feasibility grant (to T. J. B.); and American Heart Association Predoctoral Fellowship 14PRE19970008 (to Z. Y.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Fig. S1.
- CaCC
- calcium-activated chloride channel
- CF
- cystic fibrosis
- CLCA
- calcium-activated chloride channel regulator
- ICaCC
- calcium-dependent chloride current
- VWA
- von Willebrand factor type A
- MIDAS
- metal ion-dependent adhesion site
- CaV
- voltage-gated Ca2+ channel
- GPI
- glycosylphosphatidylinositol
- FnIII
- fibronectin type III
- CFTR
- cystic fibrosis transmembrane conductance regulator
- WGA
- wheat germ agglutinin
- pF
- picofarad
- ANOVA
- analysis of variance
- CAT
- metalloprotease catalytic domain
- F
- F test statistic.
References
- 1. Pedemonte N., and Galietta L. J. (2014) Structure and function of TMEM16 proteins (anoctamins). Physiol. Rev. 94, 419–459 [DOI] [PubMed] [Google Scholar]
- 2. Picollo A., Malvezzi M., and Accardi A. (2015) TMEM16 proteins: unknown structure and confusing functions. J. Mol. Biol. 427, 94–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Caputo A., Caci E., Ferrera L., Pedemonte N., Barsanti C., Sondo E., Pfeffer U., Ravazzolo R., Zegarra-Moran O., and Galietta L. J. (2008) TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science 322, 590–594 [DOI] [PubMed] [Google Scholar]
- 4. Schroeder B. C., Cheng T., Jan Y. N., and Jan L. Y. (2008) Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell 134, 1019–1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yang Y. D., Cho H., Koo J. Y., Tak M. H., Cho Y., Shim W. S., Park S. P., Lee J., Lee B., Kim B. M., Raouf R., Shin Y. K., and Oh U. (2008) TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature 455, 1210–1215 [DOI] [PubMed] [Google Scholar]
- 6. Huang F., Zhang H., Wu M., Yang H., Kudo M., Peters C. J., Woodruff P. G., Solberg O. D., Donne M. L., Huang X., Sheppard D., Fahy J. V., Wolters P. J., Hogan B. L., Finkbeiner W. E., et al. (2012) Calcium-activated chloride channel TMEM16A modulates mucin secretion and airway smooth muscle contraction. Proc. Natl. Acad. Sci. U.S.A. 109, 16354–16359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Scudieri P., Caci E., Bruno S., Ferrera L., Schiavon M., Sondo E., Tomati V., Gianotti A., Zegarra-Moran O., Pedemonte N., Rea F., Ravazzolo R., and Galietta L. J. (2012) Association of TMEM16A chloride channel overexpression with airway goblet cell metaplasia. J. Physiol. 590, 6141–6155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mall M. A., and Galietta L. J. (2015) Targeting ion channels in cystic fibrosis. J. Cyst. Fibros. 14, 561–570 [DOI] [PubMed] [Google Scholar]
- 9. Sala-Rabanal M., Yurtsever Z., Berry K. N., and Brett T. J. (2015) Novel roles for chloride channels, exchangers, and regulators in chronic inflammatory airway diseases. Mediators Inflamm. 2015, 497387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cunningham S. A., Awayda M. S., Bubien J. K., Ismailov I. I., Arrate M. P., Berdiev B. K., Benos D. J., and Fuller C. M. (1995) Cloning of an epithelial chloride channel from bovine trachea. J. Biol. Chem. 270, 31016–31026 [DOI] [PubMed] [Google Scholar]
- 11. Gibson A., Lewis A. P., Affleck K., Aitken A. J., Meldrum E., and Thompson N. (2005) hCLCA1 and mCLCA3 are secreted non-integral membrane proteins and therefore are not ion channels. J. Biol. Chem. 280, 27205–27212 [DOI] [PubMed] [Google Scholar]
- 12. Hamann M., Gibson A., Davies N., Jowett A., Walhin J. P., Partington L., Affleck K., Trezise D., and Main M. (2009) Human ClCa1 modulates anionic conduction of calcium-dependent chloride currents. J. Physiol. 587, 2255–2274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sala-Rabanal M., Yurtsever Z., Nichols C. G., and Brett T. J. (2015) Secreted CLCA1 modulates TMEM16A to activate Ca2+-dependent chloride currents in human cells. eLife 4, e05875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yurtsever Z., Sala-Rabanal M., Randolph D. T., Scheaffer S. M., Roswit W. T., Alevy Y. G., Patel A. C., Heier R. F., Romero A. G., Nichols C. G., Holtzman M. J., and Brett T. J. (2012) Self-cleavage of human CLCA1 protein by a novel internal metalloprotease domain controls calcium-activated chloride channel activation. J. Biol. Chem. 287, 42138–42149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Patel A. C., Brett T. J., and Holtzman M. J. (2009) The role of CLCA proteins in inflammatory airway disease. Annu. Rev. Physiol. 71, 425–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Alevy Y. G., Patel A. C., Romero A. G., Patel D. A., Tucker J., Roswit W. T., Miller C. A., Heier R. F., Byers D. E., Brett T. J., and Holtzman M. J. (2012) IL-13-induced airway mucus production is attenuated by MAPK13 inhibition. J. Clin. Invest. 122, 4555–4568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Young F. D., Newbigging S., Choi C., Keet M., Kent G., and Rozmahel R. F. (2007) Amelioration of cystic fibrosis intestinal mucous disease in mice by restoration of mCLCA3. Gastroenterology 133, 1928–1937 [DOI] [PubMed] [Google Scholar]
- 18. van der Doef H. P., Slieker M. G., Staab D., Alizadeh B. Z., Seia M., Colombo C., van der Ent C. K., Nickel R., Witt H., and Houwen R. H. (2010) Association of the CLCA1 p.S357N variant with meconium ileus in European patients with cystic fibrosis. J. Pediatr. Gastroenterol. Nutr. 50, 347–349 [DOI] [PubMed] [Google Scholar]
- 19. Kunzelmann K., Kongsuphol P., Aldehni F., Tian Y., Ousingsawat J., Warth R., and Schreiber R. (2009) Bestrophin and TMEM16-Ca2+ activated Cl− channels with different functions. Cell Calcium 46, 233–241 [DOI] [PubMed] [Google Scholar]
- 20. Pritchard H. A., Leblanc N., Albert A. P., and Greenwood I. A. (2014) Inhibitory role of phosphatidylinositol 4,5-bisphosphate on TMEM16A-encoded calcium-activated chloride channels in rat pulmonary artery. Br. J. Pharmacol. 171, 4311–4321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Whittaker C. A., and Hynes R. O. (2002) Distribution and evolution of von Willebrand/integrin A domains: widely dispersed domains with roles in cell adhesion and elsewhere. Mol. Biol. Cell 13, 3369–3387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Choi W. S., Khurana A., Mathur R., Viswanathan V., Steele D. F., and Fedida D. (2005) Kv1.5 surface expression is modulated by retrograde trafficking of newly endocytosed channels by the dynein motor. Circ. Res. 97, 363–371 [DOI] [PubMed] [Google Scholar]
- 23. Vossenkämper A., Nedvetsky P. I., Wiesner B., Furkert J., Rosenthal W., and Klussmann E. (2007) Microtubules are needed for the perinuclear positioning of aquaporin-2 after its endocytic retrieval in renal principal cells. Am. J. Physiol. Cell Physiol. 293, C1129–1138 [DOI] [PubMed] [Google Scholar]
- 24. Zadeh A. D., Xu H., Loewen M. E., Noble G. P., Steele D. F., and Fedida D. (2008) Internalized Kv1.5 traffics via Rab-dependent pathways. J. Physiol. 586, 4793–4813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Horii K., Okuda D., Morita T., and Mizuno H. (2004) Crystal structure of EMS16 in complex with the integrin α2-I domain. J. Mol. Biol. 341, 519–527 [DOI] [PubMed] [Google Scholar]
- 26. Kamata T., and Takada Y. (1994) Direct binding of collagen to the I domain of integrin α 2 β 1 (VLA-2, CD49b/CD29) in a divalent cation-independent manner. J. Biol. Chem. 269, 26006–26010 [PubMed] [Google Scholar]
- 27. Song G., and Springer T. A. (2014) Structures of the Toxoplasma gliding motility adhesin. Proc. Natl. Acad. Sci. U.S.A. 111, 4862–4867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dolphin A. C. (2016) Voltage-gated calcium channels and their auxiliary subunits: physiology and pathophysiology and pharmacology. J. Physiol. 594, 5369–5390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jay S. D., Sharp A. H., Kahl S. D., Vedvick T. S., Harpold M. M., and Campbell K. P. (1991) Structural characterization of the dihydropyridine-sensitive calcium channel α 2-subunit and the associated δ peptides. J. Biol. Chem. 266, 3287–3293 [PubMed] [Google Scholar]
- 30. Cassidy J. S., Ferron L., Kadurin I., Pratt W. S., and Dolphin A. C. (2014) Functional exofacially tagged N-type calcium channels elucidate the interaction with auxiliary α2δ-1 subunits. Proc. Natl. Acad. Sci. U.S.A. 111, 8979–8984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wu J., Yan Z., Li Z., Yan C., Lu S., Dong M., and Yan N. (2015) Structure of the voltage-gated calcium channel Cav1.1 complex. Science 350, aad2395. [DOI] [PubMed] [Google Scholar]
- 32. Cantí C., Nieto-Rostro M., Foucault I., Heblich F., Wratten J., Richards M. W., Hendrich J., Douglas L., Page K. M., Davies A., and Dolphin A. C. (2005) The metal-ion-dependent adhesion site in the von Willebrand factor-A domain of α2δ subunits is key to trafficking voltage-gated Ca2+ channels. Proc. Natl. Acad. Sci. U.S.A. 102, 11230–11235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hoppa M. B., Lana B., Margas W., Dolphin A. C., and Ryan T. A. (2012) α2δ expression sets presynaptic calcium channel abundance and release probability. Nature 486, 122–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Calderón-Rivera A., Andrade A., Hernández-Hernández O., González-Ramírez R., Sandoval A., Rivera M., Gomora J. C., and Felix R. (2012) Identification of a disulfide bridge essential for structure and function of the voltage-gated Ca2+ channel α2δ-1 auxiliary subunit. Cell Calcium 51, 22–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Alvarez-Laviada A., Kadurin I., Senatore A., Chiesa R., and Dolphin A. C. (2014) The inhibition of functional expression of calcium channels by prion protein demonstrates competition with α2δ for GPI-anchoring pathways. Biochem. J. 458, 365–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Davies A., Kadurin I., Alvarez-Laviada A., Douglas L., Nieto-Rostro M., Bauer C. S., Pratt W. S., and Dolphin A. C. (2010) The α2δ subunits of voltage-gated calcium channels form GPI-anchored proteins, a posttranslational modification essential for function. Proc. Natl. Acad. Sci. U.S.A. 107, 1654–1659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dolphin A. C. (2013) The α2δ subunits of voltage-gated calcium channels. Biochim. Biophys. Acta 1828, 1541–1549 [DOI] [PubMed] [Google Scholar]
- 38. Lim N. K., Lam A. K., and Dutzler R. (2016) Independent activation of ion conduction pores in the double-barreled calcium-activated chloride channel TMEM16A. J. Gen. Physiol. 148, 375–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jeng G., Aggarwal M., Yu W. P., and Chen T. Y. (2016) Independent activation of distinct pores in dimeric TMEM16A channels. J. Gen. Physiol. 148, 393–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hartzell H. C., and Whitlock J. M. (2016) TMEM16 chloride channels are two-faced. J. Gen. Physiol. 148, 367–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sheridan J. T., Worthington E. N., Yu K., Gabriel S. E., Hartzell H. C., and Tarran R. (2011) Characterization of the oligomeric structure of the Ca2+-activated Cl− channel Ano1/TMEM16A. J. Biol. Chem. 286, 1381–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tien J., Lee H. Y., Minor D. L. Jr, Jan Y. N., and Jan L. Y. (2013) Identification of a dimerization domain in the TMEM16A calcium-activated chloride channel (CaCC). Proc. Natl. Acad. Sci. U.S.A. 110, 6352–6357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Terashima H., Picollo A., and Accardi A. (2013) Purified TMEM16A is sufficient to form Ca2+-activated Cl− channels. Proc. Natl. Acad. Sci. U.S.A. 110, 19354–19359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Brunner J. D., Lim N. K., Schenck S., Duerst A., and Dutzler R. (2014) X-ray structure of a calcium-activated TMEM16 lipid scramblase. Nature 516, 207–212 [DOI] [PubMed] [Google Scholar]
- 45. Dolphin A. C. (2012) Calcium channel auxiliary α2δ and β subunits: trafficking and one step beyond. Nat. Rev. Neurosci. 13, 542–555 [DOI] [PubMed] [Google Scholar]
- 46. Macabuag N., and Dolphin A. C. (2015) Alternative splicing in Ca(V)2.2 regulates neuronal trafficking via adaptor protein complex-1 adaptor protein motifs. J. Neurosci. 35, 14636–14652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bernstein G. M., and Jones O. T. (2007) Kinetics of internalization and degradation of N-type voltage-gated calcium channels: role of the α2/δ subunit. Cell Calcium 41, 27–40 [DOI] [PubMed] [Google Scholar]
- 48. Brett T. J. (2015) CLCA1 and TMEM16A: the link towards a potential cure for airway diseases. Expert Rev. Respir. Med. 9, 503–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Patel A. C., Morton J. D., Kim E. Y., Alevy Y., Swanson S., Tucker J., Huang G., Agapov E., Phillips T. E., Fuentes M. E., Iglesias A., Aud D., Allard J. D., Dabbagh K., Peltz G., and Holtzman M. J. (2006) Genetic segregation of airway disease traits despite redundancy of calcium-activated chloride channel family members. Physiol. Genomics 25, 502–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Forrest A. S., Joyce T. C., Huebner M. L., Ayon R. J., Wiwchar M., Joyce J., Freitas N., Davis A. J., Ye L., Duan D. D., Singer C. A., Valencik M. L., Greenwood I. A., and Leblanc N. (2012) Increased TMEM16A-encoded calcium-activated chloride channel activity is associated with pulmonary hypertension. Am. J. Physiol. Cell Physiol. 303, C1229–C1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Carter S. C., and McKone E. F. (2016) Pharmacogenetics of cystic fibrosis treatment. Pharmacogenomics 17, 1453–1463 [DOI] [PubMed] [Google Scholar]
- 52. Brodsky J. L., and Frizzell R. A. (2015) A combination therapy for cystic fibrosis. Cell 163, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Aricescu A. R., Lu W., and Jones E. Y. (2006) A time- and cost-efficient system for high-level protein production in mammalian cells. Acta Crystallogr. D Biol. Crystallogr. 62, 1243–1250 [DOI] [PubMed] [Google Scholar]
- 54. Kelley L. A., Mezulis S., Yates C. M., Wass M. N., and Sternberg M. J. (2015) The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.