

Abstract
Human leukocyte antigen (HLA)-DQ2.5 (DQA1*05/DQB1*02) is a class-II major histocompatibility complex protein associated with both type 1 diabetes and celiac disease. One unusual feature of DQ2.5 is its high class-II-associated invariant chain peptide (CLIP) content. Moreover, HLA-DQ2.5 preferentially binds the non-canonical CLIP2 over the canonical CLIP1. To better understand the structural basis of HLA-DQ2.5's unusual CLIP association characteristics, better insight into the HLA-DQ2.5·CLIP complex structures is required. To this end, we determined the X-ray crystal structure of the HLA-DQ2.5· CLIP1 and HLA-DQ2.5·CLIP2 complexes at 2.73 and 2.20 Å, respectively. We found that HLA-DQ2.5 has an unusually large P4 pocket and a positively charged peptide-binding groove that together promote preferential binding of CLIP2 over CLIP1. An α9-α22-α24-α31-β86-β90 hydrogen bond network located at the bottom of the peptide-binding groove, spanning from the P1 to P4 pockets, renders the residues in this region relatively immobile. This hydrogen bond network, along with a deletion mutation at α53, may lead to HLA-DM insensitivity in HLA-DQ2.5. A molecular dynamics simulation experiment reported here and recent biochemical studies by others support this hypothesis. The diminished HLA-DM sensitivity is the likely reason for the CLIP-rich phenotype of HLA-DQ2.5.
Keywords: autoimmune disease, major histocompatibility complex (MHC), molecular dynamics, type 1 diabetes, X-ray crystallography, HLA-DM, HLA-DQ, celiac disease
Introduction
Class-II major histocompatibility complex (MHCII)6 proteins present foreign peptides to T cell receptors of CD4+ T cells (1). The membrane-associated MHCII proteins consist of one α-chain and one β-chain whose interface forms the peptide-binding groove. Humans express three MHCII isotypes, HLA-DR (DR), HLA-DP (DP), and HLA-DQ (DQ), all of which are encoded on chromosome 6. Newly synthesized MHCII proteins associate with a chaperone protein called the invariant chain (Ii) and form a nonameric complex (α3β3Ii3) in the endoplasmic reticulum (2). This complex formation prevents indiscriminate peptide loading onto the nascent MHCII and targets the nascent MHCII to the endosome for further processing (1–3). Once in the endosome, the MHCII-bound Ii is progressively proteolyzed until only a short fragment called class-II-associated invariant chain peptide (CLIP) remains attached to the MHCII peptide-binding groove (4). Subsequently, CLIP is catalytically released by HLA-DM (DM) and replaced with exogenous peptides for CD4+ T cell examination after transport to the cell surface (5). In addition to CLIP removal, DM also carries out peptide editing by catalyzing the release of low-affinity peptides (5, 6). Currently, three MHC-binding regions have been identified in Ii: the canonical CLIP1 (residues 83–101), non-canonical CLIP2 (residues 92–107), and non-canonical CLIP3 (residues 98–111) (Fig. 1). Most mouse and human MHCIIs associate exclusively with CLIP1 (5). So far only DQ2.2, DQ2.5, DQ7.5, and DQ8 have been shown to bind both CLIP1 and CLIP2 (7–9). DQ7.5 binds CLIP1, CLIP2, and CLIP3 (9). Interestingly, all human MHCII alleles that bind CLIP2 or CLIP3 are associated with one or more autoimmune diseases: celiac disease (DQ2.2, DQ2.5, DQ7.5, and DQ8) (10–12) and type 1 diabetes (DQ2.5 and DQ8) (11, 12).
Figure 1.

a, amino acid sequence of the human invariant chain protein. The MHCII-binding core sequence of CLIP1 is MRMATPLLM, and that of CLIP2 is PLLMQALPM. The MHCII-binding core sequence of CLIP3 is unknown. b, solution NMR structure of the truncated human invariant chain protein (residues 118–192; Protein Data Bank code 1IIE). The invariant chain protein is a homotrimer and associates with three class-II major histocompatibility complex proteins simultaneously in the endoplasmic reticulum.
DQ2.5 is associated with celiac disease, an autoimmune-like disorder caused by a harmful immune response to ingested wheat gluten and similar proteins from rye and barley (13). Approximately 95% of celiac disease patients express DQ2.5 that is encoded by the DQA1*05 and DQB1*02 genes (10). These alleles are found on the DR3-DQ2 haplotype (cis configuration) and in the heterozygous combination of DR5-DQ7/DR7-DQ2 haplotypes (trans configuration). The gluten-specific CD4+ T cells of celiac disease patients recognize a diverse set of gluten epitopes when they are presented in the context of DQ2.5 but not in the context of other MHCII molecules, and preferential binding of deamidated gluten peptides appears to be the basis for the association of celiac disease with this HLA molecule (15–18). One unusual phenotype of DQ2.5 is its high CLIP content. In DQ2.5-expressing B lymphoblastoid cells, CLIP1 and CLIP2 combined account for up to 53% of endogenous displayed peptides (7–9, 19). Generally, CLIP accounts for only about 10% of MHCII-displayed peptides (20). Moreover, DQ2.5 preferentially binds the non-canonical CLIP2 over the canonical CLIP1 (7, 8). The CLIP-rich phenotype of DQ2.5 was explained by MHCII·CLIP being poor substrates for DM (8, 21). To better understand the structural basis of the unusual CLIP association characteristics of DQ2.5, we have determined the DQ2.5·CLIP1 complex and DQ2.5·CLIP2 complex crystal structures.
Results
DQ2.5·CLIP1 and DQ2.5·CLIP2 crystal structures
The crystal structure of DQ2.5·CLIP1 (Protein Data Bank code 5KSU) and DQ2.5·CLIP2 (Protein Data Bank code 5KSV) were solved to 2.73- and 2.20-Å resolution, respectively (Fig. 2). In both structures, the β105–112 loop was not modeled due to missing electron density. In the DQ2.5·CLIP1 structure, side chains of α75, α158, α172, β22, and β135 were not modeled due to ambiguous electron density. Data collection and refinement statistics are presented in Table 1. Conformations of the DQ2.5 protein in the DQ2.5·CLIP1 and DQ2.5·CLIP2 structures are highly similar to each other (Cα r.m.s.d. of 1.09 Å) and to the DQ2.5 conformation in the DQ2.5·gliadin-α1a (Protein Data Bank code 1S9V) (15) and DQ2.5·gliadin-α2 (Protein Data Bank codes 4OZF, 4OZG, and 4OZH) (16) structures (Cα r.m.s.d. ranging from 0.57 to 1.27 Å). In the current structures, CLIP1 and CLIP2 have highly similar main chain conformations (Cα r.m.s.d. of 0.47 Å) and side chain orientations (Cβ r.m.s.d. of 0.85 Å).
Figure 2.

a, crystal structure of DQ2.5·CLIP1 (Protein Data Bank code 5KSU). b, crystal structure of DQ2.5·CLIP2 (Protein Data Bank code 5KSV). DQ2.5 α-chain and β-chain are shown in blue and pink, respectively. CLIP1 and CLIP2 peptides are drawn as a stick model (light yellow, carbon; dark yellow, sulfur; blue, nitrogen; red, oxygen). Hydrogen bond interactions are shown as red broken lines.
Table 1.
Data collection and refinement statistics
Values in parentheses are for highest resolution shell.
| DQ2.5·CLIP1 | DQ2.5·CLIP2 | |
|---|---|---|
| Data collection | ||
| Space group | C121 | I23 |
| Cell dimension | ||
| a, b, c (Å) | 128.86, 69.21, 146.69 | 137.01, 137.01, 137.01 |
| α, β, γ (°) | 90, 110.3, 90 | 90, 90, 90 |
| Resolution (Å) | 2.73 | 2.20 |
| Rmerge | 0.1 | 0.13 |
| I/σI | 11.7 (1.45) | 12.7 (1.99) |
| Completeness (%) | 93.8 (89.2) | 93.9 (99.9) |
| Redundancy | 3.5 | 6.5 |
| Refinement | ||
| Resolution (Å) | 39.16–2.73 (2.80–2.73) | 36.62–2.20 (2.30–2.20) |
| Number of reflections | 29,676 | 21,938 |
| Rwork/Rfree | 0.192/0.252 (0.29–0.37) | 0.169/0.207 (0.231–0.296) |
| Number of atoms | 6,139 | 3,178 |
| Protein | 6,023 | 3,003 |
| Water | 116 | 175 |
| B-factors (Å2) | 44.0 | 28.0 |
| Protein | 43.9 | 27.5 |
| Water | 51.3 | 36.1 |
| Root mean square deviations | ||
| Bond length (Å) | 0.01 | 0.01 |
| Bond angle (°) | 1.24 | 1.12 |
| Ramachandran favored (%) | 96.3 | 98.1 |
In the DQ2.5·CLIP1 structure, 14 residues of CLIP1 are clearly visible in the electron density map (Fig. 3a). CLIP1 residues MRMATPLLM (Ii 91–99) occupy the P1–P9 pockets of DQ2.5. This binding register is seen in all MHCII·CLIP1 crystal structures solved to date: DR1·CLIP1 (Protein Data Bank code 3PDO), DR3·CLIP1 (Protein Data Bank code 1A6A), and I-Ab·CLIP1 (Protein Data Bank code 1MUJ) (22–24). In the DQ2.5·CLIP2 structure, 12 residues of CLIP2 are clearly visible in the electron density map (Fig. 3b). CLIP2 residues PLLMQALPM (Ii 96–104) occupy the P1–P9 pockets of DQ2.5, which is in agreement with the biochemically determined binding register of CLIP2 (7, 8).
Figure 3.

2Fo − Fc electron density map of CLIP1 (a) and CLIP2 (b), both contoured at 1.0σ. CLIP1 and CLIP2 peptides are shown in stick representation (light yellow, carbon; dark yellow, sulfur; blue, nitrogen; red, oxygen).
CLIP1 makes 12 direct and four water-mediated hydrogen bonds with DQ2.5, whereas CLIP2 makes 14 direct and six water-mediated hydrogen bonds with DQ2.5 (Fig. 2). There are two hydrogen bond interactions that are present in DQ2.5· CLIP1 but missing in DQ2.5·CLIP2: main chain-main chain interaction between the N-H group of CLIP1 P1 (Ii Met-91) and C=O group of DQ2.5 Asn-α52 (NMet-P1–OAsn-α52) and side chain-main chain interaction between the Nη1 group of CLIP1 P2 (Ii Arg-92) and C=O group of DQ2.5 Arg-β77 (NArg-P2η1–OArg-β77). The first interaction is not possible in DQ2.5·CLIP2 because CLIP2 has a Pro at P1. There are three hydrogen bond interactions that are present in DQ2.5·CLIP2 but missing in DQ2.5·CLIP1 (OArg-P3–NArg-β88η2, OGln-P5ϵ2–NArg-β70η2, and NAla-P6–OAsn-β62δ1). Equivalent interactions are not possible in DQ2.5·CLIP1 because the main chain C=O group of CLIP1 P−3 is rotated away from DQ2.5 β88 and because the P5 and P6 residues of CLIP1 are different from those of CLIP2. Overall, the DQ2.5-binding energies for CLIP1 and CLIP2 appear to be similar as indicated by the experimentally measured dissociation times for DQ2.5·CLIP1 (140 h) and DQ2.5·CLIP2 (140 h) in the absence of DM (8).
Structural basis for the CLIP2 preference of DQ2.5 over CLIP1
Four MHCII·CLIP1 complex crystal structures have been reported to date: DQ2.5·CLIP1 (Protein Data Bank code 5KSU), DR1·CLIP1 (Protein Data Bank code 3PDO) (23), DR3·CLIP1 (Protein Data Bank code 1A6A) (22), and I-Ab·CLIP1 (Protein Data Bank code 1MUJ) (24). Among DQ2.5, DR1, DR3, and I-Ab, only DQ2.5 has been observed to bind CLIP2 (7, 8). DQ2.5 has two structural features that may explain why only it is able to bind CLIP2. First, the P4 pocket of DQ2.5 is significantly deeper and broader than that of DR1, DR3, and I-Ab due to polymorphism at β13, β26, and β78 (Fig. 4). The calculated volume of the P4 pocket in DQ2.5 is 566 Å3, whereas those of DR1, DR3, and I-Ab range from 364 to 417 Å3. In DQ2.5, the P4 pocket-lining residues are Gly-β13, Leu-β26, and Val-β78. In DR1, they are Phe-β13, Leu-β26, and Tyr-β78. In DR3, they are Ser-β13, Tyr-β26, and Tyr-β78. In I-Ab, they are Gly-β13, Tyr-β26, and Val-β78. Therefore, it is not surprising that CLIP1, which has Ala at P4, binds to all four MHCII proteins, whereas CLIP2, which has Met at P4, only binds to DQ2.5. Second, DQ2.5 has a positively charged peptide-binding groove (due to Arg-β70, Lys-β71, and Arg-β77), whereas DR1, DR3, and I-Ab have a negatively charged peptide-binding groove (due to Asp-β57, Asp-β66, and Glu-α55 in DR1/DR3 and Asp-β57, Glu-β66, and Asp-α55 in I-Ab) (Fig. 5). CLIP1 is positively charged (due to Lys-P−1 and Arg-P2), whereas CLIP2 does not contain any charged amino acid residues. Because long-range electrostatic interactions between charged amino acids are important for initial protein-protein complex formation (25–29), DQ2.5 is expected to interact more favorably with CLIP2 than with CLIP1. Indeed, previous biochemical studies have shown that CLIP2 binds to DQ2.5 with higher affinity than CLIP1 (IC50 of 6.0 versus 82.5 μm) (19, 30). Among all MHCII proteins whose three-dimensional structures have been solved, DQ8 is the only other MHCII that associates with CLIP2, and like DQ2.5, DQ8 has a large P4 pocket and a positively charged peptide-binding groove similar to DQ2.5 (Fig. 5).
Figure 4.

Close-up views of the P4 pocket in DQ2.5·CLIP1 (Protein Data Bank code 5KSU) (a), DR1·CLIP1 (Protein Data Bank code 3PDO) (b), DR3·CLIP1 (Protein Data Bank code 1A6A) (c), and I-Ab·CLIP1 (Protein Data Bank code 1MUJ) (d). The protein surfaces of the MHCII α-chain and β-chain are colored blue and pink, respectively. β-Chain residues that line the P4 pocket are shown as a stick model (pink, carbon; red, oxygen). CLIP1 peptide is shown in yellow.
Figure 5.

Adaptive Poisson Boltzmann Solver-generated electrostatic surface of MHC class-II proteins at pH 7.0 (red, negative; blue, positive; white, neutral). The view is from the top, looking straight into the peptide-binding groove. a, DQ2.5·CLIP1 (Protein Data Bank code 5KSU); b, DR1·CLIP1 (Protein Data Bank code 3PDO); c, DR3·CLIP1 (Protein Data Bank code 1A6A); d, I-Ab·CLIP1 (Protein Data Bank code 1MUJ); e, DR2w2a·Epstein-Barr virus DNA polymerase peptide (Protein Data Bank code 1H15); f, DR4w4·human collagen II peptide (Protein Data Bank code 2SEB); g, DR52a-integrin β3 peptide (Protein Data Bank code 2Q6W); h, DQ8·deamidated gluten peptide (Protein Data Bank code 2NNA); i, I-Ad·influenza hemagglutinin peptide (Protein Data Bank code 2IAD); j, I-Ag·hen egg lysozyme(11–27) peptide (Protein Data Bank code 3MBE); k, I-Ak·conalbumin peptide (Protein Data Bank code 1D9K); l, I-Ek (moth cytochrome c) peptide (Protein Data Bank code 3QIU). MHC-bound peptides were not included in the Adaptive Poisson Boltzmann Solver electrostatics calculations.
Structural basis for the CLIP-rich phenotype of DQ2.5
DQ2.5-expressing cells have an unusually high CLIP content (up to 53%; CLIP1 and CLIP2 combined) (7–9, 19). One possible explanation for this is that DQ2.5 binds CLIP with higher affinity compared with other MHCIIs. However, the available structural data do not support this notion. The numbers of direct hydrogen bonds formed between CLIP1 (P−1 to P9 only) and DQ2.5, DR1, DR3, and I-Ab are 11, 13, 17, and 13, respectively. Therefore, we propose that the CLIP-rich phenotype of DQ2.5 arises from an impaired interaction between DQ2.5 and the catalytic DM whose function is to displace the MHC-bound CLIP peptide. Much of the current structural and mechanistic understanding of MHCII-DM interaction is derived from the DR1·HA·DM crystal structure (Protein Data Bank code 4FQX) (31). We investigated whether DQ2.5 has all the structural elements to facilitate the DM interaction that is observed for DR1·DM. To do this, we built a homology model of DQ2.5·CLIP1·DM. We first examined the electrostatic complementarity of the contact surface areas shared by DQ2.5 and DM (Fig. 6). According to our model, two regions in DQ2.5 make direct contact with DM. The first region is located adjacent to the P1 pocket in the α1 domain, and the second region is located near the transmembrane segment in the α2 domain. DQ2.5 has better charge complementarity to DM than does DR1, and therefore we can rule out surface electrostatic charge distribution as the source of impaired DQ2.5-DM interaction.
Figure 6.

The regions of contact shared by MHCII and HLA-DM are indicated as corresponding ovals. The protein surface color represents Adaptive Poisson Boltzmann Solver-generated electrostatics surface of MHC class-II proteins at pH 5.5 (red, negative; blue, positive; white, neutral). a, DQ2.5 from DQ2.5·CLIP1 (Protein Data Bank code 5KSU); b, DR1 from DR1·HA·DM (Protein Data Bank code 4FQX); c, DM from DR1·HA·DM (Protein Data Bank code 4FQX).
Next, we examined whether DQ2.5 is able to undergo the same set of conformational changes that DR1 undergoes upon DM binding. In DR1, Phe-α51 has been identified as a key DM-binding residue (32, 33). When DR1 binds to DM, the α51–55 loop of DR1 transforms into an α-helix, which causes the side chain of Phe-α51 to move 13 Å from its initial solvent-exposed position to the P1 pocket cavity where it forms a hydrophobic cluster with Phe-α24, Ile-α31, Phe-α32, Phe-α48, and Phe-β89 (Fig. 7) (31). This hydrophobic interaction is thought to stabilize the otherwise vacant and unstable P1 pocket. DQ2.5 contains a deletion mutation at α53. DM sensitivity of DQ2.5 was found to be partially restored upon insertion of a Gly at this position (34). Because of the deletion at α53 in DQ2.5, Phe-α51 is located at a position that is inaccessible by DM. This unconventional location of Phe-α51 may compromise the DQ2.5-DM interaction. Furthermore, we suggest that the DM insensitivity of DQ2.5 is due to the presence of an extensive hydrogen bond network (involving Tyr-α9, Tyr-α22, His-α24, Gln-α31, Glu-β86, Thr-β90, and a buried water molecule) that spans the P1 to the P4 pockets of DQ2.5 (Fig. 8a). We carried out a 50-ns molecular dynamics (MD) simulations of the DQ2.5·CLIP1 complex to assess the stability of this hydrogen bond network. MD trajectories show that all hydrogen bonds in this network, with the exception of the peripheral Glu-β86 Oϵ1-Thr-β90 Oγ, are stable (Fig. 9). Furthermore, we observed that Phe-α51 does not enter the P1 pocket during the course of the DQ2.5·CLIP1·DM MD simulations, likely due to the presence of the α9-α22-α24-α31-β86-β90 hydrogen bond network (Fig. 10a). In particular, the water molecule that is hydrogen-bonded to His-α24 and Gln-α31 is directly blocking the P1 pocket. We mutated the α24, α31, and β86 residues in our DQ2.5·CLIP1·DM model to their counterpart in DR1: Eβ86G, Qα31I,Hα24F. These mutations are expected to disrupt the α9-α22-α24-α31-β86-β90 hydrogen bond network and potentially fully restore DM sensitivity in DQ2.5. Our 50-ns MD simulations of the triple mutant DQ2.5 shows that Phe-α51 does indeed occupy the P1 pocket (Fig. 10b) as seen in the DR1·HA·DM crystal structure (Figs. 10c and 11). In DR1, DM binding also causes the β85–90 region to change from an α-helix to a loop, which causes Phe-β89 to move 4.7 Å from the protein surface to the P1 pocket floor where it forms a hydrophobic cluster with several other hydrophobic residues including Phe-α51 (31) (Fig. 7b). In DQ2.5, similar rearrangement of the β85–90 region does not appear feasible as Glu-β86 and Thr-β90 are held in place by the α9-α22-α24-α31-β86-β90 hydrogen bond network (Fig. 8a). In summary, the α9-α22-α24-α31-β86-β90 hydrogen bond network in DQ2.5 prevents repositioning of Phe-α51 and Phe-β89, which is important in DR1-DM interaction.
Figure 7.

Conformation of the peptide-binding groove in DQ2.5·CLIP1 (Protein Data Bank code 5KSU) (a), DR1·CLIP1 (Protein Data Bank code 3PDO) (b), and DR1·HA·DM (Protein Data Bank code 4FQX) (c). α-Chain and β-chain of the MHCII are colored in blue and pink, respectively. The MHCII-bound peptide is omitted for clarity.
Figure 8.

Hydrogen bond network found at the bottom of the MHCII peptide-binding groove. a, DQ2.5·CLIP1 (Protein Data Bank code 5KSU); b, DR1·CLIP1 (Protein Data Bank code 3PDO); c, DR3·CLIP1 (Protein Data Bank code 1A6A); d, I-Ab·CLIP1 (Protein Data Bank code 1MUJ). MHCII α-chain and β-chain are shown in blue and pink, respectively. MHC-bound peptides are drawn as a stick model (light yellow, carbon; dark yellow, sulfur; blue, nitrogen; red, oxygen). Hydrogen bonds are shown as dotted red lines, and their distance is given in Å. The water molecule is shown as a red sphere.
Figure 9.

50-ns molecular dynamics simulation of the DQ2.5·CLIP1 complex. Left, distance trajectories of several atom pairs in the α22-α24-α31-β86-β90-α9 hydrogen bond network are shown: Oϵ2 of Glu-β86 and Oγ1 of Thr-β90 (green), Oϵ1 of Glu-β86 and Oϵ1 of Gln-α31 (cyan), Oϵ1 of Glu-β86 and Oη of Tyr-α9 (blue), Oϵ1 of Gln-α31 and water (red), Nδ1 of His-α24 and water (violet), and Nϵ2 of His-α24 and Oη of Tyr-α22 (yellow). The black horizontal line at 3.5 Å demarcates the hydrogen bond threshold distance. Right, the hydrogen bonds are color-coded, and their distances measured from the crystal structure are indicated in Å.
Figure 10.
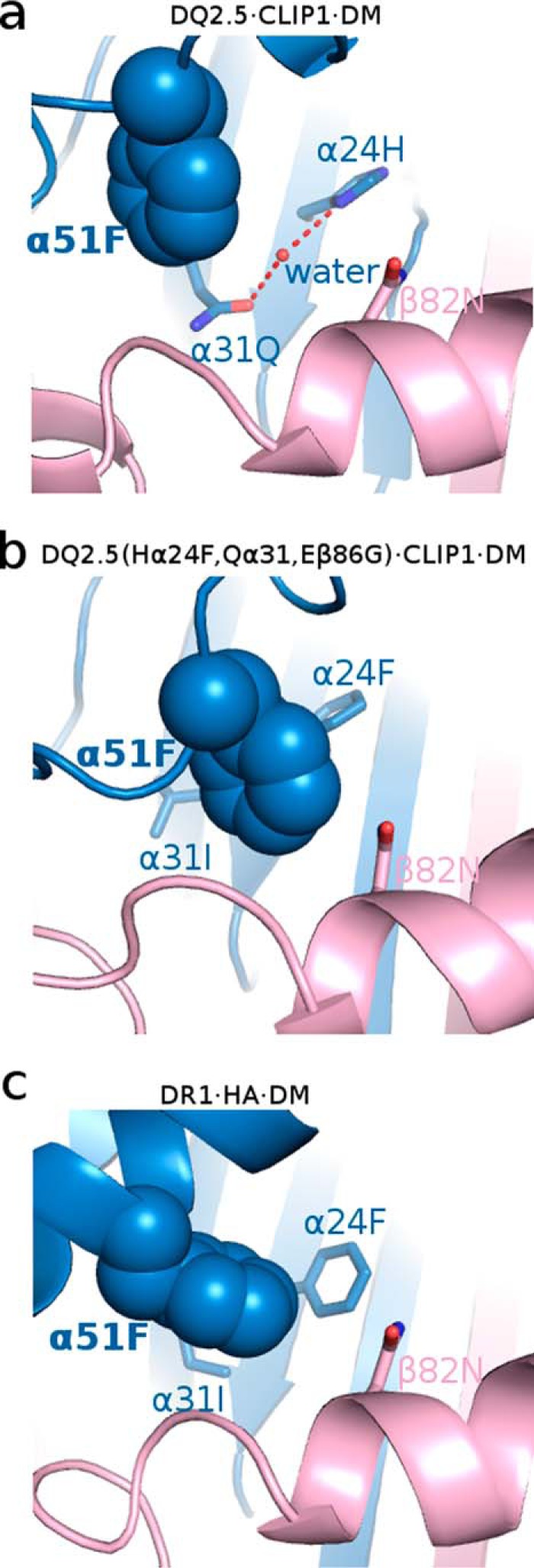
Occupancy of the P1 pocket by Phe-α51 in DQ2.5·CLIP1·DM molecular dynamics simulation model (a), mutant DQ2.5(Eβ86G, Qα31I,Hα24F)·CLIP1·DM molecular dynamics simulation model (b), and DR1·HA·DM crystal structure (Protein Data Bank code 4FQX) (c).
Figure 11.

Trajectory of the separation distance between Phe-α51 (phenyl group centroid) and Asn-β82 (Cα) in the simulated wild-type and mutant DQ2.5·CLIP1·DM complexes.
Hydrogen bond between the P1 main chain nitrogen of CLIP1 and the α52 carbonyl group of DQ2.5
All crystal structures of MHCII in complex with a peptide whose P1 residue is not Pro have a hydrogen bond between the amide nitrogen of the P1 residue and the main chain carbonyl group of MHCII α53. Interestingly, DQ2.5 has a deletion mutation at α53, and as gluten peptides binding to DQ2.5 frequently have Pro at P1, it was suggested that DQ2.5 is unable to form this hydrogen bond (34). The significance of this peptide main chain hydrogen bond has been assessed by comparing binding of peptides N-methylated at the P1 position with unmodified peptides. Whereas such substitution gave decreased affinity for peptide binding to DR1, no effect was seen for DQ2.5 (35, 36). Interestingly, our DQ2.5·CLIP1 structure shows that there is indeed a hydrogen bond between the P1 main chain nitrogen of CLIP1 and the α52 carbonyl group of DQ2.5. The gluten-derived DQ2.5-gliadin-α1a T-cell epitope (LQPFPQPELPY where the underlined residue is P1) binds to DQ2.5 with 2-fold higher affinity than the analog peptide containing norvaline (Nva) at P1 (∼25 μm) (37). Nva is a non-proteinogenic α-amino acid that is isosteric to Pro but has a primary amine group that is able to participate in hydrogen bonding. Therefore, DQ2.5-gliadin-α1a must have an overall energetic advantage over the Nva-substituted analog peptide for binding to DQ2.5 despite having a hydrogen bond deficiency at P1. We propose that DQ2.5-gliadin-α1a as well as other peptides containing Pro at P1 has an entropic advantage that compensates for the lost enthalpy associated with the P1 hydrogen bond.
Discussion
We have determined the crystal structures of DQ2.5·CLIP1 (Protein Data Bank code 5KSU) and DQ2.5·CLIP2 (Protein Data Bank code 5KSV) at 2.73 and 2.20 Å, respectively. Although crystal structures of CLIP1 in complex with DR1 (with the peptide bound in both forward and reverse directions) (23, 38), DR3 (22), and I-Ab (24) have been reported previously, no crystal structure of CLIP2 bound to an MHCII has been reported so far. DQ2.5 is unusual in that it associates with CLIP1 (Ii 83–101) as well as the non-canonical CLIP2 (Ii 92-107) (19). Our study has revealed two unique structural features of DQ2.5 that may promote its association with CLIP2. First, DQ2.5 has an unusually large P4 pocket, which can accommodate the bulky P4 Met of CLIP2. Second, DQ2.5 has a positively charged peptide-binding groove, which is electrostatically more compatible with the neutral CLIP2 compared with the positively charged CLIP1.
Another unusual characteristic of DQ2.5 is its CLIP-rich phenotype. CLIP1 and CLIP2 combined account for 53% of the eluted peptide pool (9). It was proposed that the CLIP-rich phenotype of DQ2.5 is explained by MHCII·CLIP being poor substrates for DM (8, 21). During MHC maturation, DM catalyzes release of CLIP from the nascent MHC (5). Therefore, impaired DQ2.5-DM interaction will result in DQ2.5 molecules retaining their original CLIP cargo. In contrast, DR1-expressing cells have a low abundance of CLIP (20), which suggests that DR1 is a good substrate for DM. We found two structural elements in DQ2.5 that may lower its DM sensitivity. First, α51, which is a key DM-contacting residue in DR1, is positioned internally in DQ2.5 due to the α53 deletion mutation. Second, the peptide-binding groove residues that form the α9-α22-α24-α31-β86-β90 hydrogen bond network are not as free to move about as the corresponding residues in DR1 (Fig. 8). Therefore, DQ2.5 is less predisposed to the drastic secondary structure changes that DR1 undergoes upon DM binding. Our MD study showed that the α9-α22-α24-α31-β86-β90 hydrogen bond network is stable and that Phe-α51 of DQ2.5 cannot move into the P1 pocket upon DM binding. This is due to the blockage of the P1 pocket entrance by a water molecule, which is part of the α9-α22-α24-α31-β86-β90 hydrogen bond network. To further test this idea, we disrupted the hydrogen bond network by changing α24, α31, and β86 to hydrogen bond-non-permissible residues and then repeated the MD exercise. This time, Phe-α51 did translocate to fill the P1 pocket, similar to what happens in DR1 upon DM binding. Our hypothesis that the α9-α22-α24-α31-β86-β90 hydrogen bond network leads to diminished DM sensitivity and ultimately to the CLIP-rich phenotype is further supported by a recent study by Zhou et al. (21) who showed that changing β86 of DQ8 from Glu to Ala resulted in increased DM sensitivity. DQ8 has the same α9-α22-α24-α31-β86-β90 hydrogen bond network found in DQ2.5, although it does not have the α53 deletion mutation. Our analysis, however, is based on the assumption that the DR1-DM interaction mechanism is directly applicable to DQ2.5·DM. It remains to be seen whether the DR1-DM interaction mechanism is truly universal. Even if this should prove not to be the case, the preference of bulky hydrophobic anchor residues at the P1 pocket for both DR1 (39, 40) and DQ2.5 (19, 30) indicates that these two molecules likely share the mechanistic feature of Phe-α51 translocating to fill the P1 pocket in the interaction with DM.
There are two other human MHCII alleles that have a deletion mutation at α53 like DQ2.5 and contain the same set of residues that make up the α9-α22-α24-α31-β86-β90 hydrogen bond network in DQ2.5: DQ4.4 (DQA1*04:01/DQB1*04:02) and DQ7.5 (DQA1*05:05/DQB1*03:01) (10). We predict DQ4.4 and DQ7.5 to be poor substrates for DM and to have a CLIP-rich phenotype similar to DQ2.5. Interestingly, DQ2.5, DQ4.4, and DQ7.5 are all associated with one or more human autoimmune disorders. DQ2.5 is associated with celiac disease and type 1 diabetes, DQ4.4 is associated with juvenile idiopathic arthritis (41), and DQ7.5 is associated with celiac disease (9, 42). However, there is no known mechanistic link between decreased DM sensitivity and human autoimmune disorders.
Experimental procedures
Expression and purification
DQ2.5 (DQA1*05:01/DQB1*02:01) containing covalently linked CLIP1 and CLIP2 was prepared in a manner similar to DQ2.5·αI gliadin (15, 43–45). Fos and Jun leucine zipper sequences were attached to the C terminus of the α- and β-chains, respectively, through intervening Factor Xa sites to promote heterodimer stability. The CLIP1 (PVSKMRMATPLLMQA) and CLIP2 (MATPLLMQALPMGAL) peptides were attached to the N terminus of the β-chain using a 15-residue linker. Expression of DQ2.5 molecules was done as detailed elsewhere (43) using baculovirus, ExpresSF+ insect cells, and mAb 2.12.E11 (46) for affinity purification.
Crystallization and data collection
DQ2.5·CLIP1 and DQ2.5·CLIP2 were treated with Factor Xa for 16 h at 24 °C to remove the leucine zippers from the MHCII. MHCII was then purified using anion-exchange (buffer A, 25 mm Tris, pH 8.0; buffer B, 25 mm Tris, pH 8.0, 0.5 m NaCl) and size-exclusion chromatography (buffer, 25 mm Tris, pH 8.0) and concentrated to 2 mg/ml. For DQ2.5·CLIP1, 1 μl of the protein solution and 1 μl of precipitant buffer (0.1 m ammonium sulfate, 0.1 m sodium cacodylate, pH 6.5, 25% PEG 8000, 6% glycerol) were combined in a single hanging drop and kept at 18 °C. For DQ2.5·CLIP2, 1 μl of the protein solution and 1 μl of precipitant buffer (0.1 m Bis-Tris, pH 5.5, 22% PEG 3350) was combined in a single hanging drop and kept at 18 °C. Small crystals of DQ2.5·CLIP1 and DQ2.5·CLIP2 appeared within 1 week and grew to full size in 2 weeks. Crystals were soaked in mother liquor containing 5% glycerol and then flash frozen in liquid nitrogen. X-ray diffraction data were collected at beam line 9-3 of the Stanford Synchrotron Radiation Laboratory. Diffraction data indexing and integrating were done using HKL2000 (47). The DQ2.5·CLIP1 crystal belonged to the C121 space group with cell dimensions a = 128.86, b = 69.21, and c = 146.69. The DQ2.5·CLIP2 crystal belonged to the I23 space group with cell dimensions a = 137.01, b = 137.01, and c = 137.01.
Structure determination and analysis
Both structures were determined by molecular replacement using Phaser (48, 49). DQ2.5 coordinates from the DQ2.5·gliadin structure (Protein Data Bank code 1S9V) were used as the search model. Model refinement was carried out using Refmac (50), PHENIX (51), and Coot (49). CLIP1 and CLIP2 peptides were built at the end of refinement, guided by the Fo − Fc electron density map. Bulk solvent correction and isotropic B correction were applied throughout the refinement. Water molecules were identified from residual density greater than 1.0σ in the 2Fo − Fc map. All water molecules were checked for valid geometry, environment, and density shape before conducting additional cycles of model building and refinement. The two last refinement rounds included TLS (translation, libration, and screw-rotation displacements) parameterization. Crystallographic data collection, processing, and refinement statistics are given in Table 1. The stereochemical quality of the final structures was carried out using PROCHECK (52).
Model building of DQ2.5(wild type)·CLIP1·DM and DQ2.5(Eβ86G,Qα31I,Hα24F)·CLIP1·DM
The wild-type and mutant DQ2.5·CLIP1·DM complexes were modeled using the MODELLERv9.10 suite of programs (53). The DR1·HA·DM (Protein Data Bank code 4FQX) and DQ2.5·CLIP1 (Protein Data Bank code 5KSU) crystal structures were used as templates. In this model, CLIP1 was truncated to the same length (from P2 to P10) as that of the HA peptide in the DR1·HA·DM crystal structure. A total of five models were created and evaluated using the discrete optimized protein energy statistical energy function (54). The slow refine option was applied to achieve energy-minimized models. Structure visualization and figure representation were done using Chimera (55) and PyMOL (56).
Molecular dynamics simulations
All-atom MD simulation was carried out on three systems: DQ2.5(wild type)·CLIP1, DQ2.5(wild type)·CLIP1·DM, and DQ2.5(Eβ86G,Qα31I,Hα24F)·CLIP1·DM. All crystal water molecules were included in the starting MD structures given their importance in mediating the protein-peptide interaction (57–59). The standard protonation state, at pH 7.0, is applied for all ionizable amino acid groups, i.e. Lys and Arg side chains are protonated, whereas Glu and Asp side chains are deprotonated. His side chains, by default, are singly protonated. Each system was solvated with TIP3P water box with a minimum distance of 12 Å to any protein atom and neutralized by sodium counter-ions. Periodic boundary conditions were applied on the system.
The complex was first energy-minimized using steepest descent and conjugate gradient minimization followed by heating to 300 K within 800 ps under canonical ensemble conditions. The system was then equilibrated under isothermal-isobaric ensemble conditions within 1 ns. The MD simulations under microcanonical ensemble conditions were carried out for 50 ns.
SHAKE was turned on for all bonds involving hydrogen. All simulations were carried out with the AMBER 12 program (60) together with the ff99SB (61) force fields. The particle mesh Ewald (62) algorithm was used to calculate long-range interactions, and short-range interactions were truncated at 10.0 Å. The integration time step was set to 1 fs. Resulting trajectories were analyzed using a combination of indigenously developed Python scripts and the Ptraj/Cpptraj module of AMBER 12.
Pocket volume calculation
The volume of the P4 pocket in DQ2.5, DR1, DR3, and I-Ab was calculated using an in-house script based on the Voronoi algorithm (14).
Author contributions
C.-Y. K. and L. M. S. conceived and coordinated the study. C.-Y. K., T.-B. N., and L. M. S. wrote the paper. E. B. carried out recombinant protein expression and purification. T.-B. N. and P. J. conducted X-ray crystallography experiments. T.-B. N. and M. S. M. conducted molecular dynamics simulation experiments. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
Data collection was performed at the Stanford Synchrotron Radiation Lightsource (SSRL), SLAC National Accelerator Laboratory, which is supported by the United States Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the United States Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including Grant P41GM103393). We thank Dr. I. I. Mathews at SSRL for assistance with X-ray data collection and processing and Dr. Chandra Verma at the Bioinformatics Institute for discussion on molecular dynamics simulation.
This work was supported in part by the Faculty Science and Technology Acquisition and Retention Program of University of Texas System and institutional funding from The University of Texas at El Paso (to C.-Y. K.). The work in the laboratory of L. M. S. was supported by the Research Council of Norway through its Centres of Excellence funding scheme, Project 179573/V40, as well as by the South-Eastern Norway Regional Health Authority. The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (codes 5KSU and 5KSV) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- MHCII
- class-II major histocompatibility complex
- CLIP
- class-II-associated invariant chain peptide
- Ii
- invariant chain
- DQ
- HLA-DQ
- DP
- HLA-DP
- DR
- HLA-DR
- DM
- HLA-DM
- MD
- molecular dynamics
- Nva
- norvaline
- Bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
References
- 1. Cresswell P. (1994) Assembly, transport, and function of MHC class II molecules. Annu. Rev. Immunol. 12, 259–293 [DOI] [PubMed] [Google Scholar]
- 2. Roche P. A., Marks M. S., and Cresswell P. (1991) Formation of a nine-subunit complex by HLA class II glycoproteins and the invariant chain. Nature 354, 392–394 [DOI] [PubMed] [Google Scholar]
- 3. Bakke O., and Dobberstein B. (1990) MHC class II-associated invariant chain contains a sorting signal for endosomal compartments. Cell 63, 707–716 [DOI] [PubMed] [Google Scholar]
- 4. Riberdy J. M., Newcomb J. R., Surman M. J., Barbosa J. A., and Cresswell P. (1992) HLA-DR molecules from an antigen-processing mutant cell line are associated with invariant chain peptides. Nature 360, 474–477 [DOI] [PubMed] [Google Scholar]
- 5. Sette A., Southwood S., Miller J., and Appella E. (1995) Binding of major histocompatibility complex class II to the invariant chain-derived peptide, CLIP, is regulated by allelic polymorphism in class II. J. Exp. Med. 181, 677–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sloan V. S., Cameron P., Porter G., Gammon M., Amaya M., Mellins E., and Zaller D. M. (1995) Mediation by HLA-DM of dissociation of peptides from HLA-DR. Nature 375, 802–806 [DOI] [PubMed] [Google Scholar]
- 7. Wiesner M., Stepniak D., de Ru A. H., Moustakis A. K., Drijfhout J. W., Papadopoulos G. K., van Veelen P. A., and Koning F. (2008) Dominance of an alternative CLIP sequence in the celiac disease associated HLA-DQ2 molecule. Immunogenetics 60, 551–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fallang L. E., Roh S., Holm A., Bergseng E., Yoon T., Fleckenstein B., Bandyopadhyay A., Mellins E. D., and Sollid L. M. (2008) Complexes of two cohorts of CLIP peptides and HLA-DQ2 of the autoimmune DR3-DQ2 haplotype are poor substrates for HLA-DM. J. Immunol. 181, 5451–5461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bergseng E., Dørum S., Arntzen M. Ø., Nielsen M., Nygård S., Buus S., de Souza G. A., and Sollid L. M. (2015) Different binding motifs of the celiac disease-associated HLA molecules DQ2.5, DQ2.2, and DQ7.5 revealed by relative quantitative proteomics of endogenous peptide repertoires. Immunogenetics 67, 73–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Karell K., Louka A. S., Moodie S. J., Ascher H., Clot F., Greco L., Ciclitira P. J., Sollid L. M., Partanen J., and European Genetics Cluster on Celiac Disease (2003) HLA types in celiac disease patients not carrying the DQA1*05-DQB1*02 (DQ2) heterodimer: results from the European Genetics Cluster on Celiac Disease. Hum. Immunol. 64, 469–477 [DOI] [PubMed] [Google Scholar]
- 11. Sollid L. M., Markussen G., Ek J., Gjerde H., Vartdal F., and Thorsby E. (1989) Evidence for a primary association of celiac disease to a particular HLA-DQ α/β heterodimer. J. Exp. Med. 169, 345–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Todd J. A., Bell J. I., and McDevitt H. O. (1987) HLA-DQ β gene contributes to susceptibility and resistance to insulin-dependent diabetes mellitus. Nature 329, 599–604 [DOI] [PubMed] [Google Scholar]
- 13. Trier J. S. (1991) Celiac sprue. New Engl. J. Med. 325, 1709–1719 [DOI] [PubMed] [Google Scholar]
- 14. Richards F. M. (1974) The interpretation of protein structures: total volume, group volume distributions and packing density. J. Mol. Biol. 82, 1–14 [DOI] [PubMed] [Google Scholar]
- 15. Kim C. Y., Quarsten H., Bergseng E., Khosla C., and Sollid L. M. (2004) Structural basis for HLA-DQ2-mediated presentation of gluten epitopes in celiac disease. Proc. Natl. Acad. Sci. U.S.A. 101, 4175–4179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Petersen J., Montserrat V., Mujico J. R., Loh K. L., Beringer D. X., van Lummel M., Thompson A., Mearin M. L., Schweizer J., Kooy-Winkelaar Y., van Bergen J., Drijfhout J. W., Kan W. T., La Gruta N. L., Anderson R. P., et al. (2014) T-cell receptor recognition of HLA-DQ2-gliadin complexes associated with celiac disease. Nat. Struct. Mol. Biol. 21, 480–488 [DOI] [PubMed] [Google Scholar]
- 17. Stepniak D., Wiesner M., de Ru A. H., Moustakas A. K., Drijfhout J. W., Papadopoulos G. K., van Veelen P. A., and Koning F. (2008) Large-scale characterization of natural ligands explains the unique gluten-binding properties of HLA-DQ2. J. Immunol. 180, 3268–3278 [DOI] [PubMed] [Google Scholar]
- 18. Busch R., De Riva A., Hadjinicolaou A. V., Jiang W., Hou T., and Mellins E. D. (2012) On the perils of poor editing: regulation of peptide loading by HLA-DQ and H2-A molecules associated with celiac disease and type 1 diabetes. Expert Rev. Mol. Med. 14, e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vartdal F., Johansen B. H., Friede T., Thorpe C. J., Stevanović S., Eriksen J. E., Sletten K., Thorsby E., Rammensee H.-G., and Sollid L. M. (1996) The peptide binding motif of the disease associated HLA-DQ (α1* 0501, β1* 0201) molecule. Eur. J. Immunol. 26, 2764–2772 [DOI] [PubMed] [Google Scholar]
- 20. Chicz R. M., Urban R. G., Lane W. S., Gorga J. C., Stern L. J., Vignali D. A., and Strominger J. L. (1992) Predominant naturally processed peptides bound to HLA-DR1 are derived from MHC-related molecules and are heterogeneous in size. Nature 358, 764–768 [DOI] [PubMed] [Google Scholar]
- 21. Zhou Z., Reyes-Vargas E., Escobar H., Rudd B., Rockwood A. L., Delgado J. C., He X., and Jensen P. E. (2016) Type 1 diabetes associated HLA-DQ2 and DQ8 molecules are relatively resistant to HLA-DM mediated release of invariant chain-derived CLIP peptides. Eur. J. Immunol. 46, 834–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ghosh P., Amaya M., Mellins E., and Wiley D. C. (1995) The structure of an intermediate in class II MHC maturation: CLIP bound to HLA-DR3. Nature 378, 457–462 [DOI] [PubMed] [Google Scholar]
- 23. Günther S., Schlundt A., Sticht J., Roske Y., Heinemann U., Wiesmüller K. H., Jung G., Falk K., Rötzschke O., and Freund C. (2010) Bidirectional binding of invariant chain peptides to an MHC class II molecule. Proc. Natl. Acad. Sci. U.S.A. 107, 22219–22224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhu Y., Rudensky A. Y., Corper A. L., Teyton L., and Wilson I. A. (2003) Crystal structure of MHC class II I-Ab in complex with a human CLIP peptide: prediction of an I-Ab peptide-binding motif. J. Mol. Biol. 326, 1157–1174 [DOI] [PubMed] [Google Scholar]
- 25. Camacho C. J., Weng Z., Vajda S., and DeLisi C. (1999) Free energy landscapes of encounter complexes in protein-protein association. Biophys. J. 76, 1166–1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Drozdov-Tikhomirov L. N., Linde D. M., Poroikov V. V., Alexandrov A. A., and Skurida G. I. (2001) Molecular mechanisms of protein-protein recognition: whether the surface placed charged residues determine the recognition process? J. Biomol. Struct. Dyn. 19, 279–284 [DOI] [PubMed] [Google Scholar]
- 27. Chu X., Wang Y., Gan L., Bai Y., Han W., Wang E., and Wang J. (2012) Importance of electrostatic interactions in the association of intrinsically disordered histone chaperone Chz1 and histone H2A.Z-H2B. PLoS Comput. Biol. 8, e1002608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kumar V., Dixit N., Zhou L. L., and Fraunhofer W. (2011) Impact of short range hydrophobic interactions and long range electrostatic forces on the aggregation kinetics of a monoclonal antibody and a dual-variable domain immunoglobulin at low and high concentrations. Int. J. Pharm. 421, 82–93 [DOI] [PubMed] [Google Scholar]
- 29. Uchikoga N., Takahashi S. Y., Ke R., Sonoyama M., and Mitaku S. (2005) Electric charge balance mechanism of extended soluble proteins. Protein Sci. 14, 74–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. van de Wal Y., Kooy Y. M., Drijfhout J. W., Amons R., and Koning F. (1996) Peptide binding characteristics of the coeliac disease-associated DQ(α1*0501, β1*0201) molecule. Immunogenetics 44, 246–253 [DOI] [PubMed] [Google Scholar]
- 31. Pos W., Sethi D. K., Call M. J., Schulze M. S., Anders A. K., Pyrdol J., and Wucherpfennig K. W. (2012) Crystal structure of the HLA-DM-HLA-DR1 complex defines mechanisms for rapid peptide selection. Cell 151, 1557–1568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Doebele R. C., Busch R., Scott H. M., Pashine A., and Mellins E. D. (2000) Determination of the HLA-DM interaction site on HLA-DR molecules. Immunity 13, 517–527 [DOI] [PubMed] [Google Scholar]
- 33. Painter C. A., Negroni M. P., Kellersberger K. A., Zavala-Ruiz Z., Evans J. E., and Stern L. J. (2011) Conformational lability in the class II MHC 310 helix and adjacent extended strand dictate HLA-DM susceptibility and peptide exchange. Proc. Natl. Acad. Sci. U.S.A. 108, 19329–19334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hou T., Macmillan H., Chen Z., Keech C. L., Jin X., Sidney J., Strohman M., Yoon T., and Mellins E. D. (2011) An insertion mutant in DQA1*0501 restores susceptibility to HLA-DM: implications for disease associations. J. Immunol. 187, 2442–2452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Stratikos E., Wiley D. C., and Stern L. J. (2004) Enhanced catalytic action of HLA-DM on the exchange of peptides lacking backbone hydrogen bonds between their N-terminal region and the MHC class II alpha-chain. J. Immunol. 172, 1109–1117 [DOI] [PubMed] [Google Scholar]
- 36. Yin L., Trenh P., Guce A., Wieczorek M., Lange S., Sticht J., Jiang W., Bylsma M., Mellins E. D., Freund C., and Stern L. J. (2014) Susceptibility to HLA-DM protein is determined by a dynamic conformation of major histocompatibility complex class II molecule bound with peptide. J. Biol. Chem. 289, 23449–23464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bergseng E., Xia J., Kim C. Y., Khosla C., and Sollid L. M. (2005) Main chain hydrogen bond interactions in the binding of proline-rich gluten peptides to the celiac disease-associated HLA-DQ2 molecule. J. Biol. Chem. 280, 21791–21796 [DOI] [PubMed] [Google Scholar]
- 38. Schlundt A., Günther S., Sticht J., Wieczorek M., Roske Y., Heinemann U., and Freund C. (2012) Peptide linkage to the α-subunit of MHCII creates a stably inverted antigen presentation complex. J. Mol. Biol. 423, 294–302 [DOI] [PubMed] [Google Scholar]
- 39. Jardetzky T. S., Gorga J. C., Busch R., Rothbard J., Strominger J. L., and Wiley D. C. (1990) Peptide binding to HLA-DR1: a peptide with most residues substituted to alanine retains MHC binding. EMBO J. 9, 1797–1803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Falk K., Rötzschke O., Stevanović S., Jung G., and Rammensee H. G. (1994) Pool sequencing of natural HLA-DR, DQ, and DP ligands reveals detailed peptide motifs, constraints of processing, and general rules. Immunogenetics 39, 230–242 [DOI] [PubMed] [Google Scholar]
- 41. Volz T., Schwarz G., Fleckenstein B., Schepp C. P., Haug M., Roth J., Wiesmüller K. H., and Dannecker G. E. (2004) Determination of the peptide binding motif and high-affinity ligands for HLA-DQ4 using synthetic peptide libraries. Hum. Immunol. 65, 594–601 [DOI] [PubMed] [Google Scholar]
- 42. Tinto N., Cola A., Piscopo C., Capuano M., Galatola M., Greco L., and Sacchetti L. (2015) High frequency of haplotype HLA-DQ7 in celiac disease patients from South Italy: retrospective evaluation of 5,535 subjects at risk of celiac disease. PLoS One 10, e0138324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Quarsten H., McAdam S. N., Jensen T., Arentz-Hansen H., Molberg Ø., Lundin K. E., and Sollid L. M. (2001) Staining of celiac disease-relevant T cells by peptide-DQ2 multimers. J. Immunol. 167, 4861–4868 [DOI] [PubMed] [Google Scholar]
- 44. Kozono H., White J., Clements J., Marrack P., and Kappler J. (1994) Production of soluble MHC class II proteins with covalently bound single peptides. Nature 369, 151–154 [DOI] [PubMed] [Google Scholar]
- 45. Kalandadze A., Galleno M., Foncerrada L., Strominger J. L., and Wucherpfennig K. W. (1996) Expression of recombinant HLA-DR2 molecules. Replacement of the hydrophobic transmembrane region by a leucine zipper dimerization motif allows the assembly and secretion of soluble DR αβ heterodimers. J. Biol. Chem. 271, 20156–20162 [DOI] [PubMed] [Google Scholar]
- 46. Viken H. D., Paulsen G., Sollid L. M., Lundin K. E., Tjønnfjord G. E., Thorsby E., and Gaudernack G. (1995) Characterization of an HLA-DQ2-specific monoclonal antibody. Influence of amino acid substitutions in DQ β1*0202. Hum. Immunol. 42, 319–327 [DOI] [PubMed] [Google Scholar]
- 47. Otwinowski Z., and Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 48. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Murshudov G. N., Vagin A. A., and Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 51. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Laskowski R. A., MacArthur M. W., Moss D. S., and Thornton J. M. (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291 [Google Scholar]
- 53. Eswar N., Webb B., Marti-Renom M. A., Madhusudhan M. S., Eramian D., Shen M. Y., Pieper U., and Sali A. (2007) Comparative protein structure modeling using MODELLER. Curr. Protoc. Protein Sci. Chapter 2, Unit 2.9 [DOI] [PubMed] [Google Scholar]
- 54. Shen M. Y., and Sali A. (2006) Statistical potential for assessment and prediction of protein structures. Protein Sci. 15, 2507–2524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 56. DeLano W. L. (2012) The PyMOL Molecular Graphics System, version 1.7, Schrödinger, LLC, New York [Google Scholar]
- 57. Li Y., Yang Y., He P., and Yang Q. (2009) QM/MM study of epitope peptides binding to HLA-A*0201: the roles of anchor residues and water. Chem. Biol. Drug Des. 74, 611–618 [DOI] [PubMed] [Google Scholar]
- 58. Petrone P. M., and Garcia A. E. (2004) MHC-peptide binding is assisted by bound water molecules. J. Mol. Biol. 338, 419–435 [DOI] [PubMed] [Google Scholar]
- 59. Ogata K., and Wodak S. J. (2002) Conserved water molecules in MHC class-I molecules and their putative structural and functional roles. Protein Eng. 15, 697–705 [DOI] [PubMed] [Google Scholar]
- 60. Case D. A., Darden T. A., Cheatham T. E., Simmerling C. L., Wang J., Duke R. E., Luo R., Walker R. C., Zhang W., Merz K. M., Roberts B., Hayik S., Roitberg A., Seabra G., Swails J., et al. (2012) AMBER 12, University of California, San Francisco [Google Scholar]
- 61. Hornak V., Abel R., Okur A., Strockbine B., Roitberg A., and Simmerling C. (2006) Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 65, 712–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Darden T., York D., and Pedersen L. (1993) Particle mesh Ewald: an N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 [Google Scholar]