

Abstract
Dominant mutations in voltage-gated sodium channel NaV1.7 cause inherited erythromelalgia, a debilitating pain disorder characterized by severe burning pain and redness of the distal extremities. NaV1.7 is preferentially expressed within peripheral sensory and sympathetic neurons. Here, we describe a novel NaV1.7 mutation in an 11-year-old male with underdevelopment of the limbs, recurrent attacks of burning pain with erythema, and swelling in his feet and hands. Frequency and duration of the episodes gradually increased with age, and relief by cooling became less effective. The patient's sister had short stature and reported similar complaints of erythema and burning pain, but with less intensity. Genetic analysis revealed a novel missense mutation in NaV1.7 (2567G>C; p.Gly856Arg) in both siblings. The G856R mutation, located within the DII/S4-S5 linker of the channel, substitutes a highly conserved non-polar glycine by a positively charged arginine. Voltage-clamp analysis of G856R currents revealed that the mutation hyperpolarized (−11.2 mV) voltage dependence of activation and slowed deactivation but did not affect fast inactivation, compared with wild-type channels. A mutation of Gly-856 to aspartic acid was previously found in a family with limb pain and limb underdevelopment, and its functional assessment showed hyperpolarized activation, depolarized fast inactivation, and increased ramp current. Structural modeling using the Rosetta computational modeling suite provided structural clues to the divergent effects of the substitution of Gly-856 by arginine and aspartic acid. Although the proexcitatory changes in gating properties of G856R contribute to the pathophysiology of inherited erythromelalgia, the link to limb underdevelopment is not well understood.
Keywords: electrophysiology, ion channel, molecular modeling, pain, sodium channel, erythromelalgia
Introduction
The NaV1.72 sodium channel is preferentially expressed in sensory and sympathetic neurons (1–4) and their axons (5). NaV1.7 is expressed in small neurons in dorsal root ganglion (DRG), giving rise to unmyelinated peripheral nerve fibers, which have a functional role in pain signal propagation from receptive fields to the first synapse in the dorsal horn of the spinal cord (3, 4). NaV1.7 is considered a threshold channel for its ability to boost subthreshold stimuli and set the gain of DRG neurons (6). More than 20 dominant gain-of-function mutations in SCN9A encoding NaV1.7 have been linked to inherited erythromelalgia (IEM) with the vast majority hyperpolarizing activation, slowing deactivation, slowing rates of closed-state inactivation, and increasing the response to slow ramp depolarizations (7–10). Gain-of-function NaV1.7 mutations have also been linked to small-fiber neuropathy (11–14), which is characterized by neuropathic pain and autonomic dysfunction (15). In contrast, recessive loss-of-function mutations of NaV1.7 underlie congenital insensitivity to pain (16–18). These studies provide compelling evidence that NaV1.7 channels are critical for pain-signal transmission.
IEM is characterized by episodic reddening and burning pain of the hands and feet, triggered by mild warmth or exercise (19, 20). Most subjects manifest painful symptoms early in childhood (infancy to 6 years of age); however, occasionally some families show late-onset of symptoms (19–21). Gain-of-function mutations of the NaV1.7 channel have been shown to render DRG neurons hyperexcitable, providing the cellular correlate for neuropathic pain in patients with IEM (7, 10, 19). A mutation of NaV1.7, G856D, was previously found in a family with IEM-like symptoms, dysautonomia, and small hands and small feet (14). Functional assessment of G856D mutant channels showed hyperpolarized activation and depolarized fast inactivation, and the expression of G856D channels in DRG neurons rendered these neurons hyperexcitable, suggesting that in addition to causing pain symptoms, peripheral nerve dysfunction may contribute to limb underdevelopment in this novel syndrome (14).
In this study, we describe a kindred with IEM symptoms including erythema, burning pain in the distal extremities, and underdevelopment of the limbs. We report a novel mutation in SCN9A (c.2567G>C; p.Gly856Arg) that produces functional changes including hyperpolarized voltage dependence of activation and slower deactivation but does not affect fast inactivation. We also report results from the Rosetta computational modeling suite to gain structural clues to the effects of the substitution of Gly-856 by arginine and aspartic acid.
Results
Clinical phenotype and identification of the G856R mutation
An 11-year-old male presented with limb pain and underdevelopment of the limbs. He experienced recurrent attacks of bilateral and symmetrical intense warmth, redness, pain, and swelling involving the hands and feet. Almost all of his anthropometric measurements fell below the age- and gender-matched 25th percentile cutoff with several measurements below the 5th percentile cutoff (Table 1). At the age of 5, he noted episodes of burning pain with erythema and swelling in his feet, but his hands were less affected. The symptoms were exacerbated during the summer. Prolonged sock- or shoe-wearing triggered burning pain attacks. As the proband got older, there was a gradual increase in frequency and duration of the episodes, and the burning pain could not be effectively relieved by cooling. Immersion of his feet in cold water for an extended period of time led to painful skin lesions. There were no signs of large fiber involvement. Repeated laboratory tests demonstrated thrombocytosis (platelet count 5.90 ∼ 6.10 × 1012/liter). Treatment with aspirin, propranolol, carbamazepine, gabapentin, pregabalin, misoprostol, and venlafaxine were not effective.
Table 1.
Anthropometric measurements
The asterisk symbols (*) are <5th percentile cut-off normative values.
| Measure | Mean (11–12-year-old male) | 5th percentile cut-off (11–12 years male) | Brother/% value | Mean (16–17-year-old female) | 5th percentile cut-off (16–17 years female) | Sister/% value |
|---|---|---|---|---|---|---|
| mm | mm | mm | mm | |||
| Stature | 1466 | 1350 | 1420/25 | 1590 | 1501 | 1410/<1* |
| Shoulder height sitting | 498 | 448 | 470/20 | 567 | 524 | 445/<1* |
| Elbow height sitting | 206 | 170 | 160/2* | 249 | 209 | 170/<1* |
| Forearm length | 202 | 177 | 215/78 | 217 | 194 | 195/6 |
| Hand length | 160 | 145 | 150/20 | 170 | 157 | 150/<1* |
| Hand width (without thumb) | 72 | 65 | 66/7.5 | 74 | 68 | 67/3* |
| Buttock-popliteal distance | 367 | 416 | 360/2.5* | 459 | 410 | 380/<1* |
| Buttock-knee distance | 500 | 448 | 430/1* | 548 | 509 | 440/<1* |
| Popliteal height | 367 | 324 | 395/85 | 379 | 346 | 380/51 |
| Foot length | 228 | 206 | 223/37.5 | 231 | 213 | 195/<1* |
| Foot width | 83 | 69 | 78/25 | 83 | 70 | 80/40 |
The patient's sister (aged 17 years at the time of examination) presented with more severe underdevelopment of the limbs and similar symptoms (Fig. 1A). The majority of her anthropometric measurements, including hand and foot length, fell below the age- and gender-matched 1st percentile (Table 1). The erythema and burning pain were not as intense as in the proband. However, her physical development significantly lagged behind her typical peers, and she was of small stature (Table 1). She had primary amenorrhea, and secondary sexual characteristics were absent. Her serum levels of luteinizing hormone, follicle-stimulating hormone, estradiol, and progesterone were similar to follicular-phase hormone levels of a healthy adult woman. Other laboratory examinations including growth hormone and thyroid hormone levels tests were unremarkable.
Figure 1.
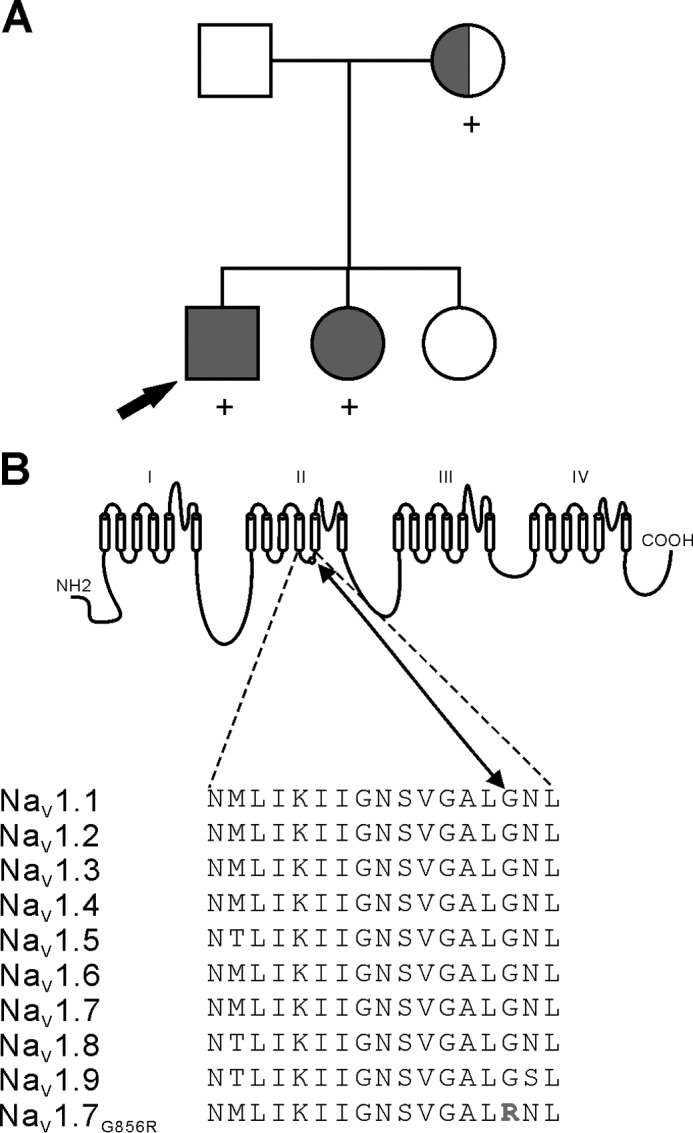
G856R mutation in DII/S4-S5 linker of NaV1.7 in IEM. A, family pedigree showing affected siblings with asymptomatic mother. Squares denote males; circles denote females. Filled symbols denote clinically affected individuals with erythromelalgia. The half-filled circle indicates the mother who experienced transient burning sensation as a teenager. Subjects identified with the novel G856R mutation are denoted with a (+) symbol. The proband is indicated by an arrow. B, sequence alignment and position of the Gly-856 residue within the S4-S5 linker domain II of the NaV1.7 channel. The G856R mutation substitutes a highly conserved glycine to an arginine.
The parents and the eldest sister did not manifest any pain symptoms, and their physical examination was unremarkable. The same examiner of the patients conducted anthropometric measurements of the parents, and the results fell within range of ethnically matched adults. However, the mother recalled transient burning sensations during her teenage years. DNA analysis showed a new SCN9A variant (c.2567G>C) in both affected siblings. Sequence analysis of the mother (blood and saliva) suggested a possible mosaicism for this mutation. This c.2567G>C variant substitutes a highly conserved non-polar glycine with an arginine at position 856 of NaV1.7 (p.Gly856Arg) within the DII/S4-S5 linker (Fig. 1B).
G856R mutation hyperpolarized the voltage dependence of activation
The effects of the mutation on NaV1.7-gating properties were assessed using voltage-clamp recordings after transient transfection of HEK293 cells with WT or G856R mutant channel together with hβ1 and hβ2 subunits. Representative inward sodium current trace families from cells expressing WT and G856R are shown in Fig. 2A. Fig. 2B shows that the average peak current density of G856R channels (557 ± 65 pA/pF; n = 22) was significantly greater than WT channels (366 ± 48 pA/pF; n = 19, p < 0.05). G856R mutant channels activated and reached maximal amplitudes at more hyperpolarized potentials compared with WT channels (Fig. 2C). The midpoint of the voltage dependence of activation of G856R mutant channels (G856R: −28.4 ± 1.3 mV, n = 18, p < 0.05) is significantly shifted in the hyperpolarized direction compared with WT channels (WT: −17.2 ± 1.1 mV, n = 17; Fig. 2D) without a change in the slope (WT: 8.2 ± 0.3, n = 17; G856R: 8.3 ± 0.3, n = 18; p > 0.05).
Figure 2.

The G856R mutation increases current density and alters activation of NaV1.7 in HEK293 cells. A, representative current traces recorded from HEK293 cells expressing WT or G856R. The cells were held at −120 mV, and inward currents were evoked by voltage steps from −80 to +40 mV for 100 ms in 5-mV increments. B, comparison of current density between WT (black) and G856R (red) channels. Current density was measured by normalizing maximal peak currents with cell capacitance. C, normalized peak current-membrane voltage relationship for activation of WT (black square; n = 17) and G856R (red circle; n = 18) channels. D, comparison of voltage dependence of activation for WT and G856R channels. G856R mutation shifts activation by 11.2 mV in a hyperpolarizing direction. Steady-state inactivation was determined by applying 500-ms prepulses (varying from −140 to 0 mV) to open the channels, followed by a 20-ms test pulse to −10 mV. The peak current of the test pulse was normalized to the maximal response amplitude, plotted as a function of the prepulse membrane potential. The voltage dependence of activation and steady-state inactivation were fitted with a Boltzmann function.
The voltage dependence of steady-state fast inactivation of G856R mutant channels was not significantly different from WT channels (WT: −80.2 ± 1.4 mV, n = 23; G856R: −82.7 ± 1.1 mV, n = 17; Fig. 2D). The slope of the steady-state inactivation curve for G856R (8.1 ± 0.3, n = 17) was not significantly different from the slope for WT channels (7.9 ± 0.2, n = 23; Fig. 2D).
Deactivation reflects the transition from the open state to the closed state of WT and G856R channels and was examined by eliciting tail currents at a range of voltages (−120 to −40 mV) after briefly depolarizing to −10 mV for 0.5 ms. The G856R mutation significantly slows the deactivation rate of sodium currents at all tested potentials (Fig. 3A). The time constant of deactivation of the mutant channel at −40 mV was almost 5-fold larger than that of WT channels (WT: 0.22 ± 0.02 ms, n = 20; G856R: 1.09 ± 0.08 ms, n = 15; p < 0.05).
Figure 3.

Deactivation is slowed by the G856R mutation. A, deactivation time constants for WT and G856R channels. Tail currents were elicited by a brief 0.5-ms depolarization to −10 mV followed by a series of hyperpolarizing pulses ranging from −120 to −40 mV from a holding potential of −120 mV. Current decay of the tail current was fitted with a single exponential equation and plotted as a function of the hyperpolarizing step. G856R channels (red circles; n = 15) deactivate significantly slower than WT channels (black squares; n = 20). Inset, superimposed tail current traces of WT (black) and G856R (red) channels at −40 mV. B, comparison of steady-state slow inactivation for WT (black squares; n = 12) and G856R (red circles; n = 11) channels. Slow inactivation was assessed with 30-s prepulses that varied from −130 to +10 mV, followed by a brief hyperpolarization (−120 mV for 100 ms) to remove fast inactivation and a depolarizing test pulse to −10 mV to determine the fraction of available current. Peak inward currents were normalized and plotted as a function of the prepulse membrane potential and fit with a Boltzmann function. C, similar time constants for development of closed-state inactivation between WT (black squares) and G856R (red circles) channels. D, representative ramp currents in response to slow depolarizations (0.2 mV/ms) that were normalized to the peak current acquired during the activation protocol and plotted as a function of membrane potential for WT (black squares) and G856R (red circles) channels.
We assessed the effect of the G856R mutation on slow inactivation by stepping from a membrane potential of −120 mV to a series of 30-s prepulses (−130 to +10 mV) followed by a 100-ms pulse to −120 mV to allow recovery from fast inactivation and then applying a 20-ms depolarizing step to −10 mV to determine the fraction of available current. Slow inactivation was enhanced for G856R mutant channels (Fig. 3B), with the midpoint significantly hyperpolarized by 9.2 mV (WT: −76.9 ± 1.6 mV, n = 12; G856R: −86.1 ± 1.6 mV, n = 11; p < 0.05). The slope for G856R (7.6 ± 0.4, n = 11; p < 0.05) was significantly steeper than WT (12.1 ± 0.7, n = 12).
Recovery from fast inactivation (repriming) of WT and G856R mutant channels was assessed using a two-pulse protocol with varied interpulse interval (1–2049 ms) at different recovery potentials (−120, −110, −100, −90, and −80 mV). The repriming rate of G856R mutant channels did not change at the test potentials when compared with WT channels (data not shown). The development of closed-state inactivation was examined at different potentials (−80, −70, −60, and −50 mV), and the time constants were found to be similar between WT and G856R mutant channels (Fig. 3C).
Slower closed-state inactivation kinetics has been suggested to contribute to enhanced peak ramp currents (22) and has previously shown to be the case with G856D mutant channels (14). We examined the currents elicited in WT and G856R channels by slow ramp depolarizations (0.2 mV/ms: depolarization from −120 to 0 mV over 600 ms). As expected, the similar closed-state inactivation kinetics observed for WT and G856R mutant channels did not lead to altered peak ramp currents. The ramp current, expressed as a percentage of peak current, was not significantly different between WT and G856R channels (WT: 0.68 ± 0.10%, n = 18; G856R: 0.89 ± 0.12%, n = 21) (Fig. 3D).
A notable feature of a previously described pain causing mutation, NaV1.7-G856D, was enhanced persistent current amplitude (14). We assessed the effect of the G856R mutation on persistent currents and found no significant change in persistent current amplitude at all measured voltages (Table 2).
Table 2.
Effect of closed-state inactivation, ramp-current and persistent-current properties of mutant NaV1.7 channels in HEK293 cells
The G856D data were published by Hoeijmakers et al. (14).
| Nav1.7 | Closed-state inactivation |
Ramp current |
Persistent current |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| −80 mV | −70 mV | −60 mV | −50 mV | n | % of peak | Peak voltage | n | % of peak (at −20 mV) | % of peak (at −55 mV) | n | |
| ms | mV | ||||||||||
| WT | 83.1 ± 8.7 | 61.0 ± 7.6 | 33.6 ± 7.0 | 17.8 ± 3.5 | 8 | 0.68 ± 0.10 | −31.6 ± 4.2 | 18 | 0.39 ± 0.11 | 0.34 ± 0.08 | 17 |
| G856R | 89.4 ± 19.1 | 57.9 ± 9.7 | 27.3 ± 4.5 | 10.3 ± 0.9 | 7 | 0.89 ± 0.12 | −38.3 ± 1.9 | 21 | 0.49 ± 0.10 | 0.39 ± 0.08 | 18 |
| WT | 77.9 ± 5.4 | 65.8 ± 4.6 | 41.2 ± 4.1 | 20.7 ± 2.6 | 9 | 0.73 ± 0.1 | −39.6 ± 1.3 | 10 | 0.32 ± 0.11 | 0.13 ± 0.06 | 11 |
| G856D | 114.9 ± 6.1a | 130.9 ± 10.4a | 82.9 ± 12.6a | 25.2 ± 4.8a | 8 | 8.4 ± 0.5a | −50.6 ± 1.1a | 19 | 3.51 ± 0.91a | 1.21 ± 0.25a | 9 |
a p < 0.05 versus WT channels
Structural modeling of the wild-type and mutant NaV1.7 channels
Here, we show that the G856R mutation enhances activation and slows deactivation similar to the previously reported mutation G856D (14). However, G856R and G856D mutant channels display divergent properties in terms of current density, steady-state fast inactivation, closed-state inactivation, and amplitude of ramp current. To explore structural differences between G856D and G856R channels that might underlie these divergent gating properties, we generated a structural model of the human NaV1.7 channel using the Rosetta computational modeling suite (Fig. 4A). Our models suggest that mutant side chains at position 856 are facing toward the lipid environment (Fig. 4A). The bulky side chain of arginine in the G856R channel markedly increases the proximity of voltage-sensing domain I (VSD-I) to transmembrane segment S5 of domain II and introduces potential steric hindrance with the side chains of Met-130 and Met-133 in DI/S1 (Fig. 4, B and C). The relatively smaller side chain of aspartic acid in the G856D channel is unhindered by neighboring residues (Fig. 4, D and E). The X-ray crystallographic structure of NaVAb-hNaV1.7-VSD-IV chimera shows a phosphatidylcholine (23) bound to the same region of the structure where Gly-856 is positioned in our structural model. Interaction of the phosphodiester head groups of lipids are essential for voltage-dependent gating (24, 25), suggesting that alteration of the lipid-channel interaction may modulate NaV channel function. Taken together, structural modeling of G856R and G856D channels predicts that substitutions of Gly-856 by arginine or aspartic acid introduces divergent effects on the proximity of transmembrane segment DII/S5 and DI/S1, as well as disruption of lipid-channel interaction near Gly-856.
Figure 4.

Structural models of domain II S4-S5 linker region of WT-NaV1.7 and G856R mutant channels. A, transmembrane view of the human NaV1.7-G856R structural model. B, close-up view toward the membrane of G856R. C, view from the intracellular side showing unfavorable clashes with side chains of Met-130 and Met-133 in DI/S1. D, close-up view toward the membrane of the human NaV1.7-G856D model. E, view from the intracellular side. G856R and G856D are shown in space-filling representation and labeled.
Discussion
The NaV1.7 sodium channel plays a major role in regulation of neuronal excitability of peripheral sensory and sympathetic neurons (2, 26) and has been shown to cause a spectrum of pain disorders (10, 27). We report here the second case of a pain syndrome in which subjects with burning pain in distal limbs caused by a mutation in NaV1.7 also manifest underdevelopment of hands and feet. Genetic analysis revealed a novel missense mutation in two siblings that produces a substitution of glycine with arginine (G856R) at codon 856 in the NaV1.7 sodium channel. To date, five mutations within the DII/S4-S5 linker of sodium channels have been linked to human painful disorders, including IEM and small-fiber neuropathy; however, only mutations at the Gly-856 residue are associated with limb underdevelopment (9, 14, 28–30).
IEM mutations in SCN9A reported to date hyperpolarize activation of NaV1.7 channels and produce hyperexcitable DRG neurons (7, 10, 19). Thus, we predict that hyperpolarized activation of G856R mutant channels will render DRG neurons hyperexcitable in response to graded stimuli and contribute to the pain phenotype in the two affected siblings. The mother of the proband was clinically asymptomatic but displayed genetic mosaicism for the G856R substitution and may have transmitted the mutant allele to her affected children. An individual with a mosaic genotype carries a mutation in all cells descended from the initial cell that acquired this mutation during embryonic development. Although the mother did not report complaints of pain as an adult, she recalled transient burning pain symptoms as a teenager. This suggests that she acquired the mutation late in embryogenesis and that she is carrying it in a small number of cells as an adult. Mosaic individuals carrying other NaV1.7 mutations have recalled burning pain as a youth and describe markedly improved symptoms as adults (29, 31), which suggests the presence of the mutation in a smaller number sensory neurons, compared with their affected children.
The role of sodium channels in limb development in humans is not fully understood. The subjects in this study presented with underdevelopment of the limbs together with symptoms of IEM. Remarkably, the subjects carrying the G856R mutation manifest impaired limb development, especially of the hands and feet, as previously reported in patients with the G856D mutation (14). The majority of the proband's anthropometric measurements fell below the age- and gender-matched 25th percentile cutoff with several measurements below the 5th percentile cutoff. The proband's sister presented with more severe underdevelopment of the limbs, including anthropometric measurements below the 1st percentile cutoff and short stature. Notably, previously reported patients with the G856D mutation (14) displayed small hands and feet.
Underdevelopment of the limbs has not been reported as a common feature in patients with IEM mutations, but there is no systemic anthropometric data available. One of the authors (Y. Y.) has clinical experience with patients carrying other NaV1.7 mutations associated with IEM but has not identified other families or singleton cases of IEM who manifest underdevelopment of the limbs. As is a common practice, precise anthropometric measurements are not implemented, except when the patients are noticeably beyond average for their age and gender group, as was the case here. Nevertheless, the Gly-856 residue reported in this study and by Hoeijmakers et al. (14) suggests that gain-of-function attributes of NaV1.7 at this residue may contribute to underdevelopment of the limbs.
We cannot exclude the possibility that impaired physical development in the subjects with mutations at Gly-856 is the result of a second mutation, representing a case of digenic inheritance. The simplest cases of digenic disorders typically include mutations in different subunits of a multimeric protein or mutations in genes within a biological pathway (32). Bones are innervated by sensory and sympathetic nerves to regulate local bone metabolism (33, 34). Sodium channels have been shown to be expressed in non-excitable cells, such as Schwann cells and glial cells (35–37), and expression of NaV1.2 channels has been reported in osteoblasts during embryogenesis (38). Bone resorption has been reported to be increased following sensory nerve denervation in adult rats (39). Dynamic interactions of small myelinated and unmyelinated sensory and sympathetic nerves with osteoclastic and osteoblastic cells play important roles in bone remodeling (33, 34, 40). Nerve fibers at the epiphysis of the bone has been suggested to be involved with bone growth at the epiphyseal growth plate (41, 42), thus raising the possibility that peripheral nerve dysfunction may affect limb morphogenesis during development. Because the underdevelopment of limbs is not a common feature in patients with IEM mutations, we would also have to speculate that mutations at the Gly-856 position are altering a non-canonical function of Nav1.7 or that this residue is the site of interaction with a factor that is important in the signaling cascade of limb morphogenesis.
The G856R mutation substitutes a highly conserved neutral glycine with a large positively charged arginine residue within the DII/S4-S5 linker (Fig. 1). Similar to the G856D mutation previously reported by Hoeijmakers et al. (14), G856R mutant channels hyperpolarize the voltage dependence of activation and significantly slow deactivation kinetics (Table 3), which is expected to increase the availability of channels for activation. In contrast to G856D, we show here that a change in charge residue at codon 856 did not enhance the persistent current or the response to slow ramp depolarizations (Table 2). Consistent with unchanged persistent current and peak ramp current, the rate of closed-state inactivation was unaltered in G856R mutant channels (Table 2). The divergent effects of substitutions of Gly-856 by arginine or aspartic acid on current density, fast inactivation, and persistent and ramp currents are not intuitively explained by the replacement of the neutral charge by either a positive or a negative charge.
Table 3.
The G856R mutation hyperpolarizes activation and slows deactivation kinetics but does not alter fast inactivation in HEK293 cells
The G856D data were published by Hoeijmakers et al. (14).
| Nav1.7 | Current density |
Activation |
Deactivation |
Steady-state fast inactivation |
Slow inactivation |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| pA/pF | n | V1/2,act | k | n | −40 mV | n | V1/2,fast | k | n | V1/2,slow | k | A% | n | |
| mV | ms | mV | mV | |||||||||||
| WT | 366 ± 48 | 19 | −17.2 ± 1.1 | 8.2 ± 0.3 | 17 | 0.22 ± 0.02 | 20 | −80.1 ± 1.4 | 7.9 ± 0.2 | 23 | −76.9 ± 1.6 | 12.1 ± 0.7 | 17.3 ± 0.9 | 12 |
| G856R | 557 ± 65a | 22 | −28.4 ± 1.3a | 8.3 ± 0.3 | 18 | 1.09 ± 0.08a | 15 | −82.7 ± 1.1 | 8.1 ± 0.3 | 17 | −86.1 ± 1.6a | 7.6 ± 0.4a | 3.9 ± 0.6a | 11 |
| WT | 504 ± 61 | 25 | −22.7 ± 0.8 | 6.3 ± 0.2 | 23 | 0.52 ± 0.05 | 13 | −79.0 ± 1.0 | 7.0 ± 0.2 | 22 | −62.9 ± 1.3 | 13.8 ± 0.7 | 8.7 ± 0.9 | 13 |
| G856D | 118 ± 13a | 28 | −32.0 ± 0.6a | 9.3 ± 0.2a | 27 | 3.06 ± 0.12a | 19 | −72.8 ± 0.8a | 6.5 ± 0.1 a | 27 | −78.0 ± 1.1a | 6.5 ± 0.2 a | 10.5 ± 1.0 | 16 |
a p < 0.05 versus WT channels.
The interaction between cell membrane phospholipids with voltage-gated channels has been suggested to provide energetic stability and proper functioning of these channels (23–25, 43). Rosetta structural modeling and predictions from the X-ray crystal structure of NaVAb-hNaV1.7-VSD-IV chimera show a phosphatidylcholine bound (23) to the same region of the structure where Gly-856 is positioned. G856D and G856R mutations may differentially regulate channel gating because of displacement of specific lipid molecule(s) near the S4-S5 linker and S5 at the intracellular face of the plasma membrane. Sequence alignment of human, bacterial, and insect voltage-gated sodium channels in the S4-S5 linkers highlight the conservation among species at the corresponding position of Gly-856 with hydrophobic or polar amino acids and the absence of charged residues that could potentially alter lipid-channel interaction near this residue (supplemental Fig. S1). Structural modeling of the G856R mutation predicts favorable interactions with lipid phosphate groups and/or stabilizes the channel at the plasma membrane, resulting in increased current density. By contrast, G856D is predicted to cause electrostatic repulsion with lipid phosphate groups and destabilize correct transmembrane topology, thus decreasing current density. These potential divergent effects of introducing a positive or a negative charge at the 856 position on the lipid-channel interaction may underlie the opposing effects of the G856R and G856D mutations on current density.
G856R may attract new lipid molecules that do not introduce significant effect on steady-state fast inactivation, ramp current, persistent current, or closed-state inactivation. In contrast, electrostatic repulsion of a negatively charged aspartic acid in G856D mutation may prevent lipid phosphate groups from binding near the Gly-856 region leading to destabilization of the DII/S4-S5 linker and enhanced persistent current and ramp current, as well as slower rate of closed-state inactivation (14). Destabilization of the channel-lipid interaction near DII/S4-S5 linker of G856D channels may also contribute to impairment of steady-state fast inactivation reported by Hoeijmaker et al. (14). As predicted in our model, charged residues (arginine and aspartic acid) at Gly-856 may displace a specific lipid molecule(s) near DII/S4-S5 linker and transmembrane segment S5 at the intracellular side of the membrane. This disruption of lipid interaction near Gly-856 may lower the energy barrier of the early gating process leading to a shift in voltage dependence of channel activation.
To generate the human NaV1.7 channel model, we used Rosetta structural modeling suite and bacterial NaVRh structure in a preopen or inactivated state (Protein Data Bank code 4DXW) (44) as a template (see Experimental Procedures). We compared recently published cryoEM structure of a voltage-gated sodium channel from cockroach (NaVPaS) in the closed-state (45) to our structural model based on the bacterial NaVRh crystal structure. The side chain of Val-642 in the NaVPaS structure is fully exposed to the lipid environment as predicted for Gly-856 in our hNaV1.7 model (supplemental Fig. S2). Once new NaV channel structures captured in different states become available, we may potentially gain new insights into molecular mechanism of G856D and G856R mutations.
In summary, our results indicate that this novel IEM-associated G856R mutation enhances activation and slows deactivation but imparts no change in the voltage dependence of inactivation, persistent current amplitude, or response to slow ramp depolarizations. Further studies will be needed to identify the specific lipid molecule(s) interacting with residue Gly-856 and disrupting channel gating in G856D and G856R mutant channels. Together with the mutation G856D (14), which was also associated with impaired distal limb development, these results support the conclusion that a gain-of-function mutation of NaV1.7 channels expressed in small sensory nerve fibers may adversely affect bone morphogenesis during development.
Experimental procedures
Patients
The male proband was 11 years old at the time of the first examination. He presented with recurrent attacks of bilateral and symmetrical intense warmth, redness, pain, and swelling involving the hands and the feet since the age of 5 years. The proband's sister, age 17 years at the time of examination, presented with similar symptoms and signs.
Anthropometric measurements
Eleven anthropometric body dimensions of the proband, the proband's sister, and their parents were measured strictly following the methods introduced by the Chinese national standard for measuring human dimensions of Chinese minors (46). Measurements were performed twice and conducted by one observer (E. Y. Z.). The findings were compared with the measurements of age- and gender-matched Chinese healthy minors reported in the same standard (46).
DNA analysis
Genomic DNA from the proband and his affected and unaffected family members was screened for mutations in SCN9A. Coding exons were amplified and compared with the reference NaV1.7 cDNA to identify sequence variation, as described previously (47).
Plasmid and transfection
The plasmid carrying the tetrodotoxin-resistant version of human NaV1.7 cDNA (WT; hNaV1.7R) was described previously (48). The hNaV1.7R-G856R mutation was introduced into hNaV1.7R using QuikChange XL site-directed mutagenesis (Stratagene), which is hereinafter referred to as G856R. WT or G856R mutant channels were co-transfected with plasmids pCD8-IRES-hβ1 and pEGFP-hβ2 (49), which encode the human β1 and β2 subunits, into HEK293 cells using Lipofectamine 2000 (Invitrogen). HEK293 cells were grown under standard tissue culture conditions (5% CO2; 37 °C) in DMEM/F12 supplemented with 10% fetal bovine serum for 18 h before electrophysiological assessment.
Voltage-clamp recording
Whole-cell patch-clamp recordings were obtained from on GFP-labeled HEK293 cells using an EPC10-double amplifier and Patchmaster software (HEKA Electronik) at room temperature (20 ± 1 °C). The extracellular solution contained 140 mm NaCl, 3 mm KCl, 1 mm MgCl2, 1 mm CaCl2, 10 mm HEPES, and 10 mm dextrose, pH 7.3, adjusted with NaOH (319 mOsm). tetrodotoxin (500 nm) was included in the bath to block endogenous sodium currents in HEK293 cells (27, 50). The pipette solution contained the following 140 mm CsF, 1 mm EGTA, 10 mm NaCl, 10 mm HEPES, and 10 mm dextrose, pH 7.3, adjusted with CsOH (adjusted to 311 mOsm with sucrose). Patch pipettes were fabricated from boroscillicate glass (1.65/1.1, OD/ID; World Precision Instruments) using a Sutter Instruments P-97 puller and had a resistance of 0.8–1.3 megohm. HEK293 cells were held at −120 mV for all parameters examined. To minimize voltage errors 80–90% series resistance compensation was applied. Voltage-clamp recordings were acquired 5 min after establishing whole-cell configuration to allow steady-state dialysis between the cytoplasm and pipette solution. Recordings were acquired at 2.9 kHz with low-pass Bessel filter and digitized at 50 kHz. Linear leak and residual capacitance artifacts were removed using P/6 subtraction. The data were analyzed offline with FitMaster (HEKA Electronik), Excel (Microsoft), and Origin (Microcal Software, Northhampton, MA) software.
To measure current-voltage (I-V) relationships, a range of potentials from −80 to +40 mV in 5-mV increments were applied from the holding potential (−120 mV) for 100 ms at 5-s intervals. The current density was calculated by normalizing maximal peak currents with cell capacitance. Peak inward currents obtained from activation protocols were converted to conductance values using the equation, G = I/(Vm − ENa), where G is the conductance, I is the peak inward current, Vm is the membrane potential used to elicit the response, and ENa is the reversal potential for sodium. The conductance data were normalized by the maximum conductance value and fit with a Boltzmann equation,
| (Eq. 1) |
where V½ is the midpoint of activation, and k is the slope factor.
Steady-state fast inactivation was assessed with a series of 500-ms prepulses (−140 to 0 mV in 10-mV increments) followed by a 20-ms step depolarization to −10 mV to activate the remaining non-inactivated channels. Peak inward currents from steady-state fast inactivation were normalized by the maximum current amplitude and fit with a Boltzmann equation,
| (Eq. 2) |
where I is the current amplitude measured during the test depolarization, V½ is the midpoint of fast inactivation, and k is the slope factor.
Deactivation was estimated from current decay, using a 0.5-ms short depolarization pulse to −10 mV followed by a 50-ms repolarization pulse to potentials ranging from −40 to −120 mV in 5-mV increments. Deactivation kinetics were calculated by fitting the decaying currents with a single exponential equation,
| (Eq. 3) |
where I is the current, A represents amplitude of the fit, τ is the time constant, and C is the steady-state asymptote. The time shift was manually selected by fitting the traces at the time when the currents were just starting to exponentially decrease. Inactivation time constants were determined by fitting the rising phase of the current traces with a single exponential equation. Time constant of decay (τ) were obtained from the traces at potentials −40 to +40 mV for WT and G856R channels.
Steady-state slow inactivation was assessed with 30-s prepulses at a range of potentials from −130 to +10 mV in 10-mV increments, followed by 100-ms pulse to −120 mV to allow recovery from fast-inactivation and then a 20-ms depolarizing step to −10 mV to elicit a test response, which reflects the remaining channels available for activation. Peak inward currents from steady-state slow inactivation were normalized by the maximum current amplitude and fit with a Boltzmann equation,
| (Eq. 4) |
where I is the current amplitude measured during the test depolarization, V½ is the midpoint of slow inactivation, and k is the slope factor. Closed-state inactivation was assessed by stepping from the holding potential to the inactivation potential ranging from −80 mV to −50 mV in 10-mV increments for 1–2049 ms, followed by a −10-mV test pulse for 20 ms to determine the fraction of current inactivated during the prepulse. Ramp currents were elicited with 600-ms slow depolarizations from −120 to 0 mV at a rate of 0.2 mV/ms. The ramp current was normalized to the maximal peak inward current recorded during the activation protocol for each cell.
Structural modeling of the wild-type and mutant NaV1.7 channels
We used the X-ray crystal structure of bacterial sodium channel NaVRh (Protein Data Bank code 4DXW) (44) as a template to build the homology of the NaV1.7, NaV1.7-G856D, and NaV1.7-G856R channels to assess structural changes and altered interactions near Gly-856 in S5 of domain II. The RosettaCM protocol (51, 52) was used with the RosettaMembrane scoring function (53–55) to thread the NaV1.7 sequence on the NaVRh structure. The extracellular loop regions connecting S5-P1 and S6-P2 were truncated. 10,000 models of the wild-type NaV1.7 were generated, and the top 1,000 models were selected based on total Rosetta score and then clustered to identify the most frequently sampled conformations as described previously (56). The G856R and G856D models were created using Rosetta by mutating Gly-856 in the best wild-type NaV1.7 model. The Rosetta kinematic loop modeling protocol (57) was used to optimize conformation of the backbone and side chains of 5 amino acids upstream and downstream of position 856 in G856R and G856D models. The figures of models were rendered with University of California, San Francisco Chimera software (58).
Data analysis
The electrophysiological data were analyzed using Fitmaster (HEKA Electronics), Excel, and Origin 8.5 (Northampton, MA) software and presented as means ± S.E. Statistical significance was determined by unpaired Student's t test.
Author contributions
B. S. T. designed, performed, and analyzed the electrophysiological experiments. B. S. T. prepared the figures. S. G. W. and S. D. D.-H. conceived the study and designed experiments. B. S. T., S. G. W., and S. D. D.-H wrote the paper. P. T. N. conducted structural modeling studies and analyzed results. P. T. N. and V. Y.-Y. conceived the structural modeling study, designed experiments, and wrote aspects of the paper. E. Y. Z. acquired patient samples and conducted anthropometric measurements of the proband and family members. E. Y. Z. and Y. Y. wrote aspects of the paper.
Supplementary Material
Acknowledgments
We thank Fadia Dib-Hajj and Palak Shah for technical support.
This work was supported by the Medical Research Service and Rehabilitation Research Service, the Department of Veterans Affairs (to S. D. D.-H. and S. G. W.), and University of California, Davis startup funds (to V. Y.-Y.). The Center for Neuroscience and Regeneration Research is a Collaboration of the Paralyzed Veterans of America with Yale University. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Figs. S1 and S2.
- used are: NaV
- voltage-gated sodium channel
- DRG
- dorsal root ganglion
- VSD
- voltage-sensing domain
- IEM
- inherited erythromelalgia.
References
- 1. Sangameswaran L., Fish L. M., Koch B. D., Rabert D. K., Delgado S. G., Ilnicka M., Jakeman L. B., Novakovic S., Wong K., Sze P., Tzoumaka E., Stewart G. R., Herman R. C., Chan H., Eglen R. M., et al. (1997) A novel tetrodotoxin-sensitive, voltage-gated sodium channel expressed in rat and human dorsal root ganglia. J. Biol. Chem. 272, 14805–14809 [DOI] [PubMed] [Google Scholar]
- 2. Toledo-Aral J. J., Moss B. L., He Z. J., Koszowski A. G., Whisenand T., Levinson S. R., Wolf J. J., Silos-Santiago I., Halegoua S., and Mandel G. (1997) Identification of PN1, a predominant voltage-dependent sodium channel expressed principally in peripheral neurons. Proc. Natl. Acad. Sci. U.S.A. 94, 1527–1532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Djouhri L., Newton R., Levinson S. R., Berry C. M., Carruthers B., and Lawson S. N. (2003) Sensory and electrophysiological properties of guinea-pig sensory neurones expressing Nav 1.7 (PN1) Na+ channel α subunit protein. J. Physiol. 546, 565–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Black J. A., Frézel N., Dib-Hajj S. D., and Waxman S. G. (2012) Expression of Nav1.7 in DRG neurons extends from peripheral terminals in the skin to central preterminal branches and terminals in the dorsal horn. Mol. Pain 8, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Persson A. K., Black J. A., Gasser A., Cheng X., Fischer T. Z., and Waxman S. G. (2010) Sodium-calcium exchanger and multiple sodium channel isoforms in intra-epidermal nerve terminals. Mol. Pain 6, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rush A. M., Cummins T. R., and Waxman S. G. (2007) Multiple sodium channels and their roles in electrogenesis within dorsal root ganglion neurons. J. Physiol. 579, 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dib-Hajj S. D., Cummins T. R., Black J. A., and Waxman S. G. (2010) Sodium channels in normal and pathological pain. Annu. Rev. Neurosci. 33, 325–347 [DOI] [PubMed] [Google Scholar]
- 8. Cheng X., Dib-Hajj S. D., Tyrrell L., Te Morsche R. H., Drenth J. P., and Waxman S. G. (2011) Deletion mutation of sodium channel NaV1.7 in inherited erythromelalgia: enhanced slow inactivation modulates dorsal root ganglion neuron hyperexcitability. Brain 134, 1972–1986 [DOI] [PubMed] [Google Scholar]
- 9. Cummins T. R., Dib-Hajj S. D., and Waxman S. G. (2004) Electrophysiological properties of mutant Nav1.7 sodium channels in a painful inherited neuropathy. J. Neurosci. 24, 8232–8236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dib-Hajj S. D., Yang Y., Black J. A., and Waxman S. G. (2013) The NaV1.7 sodium channel: from molecule to man. Nat. Rev. Neurosci. 14, 49–62 [DOI] [PubMed] [Google Scholar]
- 11. Faber C. G., Hoeijmakers J. G., Ahn H. S., Cheng X., Han C., Choi J. S., Estacion M., Lauria G., Vanhoutte E. K., Gerrits M. M., Dib-Hajj S., Drenth J. P., Waxman S. G., and Merkies I. S. (2012) Gain of function Nanu1.7 mutations in idiopathic small fiber neuropathy. Ann. Neurol. 71, 26–39 [DOI] [PubMed] [Google Scholar]
- 12. Han C., Hoeijmakers J. G., Ahn H. S., Zhao P., Shah P., Lauria G., Gerrits M. M., te Morsche R. H., Dib-Hajj S. D., Drenth J. P., Faber C. G., Merkies I. S., and Waxman S. G. (2012) Nav1.7-related small fiber neuropathy: impaired slow-inactivation and DRG neuron hyperexcitability. Neurology 78, 1635–1643 [DOI] [PubMed] [Google Scholar]
- 13. Han C., Hoeijmakers J. G., Liu S., Gerrits M. M., te Morsche R. H., Lauria G., Dib-Hajj S. D., Drenth J. P., Faber C. G., Merkies I. S., and Waxman S. G. (2012) Functional profiles of SCN9A variants in dorsal root ganglion neurons and superior cervical ganglion neurons correlate with autonomic symptoms in small fibre neuropathy. Brain 135, 2613–2628 [DOI] [PubMed] [Google Scholar]
- 14. Hoeijmakers J. G., Han C., Merkies I. S., Macala L. J., Lauria G., Gerrits M. M., Dib-Hajj S. D., Faber C. G., and Waxman S. G. (2012) Small nerve fibres, small hands and small feet: a new syndrome of pain, dysautonomia and acromesomelia in a kindred with a novel NaV1.7 mutation. Brain 135, 345–358 [DOI] [PubMed] [Google Scholar]
- 15. Hoeijmakers J. G., Faber C. G., Lauria G., Merkies I. S., and Waxman S. G. (2012) Small-fibre neuropathies: advances in diagnosis, pathophysiology and management. Nat. Rev. Neurol. 8, 369–379 [DOI] [PubMed] [Google Scholar]
- 16. Cox J. J., Reimann F., Nicholas A. K., Thornton G., Roberts E., Springell K., Karbani G., Jafri H., Mannan J., Raashid Y., Al-Gazali L., Hamamy H., Valente E. M., Gorman S., Williams R., et al. (2006) An SCN9A channelopathy causes congenital inability to experience pain. Nature 444, 894–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ahmad S., Dahllund L., Eriksson A. B., Hellgren D., Karlsson U., Lund P. E., Meijer I. A., Meury L., Mills T., Moody A., Morinville A., Morten J., O'Donnell D., Raynoschek C., Salter H., et al. (2007) A stop codon mutation in SCN9A causes lack of pain sensation. Hum. Mol. Genet. 16, 2114–2121 [DOI] [PubMed] [Google Scholar]
- 18. Goldberg Y. P., MacFarlane J., MacDonald M. L., Thompson J., Dube M. P., Mattice M., Fraser R., Young C., Hossain S., Pape T., Payne B., Radomski C., Donaldson G., Ives E., Cox J., et al. (2007) Loss-of-function mutations in the Nav1.7 gene underlie congenital indifference to pain in multiple human populations. Clin. Genet. 71, 311–319 [DOI] [PubMed] [Google Scholar]
- 19. Dib-Hajj S. D., Cummins T. R., Black J. A., and Waxman S. G. (2007) From genes to pain: Nav1.7 and human pain disorders. Trends Neurosci. 30, 555–563 [DOI] [PubMed] [Google Scholar]
- 20. Drenth J. P., and Waxman S. G. (2007) Mutations in sodium-channel gene SCN9A cause a spectrum of human genetic pain disorders. J. Clin. Invest. 117, 3603–3609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Burns T. M., Te Morsche R. H., Jansen J. B., and Drenth J. P. (2005) Genetic heterogeneity and exclusion of a modifying locus at 2q in a family with autosomal dominant primary erythermalgia. Br. J. Dermatol. 153, 174–177 [DOI] [PubMed] [Google Scholar]
- 22. Cummins T. R., Howe J. R., and Waxman S. G. (1998) Slow closed-state inactivation: a novel mechanism underlying ramp currents in cells expressing the hNE/PN1 sodium channel. J. Neurosci. 18, 9607–9619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ahuja S., Mukund S., Deng L., Khakh K., Chang E., Ho H., Shriver S., Young C., Lin S., Johnson J. P. Jr., Wu P., Li J., Coons M., Tam C., Brillantes B., et al. (2015) Structural basis of Nav1.7 inhibition by an isoform-selective small-molecule antagonist. Science 350, aac5464. [DOI] [PubMed] [Google Scholar]
- 24. Schmidt D., Jiang Q. X., and MacKinnon R. (2006) Phospholipids and the origin of cationic gating charges in voltage sensors. Nature 444, 775–779 [DOI] [PubMed] [Google Scholar]
- 25. Xu Y., Ramu Y., and Lu Z. (2008) Removal of phospho-head groups of membrane lipids immobilizes voltage sensors of K+ channels. Nature 451, 826–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rush A. M., Dib-Hajj S. D., Liu S., Cummins T. R., Black J. A., and Waxman S. G. (2006) A single sodium channel mutation produces hyper- or hypoexcitability in different types of neurons. Proc. Natl. Acad. Sci. U.S.A. 103, 8245–8250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dib-Hajj S. D., Black J. A., and Waxman S. G. (2009) Voltage-gated sodium channels: therapeutic targets for pain. Pain Med. 10, 1260–1269 [DOI] [PubMed] [Google Scholar]
- 28. Han C., Dib-Hajj S. D., Lin Z., Li Y., Eastman E. M., Tyrrell L., Cao X., Yang Y., and Waxman S. G. (2009) Early- and late-onset inherited erythromelalgia: genotype-phenotype correlation. Brain 132, 1711–1722 [DOI] [PubMed] [Google Scholar]
- 29. Han C., Rush A. M., Dib-Hajj S. D., Li S., Xu Z., Wang Y., Tyrrell L., Wang X., Yang Y., and Waxman S. G. (2006) Sporadic onset of erythermalgia: a gain-of-function mutation in Nav1.7. Ann. Neurol. 59, 553–558 [DOI] [PubMed] [Google Scholar]
- 30. Han C., Yang Y., de Greef B. T., Hoeijmakers J. G., Gerrits M. M., Verhamme C., Qu J., Lauria G., Merkies I. S., Faber C. G., Dib-Hajj S. D., and Waxman S. G. (2015) The domain II S4-S5 linker in Nav1.9: a missense mutation enhances activation, impairs fast inactivation, and produces human painful neuropathy. Neuromolecular Med. 17, 158–169 [DOI] [PubMed] [Google Scholar]
- 31. Eberhardt M., Nakajima J., Klinger A. B., Neacsu C., Hühne K., O'Reilly A. O., Kist A. M., Lampe A. K., Fischer K., Gibson J., Nau C., Winterpacht A., and Lampert A. (2014) Inherited pain: sodium channel Nav1.7 A1632T mutation causes erythromelalgia due to a shift of fast inactivation. J. Biol. Chem. 289, 1971–1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Stoffels M., and Kastner D. L. (2016) Old dogs, new tricks: monogenic autoinflammatory disease unleashed. Annu. Rev. Genomics Hum. Genet. 17, 245–272 [DOI] [PubMed] [Google Scholar]
- 33. Jimenez-Andrade J. M., Mantyh W. G., Bloom A. P., Xu H., Ferng A. S., Dussor G., Vanderah T. W., and Mantyh P. W. (2010) A phenotypically restricted set of primary afferent nerve fibers innervate the bone versus skin: therapeutic opportunity for treating skeletal pain. Bone 46, 306–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hurrell D. J. (1937) The nerve supply of bone. J. Anat. 72, 54–61 [PMC free article] [PubMed] [Google Scholar]
- 35. Chiu S. Y., Schrager P., and Ritchie J. M. (1984) Neuronal-type Na+ and K+ channels in rabbit cultured Schwann cells. Nature 311, 156–157 [DOI] [PubMed] [Google Scholar]
- 36. Bevan S., Chiu S. Y., Gray P. T., and Ritchie J. M. (1985) The presence of voltage-gated sodium, potassium and chloride channels in rat cultured astrocytes. Proc. R. Soc. Lond. B Biol. Sci. 225, 299–313 [DOI] [PubMed] [Google Scholar]
- 37. Black J. A., and Waxman S. G. (2013) Noncanonical roles of voltage-gated sodium channels. Neuron 80, 280–291 [DOI] [PubMed] [Google Scholar]
- 38. Black J. A., Westenbroek R. E., Catterall W. A., and Waxman S. G. (1995) Type II brain sodium channel expression in non-neuronal cells: embryonic rat osteoblasts. Brain Res. Mol. Brain Res. 34, 89–98 [DOI] [PubMed] [Google Scholar]
- 39. Ding Y., Arai M., Kondo H., and Togari A. (2010) Effects of capsaicin-induced sensory denervation on bone metabolism in adult rats. Bone 46, 1591–1596 [DOI] [PubMed] [Google Scholar]
- 40. Hukkanen M., Konttinen Y. T., Santavirta S., Paavolainen P., Gu X. H., Terenghi G., and Polak J. M. (1993) Rapid proliferation of calcitonin gene-related peptide-immunoreactive nerves during healing of rat tibial fracture suggests neural involvement in bone growth and remodelling. Neuroscience 54, 969–979 [DOI] [PubMed] [Google Scholar]
- 41. Hill E. L., and Elde R. (1991) Distribution of CGRP-, VIP-, DβH-, SP-, and NPY-immunoreactive nerves in the periosteum of the rat. Cell Tissue Res. 264, 469–480 [DOI] [PubMed] [Google Scholar]
- 42. Bjurholm A., Kreicbergs A., Brodin E., and Schultzberg M. (1988) Substance P- and CGRP-immunoreactive nerves in bone. Peptides 9, 165–171 [DOI] [PubMed] [Google Scholar]
- 43. Hite R. K., Butterwick J. A., and MacKinnon R. (2014) Phosphatidic acid modulation of Kv channel voltage sensor function. Elife 10.7554/eLife.04366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang X., Ren W., DeCaen P., Yan C., Tao X., Tang L., Wang J., Hasegawa K., Kumasaka T., He J., Wang J., Clapham D. E., and Yan N. (2012) Crystal structure of an orthologue of the NaChBac voltage-gated sodium channel. Nature 486, 130–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shen H., Zhou Q., Pan X., Li Z., Wu J., and Yan N. (2017) Structure of a eukaryotic voltage-gated sodium channel at near-atomic resolution. Science 355, eaal4326. [DOI] [PubMed] [Google Scholar]
- 46. Zhang X., Wang Y., Linghua R., Feng A., Ketai H., Liu T., and Niu J. (2011) Human dimensions of Chinese minors (GB/T 26158–2010), Standardization Administration of the People's Republic of China, Standards Press of China, Beijing: (in Chinese) [Google Scholar]
- 47. Yang Y., Wang Y., Li S., Xu Z., Li H., Ma L., Fan J., Bu D., Liu B., Fan Z., Wu G., Jin J., Ding B., Zhu X., and Shen Y. (2004) Mutations in SCN9A, encoding a sodium channel α subunit, in patients with primary erythermalgia. J. Med. Genet. 41, 171–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Herzog R. I., Cummins T. R., Ghassemi F., Dib-Hajj S. D., and Waxman S. G. (2003) Distinct repriming and closed-state inactivation kinetics of Nav1.6 and Nav1.7 sodium channels in mouse spinal sensory neurons. J. Physiol. 551, 741–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lossin C., Wang D. W., Rhodes T. H., Vanoye C. G., and George A. L. Jr. (2002) Molecular basis of an inherited epilepsy. Neuron 34, 877–884 [DOI] [PubMed] [Google Scholar]
- 50. Cummins T. R., Rush A. M., Estacion M., Dib-Hajj S. D., and Waxman S. G. (2009) Voltage-clamp and current-clamp recordings from mammalian DRG neurons. Nat. Protoc. 4, 1103–1112 [DOI] [PubMed] [Google Scholar]
- 51. Song Y., DiMaio F., Wang R. Y., Kim D., Miles C., Brunette T., Thompson J., and Baker D. (2013) High-resolution comparative modeling with RosettaCM. Structure 21, 1735–1742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rohl C. A., Strauss C. E., Misura K. M., and Baker D. (2004) Protein structure prediction using Rosetta. Methods Enzymol. 383, 66–93 [DOI] [PubMed] [Google Scholar]
- 53. Yarov-Yarovoy V., DeCaen P. G., Westenbroek R. E., Pan C. Y., Scheuer T., Baker D., and Catterall W. A. (2012) Structural basis for gating charge movement in the voltage sensor of a sodium channel. Proc. Natl. Acad. Sci. U.S.A. 109, E93–E102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yarov-Yarovoy V., Schonbrun J., and Baker D. (2006) Multipass membrane protein structure prediction using Rosetta. Proteins 62, 1010–1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Barth P., Schonbrun J., and Baker D. (2007) Toward high-resolution prediction and design of transmembrane helical protein structures. Proc. Natl. Acad. Sci. U.S.A. 104, 15682–15687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bonneau R., Strauss C. E., Rohl C. A., Chivian D., Bradley P., Malmström L., Robertson T., and Baker D. (2002) De novo prediction of three-dimensional structures for major protein families. J. Mol. Biol. 322, 65–78 [DOI] [PubMed] [Google Scholar]
- 57. Mandell D. J., Coutsias E. A., and Kortemme T. (2009) Sub-angstrom accuracy in protein loop reconstruction by robotics-inspired conformational sampling. Nat. Methods 6, 551–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera: a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.