

Abstract
Voltage-dependent anion channel-1 (VDAC1) is a highly regulated β-barrel membrane protein that mediates transport of ions and metabolites between the mitochondria and cytosol of the cell. VDAC1 co-purifies with cholesterol and is functionally regulated by cholesterol, among other endogenous lipids. Molecular modeling studies based on NMR observations have suggested five cholesterol-binding sites in VDAC1, but direct experimental evidence for these sites is lacking. Here, to determine the sites of cholesterol binding, we photolabeled purified mouse VDAC1 (mVDAC1) with photoactivatable cholesterol analogues and analyzed the photolabeled sites with both top-down mass spectrometry (MS), and bottom-up MS paired with a clickable, stable isotope-labeled tag, FLI-tag. Using cholesterol analogues with a diazirine in either the 7 position of the steroid ring (LKM38) or the aliphatic tail (KK174), we mapped a binding pocket in mVDAC1 localized to Thr83 and Glu73, respectively. When Glu73 was mutated to a glutamine, KK174 no longer photolabeled this residue, but instead labeled the nearby Tyr62 within this same binding pocket. The combination of analytical strategies employed in this work permits detailed molecular mapping of a cholesterol-binding site in a protein, including an orientation of the sterol within the site. Our work raises the interesting possibility that cholesterol-mediated regulation of VDAC1 may be facilitated through a specific binding site at the functionally important Glu73 residue.
Keywords: cholesterol, lipid binding protein, photoaffinity labeling, protein drug interaction, voltage-dependent anion channel (VDAC)
Introduction
Voltage-dependent anion channel-1 (VDAC1)3 is a highly regulated β-barrel membrane protein that mediates transport of ions and metabolites between the mitochondria and cytosol, and is implicated in the control of mitochondrial respiration and apoptosis (1, 2). Among the endogenous factors that alter VDAC1 function, membrane lipids have been the subject of several studies (3–8), in part, because of the marked changes that occur in mitochondrial lipid composition with apoptosis (9) and neurodegenerative diseases (10). Sterols and anionic phospholipids affect VDAC selectivity (6), voltage-dependent gating (3, 7), and oligomerization (4). Furthermore, sterols co-purify with VDAC from fungi (11) and bovine heart (12), and are important for the functional reconstitution of purified VDAC from eukaryotic expression systems (7).
Compared with α-helical integral membrane proteins (IMPs), relatively few structural studies of β-barrel IMPs have delineated binding sites for membrane lipids (13, 14), and this is particularly true of cholesterol (15–17). Based on a VDAC NMR study that reported chemical shifts at multiple residues in the presence of cholesterol, five cholesterol-binding sites were recently proposed by molecular modeling (5, 8). Experimental evidence for the sites of cholesterol interaction in VDAC is otherwise lacking.
One approach to map lipid-binding sites is photoaffinity labeling, in which IMPs are covalently labeled with photoactivatable lipid analogues and the sites of interaction are identified by bottom-up mass spectrometry (MS) (18, 19). In bottom-up MS, the photolabeled IMP is digested into peptides, and the photolabeled sites are determined by identifying and sequencing the associated peptide-ligand adducts. We have found this process to be particularly challenging for sterol-based ligands because the resulting peptide adducts have poor aqueous solubility and consistently undergo adduct loss with conventional collision-induced dissociation fragmentation (20). To overcome these challenges, we have employed FLI-tag, a clickable stable isotope-labeled tag, which enables peptide adducts to be identified by an MS1 level search (20). We have also utilized top-down MS to gain complementary information regarding the efficiency, stoichiometry, and sites of photolabeling. Using two analogues with a photoactivatable diazirine in different positions, we have mapped a cholesterol binding pocket with a preferred ligand orientation in mVDAC1.
Results
Cholesterol analogues photolabel mVDAC1 with variable efficiency and a stoichiometry of one
To determine cholesterol-binding sites in VDAC1, we photolabeled WT mouse VDAC1 (mVDAC1) with two cholesterol analogues that have a photoactive diazirine in either the 7-position (LKM38) (21) or the aliphatic tail (KK174) (see Fig. 1A for structures and supplemental materials for KK174 synthesis). These reagents were designed to permit mapping of cholesterol binding pockets by placing the diazirine in different positions. They also contain an alkyne allowing for the attachment of FLI-tag to facilitate identification of peptide-steroid adducts (20). Top-down MS of photolabeled mVDAC1 was first performed to characterize the efficiency and stoichiometry of labeling. The masses of mVDAC1 photolabeled by LKM38 and KK174 are listed in Table 1. First, mVDAC1 was photolabeled three times with 100 μm of cholesterol analogue added each time. LKM38, which has the diazirine in the 7-position, photolabeled mVDAC1 with an efficiency of only ∼38% (Fig. 1A). KK174, which has the diazirine in the aliphatic tail, photolabeled mVDAC1 with 100% efficiency; the absence of a feature corresponding to mVDAC1 photolabeled with two ligands is consistent with a stoichiometry of one (Fig. 1A). HCD fragmentation of intact mVDAC1 gives limited sequence coverage, and therefore top-down mass spectrometry can only localize photo-incorporation of the ligands to a general region within the protein. Fragmentation of photolabeled mVDAC1 showed a series of b ions that localize the photolabeled site to a region encompassing residues 44–120 for LKM38, and residues 44–128 for KK174 (Fig. 1B). The variable photolabeling efficiencies of these two analogues under exhaustive photolabeling conditions suggest that they may photolabel different residues within this region. We also tested KK174 photolabeling of mVDAC1 at different concentrations, which demonstrated concentration-dependent labeling that saturates above 100 μm (Fig. 2, A and B). The apparent EC50 of photolabeling is 15.3 μm (Fig. 2B), which is a function of both binding and photochemical efficiency and thus not indicative of binding affinity.
Figure 1.
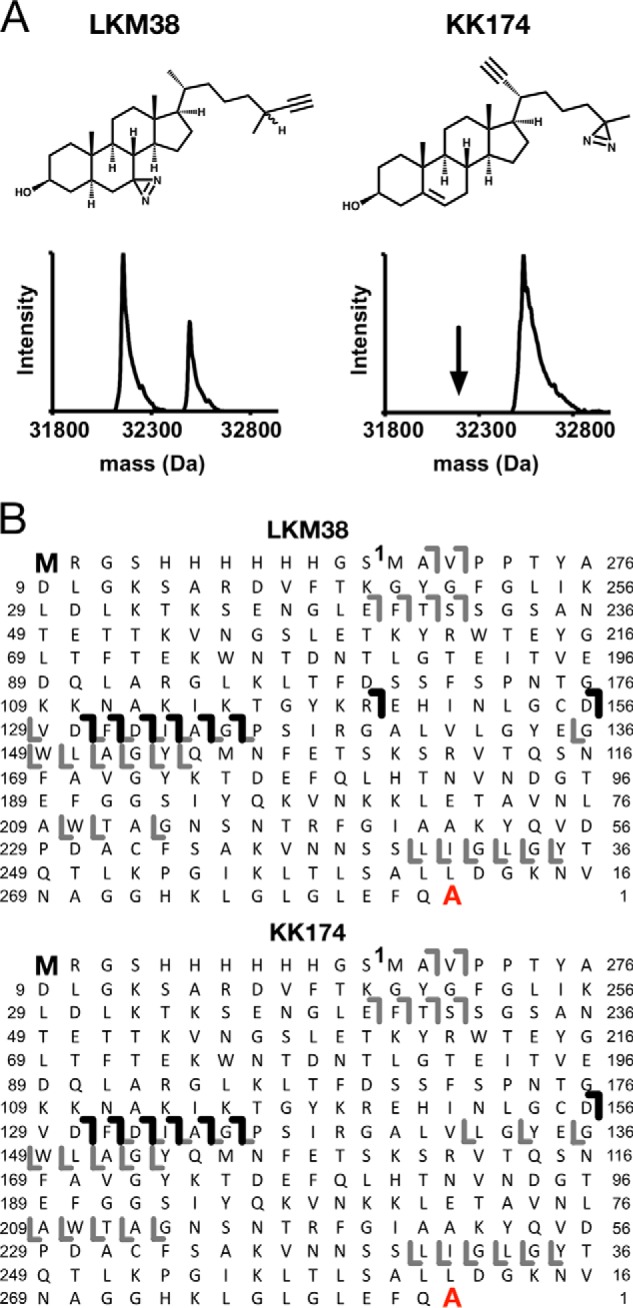
Top-down MS of mVDAC1 photolabeled with cholesterol analogues. A, chemical structures of cholesterol analogues, and associated deconvoluted spectra of mVDAC1 photolabeled with LKM38 and KK174. For LKM38, two peaks in the deconvoluted spectrum represent unlabeled and singly labeled mVDAC1 at 32,157 and 32,553 Da, respectively. For KK174, the one peak represents singly labeled mVDAC1 at 32,537 Da. B, HCD fragment ion assignments obtained by isolating the LKM38- and KK174-photolabeled species, respectively. Amino acids are numbered to exclude the first 12 residues in the N terminus that comprise the affinity tag. The gray lines represent b and y ions that do not contain a ligand adduct. The black lines represent b ions that do contain a ligand adduct and were identified by searching the fragmentation data against a database in which the ligand mass was added to the N-terminal methionine. The absence of red lines indicates an absence of y ions identified by searching against a database in which the ligand mass was added to the C-terminal alanine.
Table 1.
Masses of photolabeled mVDAC1 acquired on the Elite LTQ
| Average mass | Theoretical mass | Mass accuracy | |
|---|---|---|---|
| Da | ppm | ||
| WT LKM38 | 32553.0 | 32550.38 | 81 |
| WT KK174 | 32537.0 | 32534.35 | 82 |
| E73Q LKM38 | 32550.0 | 32549.39 | 19 |
| E73Q KK174 | 32537.0 | 32533.36 | 112 |
| E73A LKM38 | 32493.0 | 32492.34 | 20 |
| E73A KK174 | 32477.4 | 32476.31 | 34 |
Figure 2.
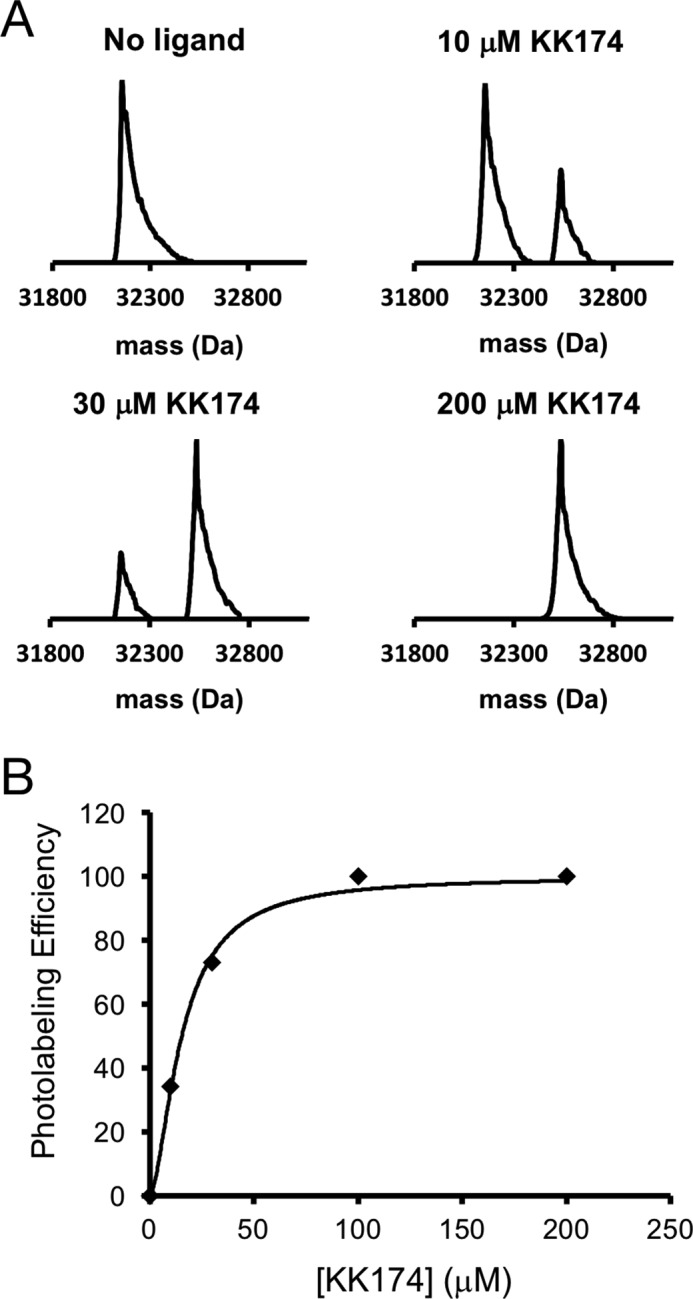
Concentration-dependent photolabeling of mVDAC1 by KK174 demonstrates saturation and a stoichiometry of one. A, deconvoluted spectra of mVDAC1 photolabeled with KK174 at 0, 10, 30, and 200 μm. The theoretical average masses of unlabeled mVDAC1, singly labeled mVDAC1, and doubly labeled mVDAC1 are 32,154, 32,534, and 32,914 Da, respectively. B, scatter plot of the photolabeling efficiency of mVDAC1 by KK174. Data are fit with a four-parameter logistic equation, which yields an apparent EC50 of 15.3 μm and Hill slope of 1.6.
LKM38 and KK174 define a cholesterol binding pocket in WT mVDAC1 by photolabeling Thr83 and Glu73
To determine the exact residues photolabeled by LKM38 and KK174, mVDAC1 was photolabeled with each reagent three times at 30 and 100 μm. The photolabeled proteins were derivatized with FLI-tag using copper-catalyzed cycloaddition. Bottom-up mass spectrometry was performed using an established protocol (22), and peptide adducts were identified using an automated pairs search followed by manual interpretation of the ETD fragmentation spectra. All photolabeled residues are listed in Table 2. We first examined KK174. A search of the MS1 data identified multiple paired features consistent with peptide-KK174 adducts (supplemental Table S1). Analysis of the ETD fragmentation spectra for each of these paired features localized the site of KK174 insertion to Glu73 (Fig. 3, A and B, supplemental Table S1). No other photolabeled residues were identified consistent with a stoichiometry of one (supplemental Table S1).
Table 2.
Summary of KK174 and LKM38 photolabeling sites in mVDAC1
| 100 μm × 3 | 30 μm | ||
|---|---|---|---|
| WT | KK174 | Glu73 | Glu73 |
| LKM38 | Thr83 | None identified | |
| E73Q | KK174 | Tyr62, Lys110, Tyr173, Asp230, Ala283 | Lys110 |
| LKM38 | None identified | None identified |
Figure 3.

Bottom-up MS of mVDAC1 photolabeled with cholesterol analogues maps a cholesterol-binding site. A, representative MS1 doublet corresponding to the peptide GLTFTEKW labeled with KK174 (z = 4) and coupled to light and heavy FLI-tag. B, fragmentation (ETD) spectra corresponding to the heavy feature in A. The site defining ions localize KK174 labeling to Glu73. C, a representative MS1 doublet corresponding to the peptide NTDNTLGTEITVEDQLARGL labeled with LKM38 (z = 4) and coupled to light and heavy FLI-tag. D, fragmentation (ETD) spectra corresponding to the light feature in C. Site defining ions localize LKM38 labeling to Thr83. E, a cholesterol-binding pose from AutoDock bound by Tyr62, Phe71, Glu73, Thr83, Ile85, and Ser101. F, same cholesterol binding pose as in E showing the aliphatic tail 3.5 Å from Glu73 and the 7 position 2.5 Å from Thr83, consistent with KK174 and LKM38 labeling of these residues.
For LKM38 we identified two paired features consistent with peptide-LKM38 adducts (supplemental Table S1). Both features localized the site of LKM38 insertion to Thr83 (Fig. 3, C and D). Interestingly, Thr83 is directly adjacent to Glu73 in the neighboring β-strand based on the mVDAC1 crystal structure (23). Despite being in such close proximity to Glu73, LKM38 selectively photolabels Thr83. No peptide adducts with Glu73 labeling were observed in the LKM38 sample (supplemental Table S1). To further explore whether these two residues form a single cholesterol-binding site, Autodock simulations were performed using cholesterol and the mVDAC1 structure (23, 24), with docking constrained to a 30 × 30-Å area centered on Glu73. After clustering 1000 poses, the two most populated clusters show cholesterol bound to a groove formed by Phe71, Glu73, Thr83, and Ile85 as well as Tyr62 and Ser101 on either end of the cholesterol molecule (Fig. 3E). The pose that most closely agrees with the experimental data shows, quite remarkably, that the end of the cholesterol aliphatic tail is 3.5 Å from Glu73 and position 7 of the steroid ring is 2.5 Å from Thr83 (Fig. 3F). In summary, using two cholesterol analogues with the photoactivatable diazirine in different positions, we have precisely mapped a cholesterol-binding site in mVDAC1.
Competitive inhibition of cholesterol photolabeling in mVDAC1
To confirm that the photolabeled site is an actual cholesterol-binding site, we photolabeled mVDAC1 with 1 μm KK174 or LKM38 in the presence of excess cholesterol. Photolabeled mVDAC1 was detected by coupling the photolabeled protein to TAMRA using click chemistry, separating the protein by SDS-PAGE, and visualizing with fluorescent imaging. Labeling by KK174 was inhibited by 30 μm cholesterol, and labeling by LKM38 showed a trend toward inhibition that was not statistically significant (Fig. 4, A and B). Competitive inhibition of photolabeling by cholesterol for KK174 is consistent with the conclusion that cholesterol binds to the site photolabeled by this analogue.
Figure 4.

Competitive inhibition of LKM38 and KK174 photolabeling of mVDAC1 by cholesterol. SDS-PAGE of WT mVDAC1 photolabeled for 1 min with 1 μm LKM38 (A) or KK174 (B) in the absence and presence of 10 or 30 μm cholesterol. The gel bands on the left show fluorescence signal with TAMRA or SYPRO Ruby. The scatter plots on the right show densitometry measurements of TAMRA signal from three separate experiments. Error bars indicate S.D. * indicates p < 0.05 and ** p < 0.01 based on an analysis of variance with Tukey HSD test.
Cholesterol analogues photolabel E73 mutants: top-down analysis
Top-down analysis of photolabeled E73Q and E73A mVDAC1 revealed reduced labeling efficiency in the mutants for both LKM38 (8% for E73Q and 13% for E73A) and KK174 (21% for E73Q and 10% for E73A) (Fig. 5, A and B). Moreover, top-down analysis with HCD fragmentation for both mutants identified photolabeled sites in two regions, compared with just one for WT (Fig. 5, C and D). LKM38 photolabeling of E73Q showed a series of b ions localizing a site or sites from residues 44–135, as well as a series of y ions localizing a site or sites from residues 133–209 (Fig. 5C). A similar pattern of altered HCD fragmentation was observed for KK174 labeling of E73Q (Fig. 5C) as well as KK174 and LKM38 photolabeling of E73A (Fig. 5D). This suggests that there is at least one additional site of cholesterol labeling in the E73Q and E73A mutants compared with WT.
Figure 5.

Top-down MS of E73Q and E73A mVDAC1 photolabeled with cholesterol analogues. A, deconvoluted spectra of E73Q mVDAC1 photolabeled with LKM38 and KK174. For LKM38, the two peaks represent unlabeled and singly labeled E73Q at 32,153 and 32,550 Da, respectively. For KK174, the two peaks represent unlabeled and singly labeled E73Q at 32,153 and 32,493 Da, respectively. B, same as A for E73A mVDAC1. For LKM38, the two peaks represent unlabeled and singly labeled E73A at 32,097 and 32,493 Da, respectively. For KK174, the two peaks represent unlabeled and singly labeled E73A at 32,097 and 32,477 Da, respectively. C, HCD fragment ion assignments obtained by isolating the LKM38- and KK174-E73Q photolabeled species, respectively. The gray lines represent b and y ions that do not contain a ligand adduct. The black lines represent b ions that contain ligand adduct identified by searching against a database in which the ligand mass was added to the N-terminal methionine. The red lines represent y ions that contain ligand adduct identified by searching against a database in which the ligand mass was added to the C-terminal alanine. D, same as C for E73A mVDAC1.
KK174 photolabels multiple residues in the E73Q mutant including Tyr62 in the cholesterol binding pocket
Bottom-up analysis was performed for LKM38 and KK174 photolabeling of E73Q. For KK174, we identified multiple peptide adducts (supplemental Table S1). Three peptide-KK174 adducts were identified in which KK174 insertion was localized to Tyr62 (Fig. 6, A and B). Interestingly, this residue is adjacent to Glu73 in the mVDAC1 structure. Based on the modeled cholesterol binding pocket, Tyr62 is 6.0 Å away from the flexible aliphatic tail of cholesterol using the most prominent docking pose (Fig. 6C). Thus, it appears that cholesterol still binds to a groove (Fig. 6D) defined by Thr83, Gln73, and Tyr62 in the E73Q mutant. However, most likely because tyrosine is more nucleophilic than glutamine, Tyr62 becomes the preferred target for KK174 over Gln73. In addition to Tyr62, four other residues were photolabeled by KK174 in the E73Q mutant: Lys110, Tyr173, Asp230, and Ala283 (Fig. 7, supplemental Table S1). KK174 labeling of the lysine residue, Lys110 (Fig. 7, A, B, and E), is observed with both exhaustive photolabeling (100 μm × 3) and photolabeling at 30 μm (supplemental Table S1). Labeling of Tyr173 (Fig. 7, C and D) is consistent with top-down fragmentation data where photolabeled sites in E73Q were localized to two regions including a C-terminal region, which was not detected in WT. Tyr173 is in close proximity to one of the sites previously identified by molecular modeling (25). We replicated this result by performing an Autodock simulation that searched the entire mVDAC1 structure, and identified a highly populated cluster within this site. The average pose of cholesterol in this site shows the flexible aliphatic tail bound by Trp149 and Val171, but also in close proximity to Tyr173 (∼8 Å) (Fig. 7F). Due to the low efficiency of LKM38 photolabeling in the mutants, we were unable to identify LKM38-labeled sites in E73Q by bottom-up mass spectrometry.
Figure 6.

Photolabeling a third residue, Tyr62, in the cholesterol binding pocket in E73Q mVDAC1. A, an MS1 doublet corresponding to the peptide TSSGSANTETTKVNGSLETKYRWTEY from E73Q mVDAC1 photolabeled with KK174 (z = 5) and coupled to light and heavy FLI-tag. B, fragmentation (ETD) spectra corresponding to the feature in A. Site defining ions localize KK174 photolabeling to Tyr62. C, the same cholesterol binding pose as in Fig. 2E showing Tyr62, Glu73, and Thr83. D, space filling view of the binding pocket mapped by KK174 and LKM38.
Figure 7.

Additional sites of KK174 photolabeling in E73Q mVDAC1. A, an MS1 doublet corresponding to the peptide TGKKNAKIKTGY labeled with KK174 (z = 4) and coupled to light and heavy FLI-tag. B, fragmentation (ETD) spectra corresponding to the heavy feature in A. Site defining ions localize KK174 labeling to Lys110. C, an MS1 doublet corresponding to the peptide AVGYKTDEF labeled with KK174 (z = 4) and coupled to light and heavy FLI-tag. D, fragmentation (ETD) spectra corresponding to the light feature in C. Site defining ions localize KK174 labeling to Tyr173. E, mVDAC1 crystal structure (PDB code 3emn) highlighting the photolabeled residue Lys110. F, a cholesterol binding pose from Autodock located within a previously identified cholesterol binding pocket (8) formed by Val171, Ala151, Leu144, Ile123, Tyr146, and Trp149. The aliphatic tail of cholesterol in this pose is near Tyr173, consistent with KK174 labeling of this residue in E73Q mVDAC1.
Mixing experiment to test for the formation of a long-lived reactive intermediate
Aliphatic diazirines are capable of forming short-lived reactive carbenes and also of rearranging to form long-lived intermediates, such as a diazo compound (26). Formation of long-lived intermediates increase the likelihood of ligand diffusion and labeling of residues remote from the binding site. To test if KK174 and LKM38 form long-lived reactive intermediates, we irradiated 30 μm of each reagent for 1 min prior to immediate mixing with mVDAC1. We then clicked the samples to TAMRA, separated the protein via SDS-PAGE, and visualized via fluorescent imaging. Irradiating LKM38 prior to mixing with mVDAC1 does not result in significant labeling, in contrast to KK174 which does (Fig. 8). This indicates that activated KK174 but not LKM38 forms a long-lived intermediate with a lifetime of seconds to minutes.
Figure 8.

Formation of a long-lived reactive intermediate for KK174 but not LKM38. SDS-PAGE of mVDAC1 photolabeled with 30 μm KK174 and LKM38. KK174 and LKM38 were UV irradiated for 1 min in the presence of mVDAC1 (UV Ligand + Protein), or in the absence of mVDAC1 followed by immediate mixing with mVDAC1 (UV Ligand). The gel bands show fluorescence signal with TAMRA (left) or SYPRO Ruby (right).
Discussion
Cholesterol is abundant and highly regulated in membranes, including the outer mitochondrial membrane where VDAC is the most abundant integral membrane protein (27, 28). The fact that multiple cholesterol molecules remain bound to VDAC after detergent solubilization and purification indicates a specific interaction (11, 12), which is likely mediating effects such as the modulation of VDAC gating (6, 11). However, the structural basis of cholesterol interaction with VDAC has not been well characterized. We utilized a photoaffinity labeling approach combined with top-down and bottom-up MS to identify and map cholesterol-binding sites in mVDAC1. Photolabeling efficiency and fragmentation patterns provided by top-down MS complemented our residue level site identification by bottom-up MS. Furthermore, derivatization with FLI-tag and subsequent ETD fragmentation enabled detection of sterol-based peptide adducts and site-defining ions, which are otherwise difficult to identify due to collision cell adduct loss (20).
Four key aspects of cholesterol photolabeling in WT mVDAC1 support the photolabeled residues being a specific cholesterol-binding site. First, residues photolabeled by two cholesterol analogues, LKM38 with the diazirine on the 7-position of the steroid ring, and KK174 with the diazirine on the aliphatic tail, map a single binding pocket in WT mVDAC1. Glu73 and Thr83, which are photolabeled by KK174 and LKM38, respectively, are separated by only 6 Å in the mVDAC1 crystal structure. KK174 photolabels Glu73 with high efficiency consistent with the photochemical preference of aliphatic diazirines for nucleophilic acidic amino acids (26). The fact that LKM38 labels Thr83, rather than the nearby, more nucleophilic glutamate residue, indicates that cholesterol is significantly constrained within this binding pocket.
A second aspect of cholesterol photolabeling that argues for specific binding is the photolabeling of E73Q mVDAC1. The conservative E73Q mutation abolishes KK174 photolabeling of residue 73, without disrupting binding at this site because KK174 then photolabels the nearby residue, Tyr62. With the diazirine at the end of a flexible aliphatic tail, we predict that KK174 photolabels Tyr62 within the constraints of the same binding pocket defined in WT mVDAC1. The orientation of cholesterol within this binding pocket agrees remarkably well with results from docking simulations.
Third, KK174 photolabels mVDAC1 with a stoichiometry of one at concentrations well above saturation (i.e. 100% mVDAC1 is photolabeled), indicating that this singular photolabeled site is highly specific. Even at concentrations greater than three times needed to saturate labeling at Glu73, a mVDAC1 species labeled with two KK174 is not observed by top-down MS. This finding is particularly remarkable in light of the fact that KK174 forms a long-lived reactive intermediate, which may be prone to diffuse and nonspecifically photolabel other sites.
Last, inhibition of KK174 photolabeling in WT mVDAC1 by excess cholesterol also argues for Glu73 being a cholesterol-binding site. Cholesterol inhibition of LKM38 photolabeling was modest and not statistically significant. We have found that competitive inhibition of ligand photolabeling also cannot be demonstrated for known ligand-receptor interactions such as the GABA receptor and neurosteroids (19). We suspect the reasons competition is difficult to demonstrate or replicate in certain cases include the low affinity of ligands such as neurosteroids or cholesterol, the irreversible nature of photolabeling combined with relatively long photolabeling times, and the possibility that photoactivatable analogues have different binding affinities than the ligands themselves (29). Thus, whereas the presence of inhibition by cholesterol of KK174 photolabeling confirms that Glu73 is a cholesterol-binding site, the lack of significant inhibition in the case of LKM38 does not lead to the opposite conclusion.
Glu73 is a uniquely positioned, charged amino acid in the transmembrane region of β-strand 4 that faces out into the membrane. Interestingly, this residue is an important determinant of VDAC1 channel activity and is implicated in hexokinase or ruthenium red-mediated inhibition of apoptosis (30). The presence of a cholesterol-binding site at Glu73 raises the interesting hypothesis that this site is mediating the effects of cholesterol on VDAC function (6, 11). Cholesterol binding at Glu73 may also be mediating interactions with other proteins, because Glu73 is critical for the interaction of hexokinase-I with VDAC (30) and potentially dimerization of VDAC itself (23).
We also observed that cholesterol analogues photolabel additional sites in E73Q compared with WT mVDAC1. Evidence for specific binding of cholesterol to these residues is limited. One site, Tyr173, is near one of five binding sites previously predicted by molecular modeling (8). Unfortunately, low photolabeling efficiency prevented the mapping of these sites using LKM38 in the E73Q mutant. Given the finding that KK174 can form a long-lived reactive intermediate, we cannot exclude the possibility that labeling of these residues is nonspecific, resulting from diffusion of a long-lived reactive intermediate. This is especially likely for Asp230, which is located in a soluble loop of the mVDAC1 protein.
We also cannot exclude the possibility that there are cholesterol sites not identified by our photolabeling experiments. Five cholesterol molecules were estimated to bind VDAC when purified from bovine heart (12). A previous molecular modeling study defined five cholesterol-binding sites based on the mVDAC1 structure and chemical shift data from an NMR study (5, 8). We recognize that our cholesterol analogues mainly photolabel nucleophilic residues and that the sites proposed in the computation study are predominantly composed of aliphatic amino acids and few hydrophilic residues. Thus, these other putative cholesterol-binding sites might not have been identified due to limitations in our photolabeling reagents. Alternatively, molecular docking to a static mVDAC1 structure or NMR data obtained in detergent micelles may not recapitulate the dynamic state of mVDAC1 in a membrane environment, where the cholesterol binding pocket we identified at Glu73 is predicted to have significant flexibility (31). Indeed, photoaffinity labeling provides an advantage over studies based on static crystal structures in that multiple protein conformations are likely sampled in a lipid environment.
In conclusion, we have identified a specific cholesterol-binding site in mVDAC1 using photoaffinity labeling. Using two cholesterol analogues with the diazirine in different positions, we have precisely mapped this site with photolabeling of Glu73, Thr83, and Tyr62. This work raises the interesting hypothesis that cholesterol regulation of VDAC1 may be mediated through a specific binding site at this functionally important residue. To our knowledge, this is the first time a sterol-binding site has been defined with photolabeling reagents in such molecular detail. This approach could be applied to precisely map sterol-binding sites in other integral membrane proteins.
Experimental procedures
Expression and purification of mVDAC1 from Escherichia coli
Protein expression and purification were performed as described previously (23). In short, mVDAC1 carrying a His6 tag was expressed using M15 E. coli cells. Inclusion bodies were isolated, solubilized, and mVDAC1 was purified on a Talon affinity column. The protein was then refolded and a size exclusion chromatography step allowed isolation of a homogeneous population.
Top-down MS of photolabeled mVDAC1
Reconstitution of mVDAC1, photolabeling, and top-down MS was performed as previously described (32–35). 120 μg of WT or mutant mVDAC1 was reconstituted by adding a 35% solution of l-α -dimyristoylphosphatidylcholine/CHAPSO bicelles in a 4:1 volume ratio of protein to bicelles and incubating on ice for 30 min. Samples were then diluted to 100 μl with 20 mm Tris buffer, pH 8, and photolabeled with cholesterol analogues at different concentrations via irradiation in a quartz cuvette with >320 nm UV light as previously described (35). 30 μg were removed for bottom-up analysis. The samples were then reduced with 250 mm DTT as previously described (36). Briefly, methanol, chloroform, and water in approximately equal volumes were added to the sample, vortexed, and centrifuged. The upper phase was removed and methanol and water were added and centrifuged three more times to further wash the sample. After the final wash, the precipitated protein was pelleted, reconstituted in 2 μl of 90% formic acid (37), and then diluted in 120 μl of chloroform/methanol/water (4:4:1). Top-down MS analysis was performed by direct injection of this sample at 3 μl/min using a Max Ion API source with a HESI-II probe into an Orbitrap Elite MS (Thermo Scientific, Waltham, MA). Full spectra of intact photolabeled mVDAC1 were acquired on the LTQ using a spray voltage of 4 kV, capillary temperature of 320 °C, in source dissociation voltage of 30 V, and otherwise standard tune parameters. The HCD/C-trap ΔP was adjusted to between 0.05 and 0.1 to optimize Orbitrap analysis of fragmentation spectra. HCD fragmentation spectra were acquired at 60,000 resolution, with an AGC target of 5 × 105, normalized energy of 10, activation time of 0.1 ms, and 4 m/z isolation window of the 31+ charge state. Deconvolution of full spectra from the LTQ was performed using MagTran (38). MASH was used for deconvolution of isotopically-resolved spectra to obtain monoisotopic masses, and to analyze top-down HCD fragmentation data (39). Fragment ions were searched using a mass accuracy of 1.1 Da to account for the “δ 1 Da” error (33). Fragment ions containing ligand were identified by adding the monoisotopic mass of the ligand to the N-terminal or C-terminal amino acids and searching for fragment ions plus ligand. Each identified fragment ion was manually verified and accepted if within 10 ppm. Fit of the concentration dependence of KK174 photolabeling efficiency was performed in Excel using a standard logistic binding equation,
| (Eq. 1) |
where max is maximum labeling efficiency set at 100%, min is minimum labeling efficiency set at 0%.
Click chemistry and endoproteolytic digestion of photolabeled mVDAC1
Aliquots of photolabeled VDAC were diluted to 100 μl and SDS was added to 1%. FLI-tag was covalently attached at sites of ligand incorporation via copper-catalyzed cyclo-addition (click chemistry) as previously described (40). Briefly, 2 mm C6H7NaO6, 0.1 mm Tris(benzyltriazolylmethyl)amine, and 1 mm CuSO4 were added and the reaction was initiated by 0.5 mm FLI-tag (in a 1:1 light to heavy ratio). The reaction was kept at room temperature overnight. As previously described (22), the protein was precipitated with a 2D SDS clean-up kit (GE Healthcare) and delipidated with 5% RapigestTM (in 20 μl of 100 mm Tris, pH 8.0, 8 m urea) at 37 °C for 1 h. The protein was precipitated a second time and resuspended in 8 m urea, 0.2% RapigestTM. The protein was then reduced with 5 mm triscarboxyethylphosphine, alkylated with 5 mm idoacetamide, and alkylation was quenched with 5 mm DTT. The protein was digested overnight at 37 °C with 2 μg of endoprotease LysC, and then diluted to 1 m urea with 100 mm Tris, pH 8.0, + 10 mm CaCl2 and digested with 2 μg of chymotrypsin for 3 or 6 h. Digestions were stopped with 1% formic acid (FA) and incubated at 37 °C for 30 min before peptides were sequentially extracted with C4 and C18 stage tips. Peptides were eluted from stage tips with 10% ACN, 1% FA, dried via vacuum centrifugation, resuspended in 10% ACN, 1% FA, and analyzed by LC/MS/MS on an Orbitrap Elite mass spectrometer.
Liquid chromatography and high resolution tandem mass spectrometry
Samples were analyzed as previously described (20). Samples were separated on a home packed C18 column (15 cm × 75 μm × 200 Å) and introduced to the mass spectrometer with a nanospray source. Samples were separated on a 102-min gradient (5 min hold at 10% ACN, 1% FA, increase to 90% ACN, 1% FA over 85 min, hold at 90% ACN, 1% FA, flow rate = 800 nl/min). Survey scans were collected in the Orbitrap (res 60,000) and MS2 (MS/MS) scans were acquired using data-dependent acquisition of the 20 most abundant features. MS2 spectra were acquired using either collision-induced dissociation (normalized collision energy = 35) or ETD (100 ms activation time). Dynamic exclusion limited MS2 collection of a given m/z to one event every 15 s. ETD MS2 collection was limited to features with z > = 3.
MS1-based pair search
Bottom-up spectra were analyzed via a pair search as previously described (20). Briefly, .raw data were converted to .mzml via MSConvert. OpenMS featurefinder multiplex tool was used to identify pairs separated by 10.008 Da and results were processed in Knime to exclude pairs with integral of intensity differences greater than 50%. The feature list created in Knime was exported to Excel and z > = 2 pairs were manually analyzed in Xcalibur for correct charge state and isotope distribution.
MS2-based software searches
ETD fragmentation spectra were searched against a database containing the mVDAC1 sequence (UniProtKB code Q60932) modified with a His6 tag and filtered at a 1% false discovery rate using PEAKS 8.0 (Bioinformatics Solution Inc.). Met oxidation and Cys carbamidomethylation were included in each sample as variable modifications. Customized modifications were created for the add weight of KK174 + FLI-tag (light m/z = 680.474, heavy m/z = 690.482) and LKM38 + FLI-tag (light m/z = 696.505, heavy m/z = 706.513).
Gel-based competition assay
mVDAC1 was photolabeled for 1 min with 1 μm LKM38 and KK174 in the absence or presence of 10 or 30 μm cholesterol. Samples were clicked to TAMRA azide (Thermo Fisher Scientific) by mixing the samples with 2 mm sodium ascorbate, 0.1 mm Tris(benzyltriazolylmethyl)amine, and 1 mm copper sulfate, followed by 100 μm TAMRA azide. The reaction proceeded overnight at room temperature. The samples were then run on a 12% Tris glycine gel and imaged using a Typhon FLA 9500 scanner (GE Healthcare) set for TAMRA fluorescence (550/580 nm excitation/emission). The gels were then stained overnight for SYPRO Ruby imaging. Densitometry was performed using ImageJ and statistical analysis was performed using analysis of variance with a Tukey HSD test.
Mixing experiment
30 μm KK174 and LKM38 were irradiated with UV for 1 min in the presence or absence of mVDAC1. Reagent irradiated in the absence of mVDAC1 was immediately mixed with protein following UV irradiation. All samples were then incubated in the dark for 5 min before being denatured with SDS and clicked to TAMRA azide. SDS-PAGE, TAMRA fluorescence imaging, and SYPRO Ruby imaging were performed as described above.
Autodock analysis of the cholesterol-binding site
Cholesterol binding poses were obtained with AutoDock 4.2 (24). Docking was performed with the mVDAC1 crystal structure (PDB code 3emn) prepared in AutoDock Tools by deleting waters, adding hydrogen, computing Gasteiger charges, and merging non-polar hydrogens. The cholesterol ligand was prepared by converting an sdf file from Pubchem into a pdb file using Open Babel (41), and partial charges and torsion angles were calculated using PRODRG set for reduced charges and energy minimization (42). The AutoDock grid box was chosen to be a 30 × 30 × 30-Å space entered on Glu73 with 0.3-Å spacing. Alternatively, docking was also performed with a grid that encompassed the entire protein with 1.0-Å spacing. Docking was performed using the genetic algorithm, 1000 runs, and otherwise default parameters. Clustering of the resulting 1000 poses at 2-Å root mean square deviation yielded multiple clusters, which were considered for comparison with photolabeled data.
Author contributions
W. W. C. and M. M. B. contributed to the conception, design, acquisition, and analysis of data, and writing of manuscript. A. S. E. contributed to the conception, design, and writing of manuscript. L. B. and J. A. purified mVDAC1 and edited the manuscript. J. P. W. and Z. C. contributed to the analysis and interpretation of data. D. F. C., L. M. M., and K. K. contributed to the synthesis of cholesterol analogues. J. W. J. assisted in the synthesis of FLI-tag. All authors reviewed and approved the content of the final manuscript.
Supplementary Material
Acknowledgment
We thank Sunjoo Lee for assistance in setting up Autodock simulations.
This work was supported, in whole or in part, by National Institutes of Health Grants R01GM108799, R01GM078844, R21AI120490–01, P01GM47969, T32-GM108539, and R01HL067773, and T32 HL007275, National Science Foundation Grant DGE-1143954, and the Taylor Family Institute for Innovative Psychiatric Research. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Table S1.
- VDAC1
- voltage-dependent anion channel-1
- IMP
- integral membrane protein
- CHAPSO
- 3-[(3-cholamidopropyl)dimethylammonio]-2-hydroxy-1-propanesulfonic acid
- FA
- formic acid
- ACN
- acetonitrile.
References
- 1. McCommis K. S., and Baines C. P. (2012) The role of VDAC in cell death: friend or foe? Biochim. Biophys. Acta 1818, 1444–1450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Colombini M. (2012) VDAC structure, selectivity, and dynamics. Biochim. Biophys. Acta 1818, 1457–1465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rostovtseva T. K., Kazemi N., Weinrich M., and Bezrukov S. M. (2006) Voltage gating of VDAC is regulated by nonlamellar lipids of mitochondrial membranes. J. Biol. Chem. 281, 37496–37506 [DOI] [PubMed] [Google Scholar]
- 4. Betaneli V., Petrov E. P., and Schwille P. (2012) The role of lipids in VDAC oligomerization. Biophys. J. 102, 523–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hiller S., Garces R. G., Malia T. J., Orekhov V. Y., Colombini M., and Wagner G. (2008) Solution structure of the integral human membrane protein VDAC-1 in detergent micelles. Science 321, 1206–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mlayeh L., Chatkaew S., Léonetti M., and Homblé F. (2010) Modulation of plant mitochondrial VDAC by phytosterols. Biophys. J. 99, 2097–2106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Popp B., Schmid A., and Benz R. (1995) Role of sterols in the functional reconstitution of water-soluble mitochondrial porins from different organisms. Biochemistry 34, 3352–3361 [DOI] [PubMed] [Google Scholar]
- 8. Weiser B. P., Salari R., Eckenhoff R. G., and Brannigan G. (2014) Computational investigation of cholesterol binding sites on mitochondrial VDAC. J. Phys. Chem. B 118, 9852–9860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Crimi M., and Esposti M. D. (2011) Apoptosis-induced changes in mitochondrial lipids. Biochim. Biophys. Acta 1813, 551–557 [DOI] [PubMed] [Google Scholar]
- 10. Aufschnaiter A., Kohler V., Diessl J., Peselj C., Carmona-Gutierrez D., Keller W., and Büttner S. (2017) Mitochondrial lipids in neurodegeneration. Cell Tissue Res. 367, 125–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Freitag H., Neupert W., and Benz R. (1982) Purification and characterisation of a pore protein of the outer mitochondrial membrane from Neurospora crassa. Eur. J. Biochem. 123, 629–636 [DOI] [PubMed] [Google Scholar]
- 12. De Pinto V., Benz R., and Palmieri F. (1989) Interaction of non-classical detergents with the mitochondrial porin: a new purification procedure and characterization of the pore-forming unit. Eur. J. Biochem. 183, 179–187 [DOI] [PubMed] [Google Scholar]
- 13. Lee A. G. (2003) Lipid–protein interactions in biological membranes: a structural perspective. Biochim. Biophys. Acta 1612, 1–40 [DOI] [PubMed] [Google Scholar]
- 14. Galdiero S., Galdiero M., and Pedone C. (2007) β-Barrel membrane bacterial proteins: structure, function, assembly and interaction with lipids. Curr. Protein Pept. Sci. 8, 63–82 [DOI] [PubMed] [Google Scholar]
- 15. Hanson M. A., Cherezov V., Griffith M. T., Roth C. B., Jaakola V. P., Chien E. Y., Velasquez J., Kuhn P., and Stevens R. C. (2008) A specific cholesterol binding site is established by the 2.8-Å structure of the human β2-adrenergic receptor. Structure 16, 897–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shinoda T., Ogawa H., Cornelius F., and Toyoshima C. (2009) Crystal structure of the sodium-potassium pump at 2.4-Å resolution. Nature 459, 446–450 [DOI] [PubMed] [Google Scholar]
- 17. Ruprecht J. J., Mielke T., Vogel R., Villa C., and Schertler G. F. (2004) Electron crystallography reveals the structure of metarhodopsin I. EMBO J. 23, 3609–3620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Xia Y., and Peng L. (2013) Photoactivatable lipid probes for studying biomembranes by photoaffinity labeling. Chem. Rev. 113, 7880–7929 [DOI] [PubMed] [Google Scholar]
- 19. Chen Z. W., Manion B., Townsend R. R., Reichert D. E., Covey D. F., Steinbach J. H., Sieghart W., Fuchs K., and Evers A. S. (2012) Neurosteroid analog photolabeling of a site in the third transmembrane domain of the β3 subunit of the GABA(A) receptor. Mol. Pharmacol. 82, 408–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Budelier M. M., Cheng W. W., Bergdoll L., Chen Z. W., Abramson J., Krishnan K., Qian M., Covey D. F., Janetka J. W., and Evers A. S. (2017) Click chemistry reagent for identification of sites of covalent ligand incorporation in integral membrane proteins. Anal. Chem. 89, 2636–2644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Castellano B. M., Thelen A. M., Moldavski O., Feltes M., van der Welle R. E., Mydock-McGrane L., Jiang X., van Eijkeren R. J., Davis O. B., Louie S. M., Perera R. M., Covey D. F., Nomura D. K., Ory D. S., and Zoncu R. (2017) Lysosomal cholesterol activates mTORC1 via an SLC38A9-Niemann-Pick C1 signaling complex. Science 355, 1306–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen Z. W., Fuchs K., Sieghart W., Townsend R. R., and Evers A. S. (2012) Deep amino acid sequencing of native brain GABAA receptors using high-resolution mass spectrometry. Mol. Cell. Proteomics 11, M111.011445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ujwal R., Cascio D., Colletier J. P., Faham S., Zhang J., Toro L., Ping P., and Abramson J. (2008) The crystal structure of mouse VDAC1 at 2.3-Å resolution reveals mechanistic insights into metabolite gating. Proc. Natl. Acad. Sci. U.S.A. 105, 17742–17747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Morris G. M., Huey R., Lindstrom W., Sanner M. F., Belew R. K., Goodsell D. S., and Olson A. J. (2009) AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 30, 2785–2791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Weiser B. P., Woll K. A., Dailey W. P., and Eckenhoff R. G. (2014) Mechanisms revealed through general anesthetic photolabeling. Curr. Anesthesiol. Rep. 4, 57–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Das J. (2011) Aliphatic diazirines as photoaffinity probes for proteins: recent developments. Chem. Rev. 111, 4405–4417 [DOI] [PubMed] [Google Scholar]
- 27. Hallermayer G., and Neupert W. (1974) Lipid composition of mitochondrial outer and inner membranes of Neurospora crassa. Hoppe-Seyler's Z. Physiol. Chem. 355, 279–288 [DOI] [PubMed] [Google Scholar]
- 28. Parsons D. F., and Yano Y. (1967) The cholesterol content of the outer and inner membranes of guinea-pig liver mitochondria. Biochim. Biophys. Acta 135, 362–364 [DOI] [PubMed] [Google Scholar]
- 29. Woll K. A., Dailey W. P., Brannigan G., and Eckenhoff R. G. (2016) Shedding light on anesthetic mechanisms: application of photoaffinity ligands. Anesth. Analg. 123, 1253–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zaid H., Abu-Hamad S., Israelson A., Nathan I., and Shoshan-Barmatz V. (2005) The voltage-dependent anion channel-1 modulates apoptotic cell death. Cell Death Differ. 12, 751–760 [DOI] [PubMed] [Google Scholar]
- 31. Villinger S., Briones R., Giller K., Zachariae U., Lange A., de Groot B. L., Griesinger C., Becker S., and Zweckstetter M. (2010) Functional dynamics in the voltage-dependent anion channel. Proc. Natl. Acad. Sci. U.S.A. 107, 22546–22551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Whitelegge J., Halgand F., Souda P., and Zabrouskov V. (2006) Top-down mass spectrometry of integral membrane proteins. Expert Rev. Proteomics 3, 585–596 [DOI] [PubMed] [Google Scholar]
- 33. Ryan C. M., Souda P., Bassilian S., Ujwal R., Zhang J., Abramson J., Ping P., Durazo A., Bowie J. U., Hasan S. S., Baniulis D., Cramer W. A., Faull K. F., and Whitelegge J. P. (2010) Post-translational modifications of integral membrane proteins resolved by top-down Fourier transform mass spectrometry with collisionally activated dissociation. Mol. Cell. Proteomics 9, 791–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ujwal R., and Abramson J. (2012) High-throughput crystallization of membrane proteins using the lipidic bicelle method. J. Vis. Exp. 59, e3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Darbandi-Tonkabon R., Hastings W. R., Zeng C. M., Akk G., Manion B. D., Bracamontes J. R., Steinbach J. H., Mennerick S. J., Covey D. F., and Evers A. S. (2003) Photoaffinity labeling with a neuroactive steroid analogue: 6-azi-pregnanolone labels voltage-dependent anion channel-1 in rat brain. J. Biol. Chem. 278, 13196–13206 [DOI] [PubMed] [Google Scholar]
- 36. Wessel D., and Flügge U. I. (1984) A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal. Biochem. 138, 141–143 [DOI] [PubMed] [Google Scholar]
- 37. Whitelegge J. P., Gundersen C. B., and Faull K. F. (1998) Electrospray-ionization mass spectrometry of intact intrinsic membrane proteins. Protein Sci. 7, 1423–1430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang Z., and Marshall A. G. (1998) A universal algorithm for fast and automated charge state deconvolution of electrospray mass-to-charge ratio spectra. J. Am. Soc. Mass Spectrom. 9, 225–233 [DOI] [PubMed] [Google Scholar]
- 39. Guner H., Close P. L., Cai W., Zhang H., Peng Y., Gregorich Z. R., and Ge Y. (2014) MASH Suite: a user-friendly and versatile software interface for high-resolution mass spectrometry data interpretation and visualization. J. Am. Soc. Mass Spectrom. 25, 464–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rostovtsev V. V., Green L. G., Fokin V. V., and Sharpless K. B. (2002) A stepwise huisgen cycloaddition process: copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. Int. Ed. Engl. 41, 2596–2599 [DOI] [PubMed] [Google Scholar]
- 41. O'Boyle N. M., Banck M., James C. A., Morley C., Vandermeersch T., and Hutchison G. R. (2011) Open Babel: an open chemical toolbox. J. Cheminform. 3, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schüttelkopf A. W., and van Aalten D. M. (2004) PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 60, 1355–1363 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.