

Abstract
The steroid hormone-activated glucocorticoid receptor (GR) regulates cellular stress pathways by binding to genomic regulatory elements of target genes and recruiting coregulator proteins to remodel chromatin and regulate transcription complex assembly. The coregulator hydrogen peroxide-inducible clone 5 (Hic-5) is required for glucocorticoid (GC) regulation of some genes but not others and blocks the regulation of a third gene set by inhibiting GR binding. How Hic-5 exerts these gene-specific effects and specifically how it blocks GR binding to some genes but not others is unclear. Here we show that site-specific blocking of GR binding is due to gene-specific requirements for ATP-dependent chromatin remodeling enzymes. By depletion of 11 different chromatin remodelers, we found that ATPases chromodomain helicase DNA-binding protein 9 (CHD9) and Brahma homologue (BRM, a product of the SMARCA2 gene) are required for GC-regulated expression of the blocked genes but not for other GC-regulated genes. Furthermore, CHD9 and BRM were required for GR occupancy and chromatin remodeling at GR-binding regions associated with blocked genes but not at GR-binding regions associated with other GC-regulated genes. Hic-5 selectively inhibits GR interaction with CHD9 and BRM, thereby blocking chromatin remodeling and robust GR binding at GR-binding sites associated with blocked genes. Thus, Hic-5 regulates GR binding site selection by a novel mechanism, exploiting gene-specific requirements for chromatin remodeling enzymes to selectively influence DNA occupancy and gene regulation by a transcription factor.
Keywords: ATPase, chromatin remodeling, glucocorticoid receptor, transcription coregulator, transcription regulation, BRM1, CHD9, Hic-5
Introduction
Transcription factors regulate gene expression by binding to specific regulatory DNA sequences, where they can either activate or repress transcription of the associated gene(s). The regulatory process directed by the transcription factor involves recruitment of numerous coregulator proteins that remodel the chromatin landscape around the transcription factor binding site and the transcription start site of the regulated gene and regulate the assembly of an active transcription complex at the transcription start site. Each coregulator contributes specific molecular functions to accomplish these complex processes in a presumably coordinated fashion, resulting in increased or decreased production of mRNA encoded by the gene (1–3). Many coregulators act in a gene-specific manner, i.e. they are required for the regulation of a subset of the genes regulated by any given transcription factor. Furthermore, a single coregulator can have different effects (i.e. activation or repression) and act by different mechanisms on different target genes, even for genes regulated by a single transcription factor within a single cell type (4–9). However, the mechanisms that specify the action of a coregulator on a given target gene are mostly unknown. Here we report a specific mechanism that directs the gene-specific actions of the protein hydrogen peroxide-inducible clone 5 (Hic-5, also known as TGFB1I1) as a coregulator for the glucocorticoid receptor (GR,2 NR3C1). GR, a member of the nuclear receptor family of ligand-activated transcription factors, regulates diverse physiological programs, including inflammation and metabolism of glucose, lipids, and proteins, by activating and repressing the transcription of specific genes. GR is activated by binding of the natural glucocorticoid (GC) hormone cortisol or various synthetic analogues, which are widely used in the treatment of many types of inflammatory diseases and cancer (10).
Hic-5 is a member of the paxillin family of molecular adaptor proteins, characterized by two types of protein interaction domains: four LD (leucine and aspartate-containing) motifs at the N terminus and four Lin11, Isl-1, and Mec-3 (LIM) domains, each composed of two adjacent zinc fingers, on the C terminus (11, 12). In the cytosol, Hic-5 has been widely studied as an adapter protein at focal adhesion complexes (13). In the nucleus, Hic-5 serves as a coregulator for a variety of transcription factors, including GR (14–20). As a coregulator of nuclear receptors, Hic-5 has been associated with many physiological and disease functions, including endometriosis through the progesterone receptor (16), epithelial cell differentiation by affecting peroxisome proliferator-activated receptor γ transcriptional activity (15), and prostate tumorigenesis and castrate responsiveness through the androgen receptor (17). We recently showed that endogenous Hic-5 modulates GC-regulated gene transcription by GR in a highly gene-specific manner, functioning as a coactivator for some GR target genes and as a corepressor for others. GC-regulated genes were categorized into three different classes with respect to their dependence on Hic-5: Hic-5-independent (ind) genes are regulated by GC independent of Hic-5 depletion; Hic-5-modulated (mod) genes are regulated by GC in the presence of Hic-5, but depletion of Hic-5 alters the magnitude of activation or repression by GC; Hic-5-blocked (block) genes are not regulated by GC until Hic-5 is depleted from the cells (9). The effects of Hic-5 depletion were observed at both the mRNA and pre-mRNA levels of mod and block genes, demonstrating that the mechanism of Hic-5 action occurs at the transcriptional level. These same Hic-5-influenced classes of genes were also observed in studies with estrogen receptor α and androgen receptor (20, 21). Mechanistic examination of selected GC-induced mod genes showed that Hic-5 is recruited to GR-binding regions (GBR) by its interaction with GR and acts at late stages of transcription complex assembly, facilitating recruitment of the Mediator complex and RNA polymerase II (9). In contrast, examination of three selected block genes indicated that Hic-5 prevented transcriptional activation by impeding GC-induced chromatin remodeling and robust GR occupancy at GBR associated with the block genes (9). Additionally, in GR chromatin immunoprecipitation sequencing analysis, Hic-5 depletion almost doubled the number of GR-occupied sites in the genome (21).
Because coregulators generally have been shown to facilitate steps of transcription complex assembly that occur subsequent to transcription factor binding to DNA, the inhibition by Hic-5 of GR occupancy at GBR that control GC regulation of a specific set of genes is an unexpected and unique observation. Here we explore the hypothesis that transcription factor occupancy and chromatin remodeling are co-dependent processes and that Hic-5 influences GR occupancy by interfering with the dynamic interaction of GR with chromatin remodeling complexes. The recruitment of chromatin remodelers by GR is thus required for chromatin remodeling, which, in turn, facilitates robust GR occupancy of the GBR and subsequent transcription complex assembly on the transcription start site of the associated GC-regulated gene. We also address the gene-specific actions of Hic-5, i.e. the mechanism that allows Hic-5 to block GR binding and transcriptional regulation at the block class of GR target genes while allowing robust GR binding and GC-regulated transcription at other GR target genes (the ind and mod classes). Our findings elucidate new mechanisms that control transcription factor binding site selection and contribute to the gene-specific actions of coregulators.
Results
Hypothesis: A dynamic model for explaining the differential effects of Hic-5 on GR binding to different classes of GBR
The chromatin conformation at GBR is dramatically altered to a more open state in response to GC treatment of cells (9), e.g. by creating a nucleosome-free region at the GBR (Fig. 1). GC-induced binding of GR to GBR is a cooperative process between GR and chromatin remodelers in which GR recognizes its specific DNA-binding motif and recruits ATP-dependent chromatin remodeling enzymes that open up the chromatin structure (22–24). Furthermore, GR interacts in a rapid, dynamic on-and-off fashion with its specific DNA motifs (25), and experimental techniques such as chromatin immunoprecipitation that are used to measure the strength of association of GR with a specific GBR (often referred to as occupancy) are simply capturing a snapshot of the steady-state dynamic interaction between GR and a given GBR. In view of the dynamic and cooperative nature of this process, we propose that the initial GR interaction with the GBR is weak but that this weak GR-GBR interaction allows GR to recruit chromatin remodelers, which open up the chromatin and thereby allow a more robust but still dynamic GR occupancy of the site (Fig. 1).
Figure 1.
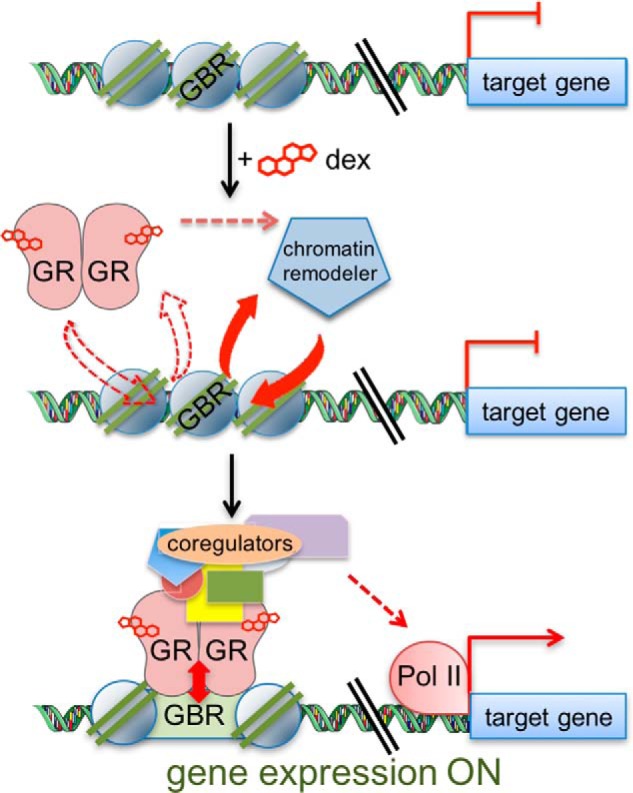
Dynamic GR-GBR interaction model. Before dexamethasone (dex) treatment, GBR is in a relatively closed chromatin conformation, and gene transcription is silent (or at a basal level). Upon binding dexamethasone, GR recognizes and initially interacts weakly with the GBR in a dynamic on/off relationship. GR recruits chromatin remodeling enzymes that subsequently remodel chromatin at the GBR to a more open state, thereby allowing a more robust but still dynamic GR occupancy, recruitment of coregulators, and recruitment of transcription machinery, including RNA polymerase II (Pol II) to the transcription start site, resulting in enhanced expression of the target gene.
In cells, Hic-5 selectively prevents robust GC-induced GR occupancy at GBR of block genes and the accompanying transition to a more open chromatin conformation (9). We therefore further hypothesize that, although GR can interact weakly with GBR of block genes in the presence of Hic-5, Hic-5 inhibits the dynamic and cooperative chromatin remodeling process by GR and chromatin remodelers at GBR of block genes, thus maintaining a chromatin conformation at the GBR of block genes that is not permissive for the robust GR interaction with the GBR required for establishing an active transcription complex. In addition, to explain why Hic-5 prevents chromatin remodeling and robust GR binding at block genes but not at mod and ind genes, we recall a previous report showing that different DNase hypersensitive sites (which frequently overlap with transcription factor-binding sites) require different combinations of chromatin remodeling complexes (26). In light of these findings we propose that different chromatin remodeling complexes are required for GR binding at block genes than at mod and ind genes. In the experiments described below, we test these hypotheses.
CHD9 and BRM chromatin remodelers selectively facilitate GC-regulated expression of representative GR target genes from the block gene class
Using the U2OS-GRα cell line, in which we previously characterized the actions of Hic-5 (9), we first determined whether specific chromatin remodeling enzymes are selectively required for GC-induced expression of representative genes from the block gene class. Eleven ATP-dependent chromatin remodeling enzymes shown previously to be involved in transcriptional activation by GR or other transcription factors (23, 27–37) were individually depleted by siRNA transfection. Because Hic-5 inhibits the expression of block genes, it was depleted simultaneously with each chromatin remodeler. Cells were treated with the synthetic GC agonist dexamethasone or an equivalent volume of the vehicle ethanol for 4 h, and GC-induced expression of representative block and ind genes identified in our previous study (9) was measured using quantitative RT-PCR (Fig. 2). Immunoblots verified successful depletion of the chromatin remodeling enzymes and Hic-5 (Fig. 2C). Depletion of the chromatin remodelers did not affect the expression of GR or Hic-5, and depletion of Hic-5 did not alter the expression of GR or the chromatin remodelers (Fig. 2C). As shown previously (9), depletion of Hic-5 alone resulted in dexamethasone-regulated expression of the block genes RP1L1 and HOXD1, whereas these genes were not induced by dexamethasone in the nonspecific siRNA control sample (Fig. 2A). For most of the chromatin remodelers, the double depletion of a chromatin remodeler and Hic-5 did not eliminate dexamethasone-induced expression of the block genes. However, the double depletion of chromodomain helicase DNA-binding protein 9 (CHD9) and Hic-5 or of Brahma homologue (BRM, a product of the SMARCA2 gene) and Hic-5 eliminated a significant increase in mRNA for the block genes RP1L1 and HOXD1 upon dexamethasone treatment, indicating that CHD9 and BRM are required for dexamethasone-regulated expression of these two block genes. In contrast, dexamethasone treatment induced expression of the ind genes IGFBP1 and MSX2 in the presence and absence of Hic-5, and depletion of each of the 11 chromatin remodelers along with Hic-5 had little or no effect on their dexamethasone-regulated expression (Fig. 2B).
Figure 2.

CHD9 and BRM are selectively required for dexamethasone-induced expression of representative block genes. A and B, cells were transfected with the indicated siRNA combinations, and after 48 h, relative mRNA levels for the indicated block genes (A) and ind genes (B) were measured by quantitative RT-PCR following 4 h of ethanol (etoh) or dexamethasone (dex) treatment. Relative mRNA levels for each gene were normalized to GAPDH mRNA levels. Data shown are the mean ± S.D. of three biological replicates. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ns, not significant; paired t test. C, siRNA depletion of chromatin remodelers (CR) and Hic-5. Immunoblots for the indicated chromatin remodelers, GR, and Hic-5 are shown, with GAPDH as an internal control.
Similar results were observed when expression of genes was examined at 2, 4, and 8 h of dexamethasone treatment. Dexamethasone-induced expression of the block genes RP1L1, GRAMD4, and HOXD1 was observed after depletion of Hic-5 alone but was eliminated by double depletion of Hic-5 and CHD9 or of Hic-5 and BRM (supplemental Fig. S1A). There was no dexamethasone-induced expression of the three block genes when Hic-5 was present in cells and BRM only or CHD9 only was depleted. In contrast, the temporal expression profiles of three representative ind genes, IGFBP1, MSX2, and TIPARP, were unaffected by the depletion of Hic-5, CHD9, or BRM individually or in combination (supplemental Fig. S1B). Similarly, CHD9 and BRM were not required for dexamethasone-induced expression of two representative mod genes, SCNN1A and SLN (supplemental Fig. S1C). As shown previously (9), these genes were activated by dexamethasone in the presence but not in the absence of Hic-5. When a second siRNA for CHD9 and BRM was used, similar results were obtained for these representative block, ind, and mod genes (supplemental Fig. S2), thus eliminating concerns about off-target effects of the first set of siRNAs used. Therefore, CHD9 and BRM chromatin remodelers are necessary for the dexamethasone-induced expression of the block genes but not the ind and mod genes.
Genome-wide analysis of CHD9 and BRM requirements for dexamethasone-regulated expression of block, ind, and mod GR target genes
Because CHD9 and BRM were selectively required for dexamethasone-induced expression of representative genes from the block gene class, we employed RNA sequencing to explore whether this association applies genome-wide. RNA was prepared from cells transfected with six different siRNA combinations: nonspecific siRNA control (siNS/siNS), depletion of Hic-5 (siHic5/siNS), depletion of CHD9 (siCHD9/siNS), double depletion of Hic-5 and CHD9 (siCHD9/siHic5), depletion of BRM (siBRM/siNS), and double depletion of Hic-5 and BRM (siBRM/siHic5). Cells in each category were treated with either 100 nm dexamethasone or ethanol for 8 h, making a total of 12 conditions, and three biological replicates were performed on different days. The differentially expressed genes reported here are defined as genes with at least a 1.3-fold change in expression and a false discovery rate (FDR)-adjusted p ≤ 0.05. These relatively low-stringency values were chosen to include reasonably large numbers of genes in the block, ind, and mod gene classes and thus provide more statistical power for subsequent analysis of CHD9 and BRM effects on these gene classes. Assignment of genes to the block, ind, and mod classes involved an algorithm developed previously (6), described in detail under “Experimental Procedures.” In total, there were 105 ind genes (Fig. 3A, blue region), 364 mod genes (Fig. 3A, green region), and 534 block genes (Fig. 3A, red region). These categories include genes that were up-regulated or down-regulated by dexamethasone and where Hic-5 could have a positive or negative effect, as illustrated by the hypothetical examples for each of the three gene classes in Fig. 3A.
Figure 3.

Genome-wide selective requirement of CHD9 and BRM for dexamethasone-regulated expression by block genes versus ind and mod genes. Genome-wide RNA sequencing analysis was performed to evaluate the genes dependent on CHD9 and BRM for dexamethasone (dex)-regulated expression. A, the numbers of block (red), ind (blue), and mod (green) genes were determined by overlapping three gene sets (supplemental Fig. 3A) according to the algorithms described under “Experimental Procedures.” Theoretical examples of activated and repressed gene expression profiles are provided as bar graphs for each gene class. B, block, ind, and mod genes that require CHD9 for dexamethasone-regulated expression. The block gene set was overlapped with gene sets a and b (supplemental Fig. 3B). The ind and mod gene sets were each overlapped with gene sets c and d (supplemental Fig. 3B). Overlapping was performed according to the algorithms under “Experimental Procedures.” Dark-colored regions show the number and percentage of genes in each class (block, ind, and mod) that are dependent on CHD9 for dexamethasone-regulated expression. C, block, ind, and mod genes that require BRM for dexamethasone-regulated expression. BRM-dependent genes in each class were determined as in B, using data from cells with depletion of BRM instead of CHD9. D, the percentages of genes from each class that are dependent on CHD9 and/or BRM. E, the decrease in dexamethasone regulation caused by depletion of CHD9 (top panel) or BRM (bottom panel) is shown as the distribution of the depletion effects for all block, ind, and mod genes, using a box plot that divides genes for each class into quartiles. The y axis values represent the percent decrease in dexamethasone effect, calculated with the formula specified under “Experimental Procedures.” The median decrease caused by depletion of CHD9 or BRM is indicated by thick horizontal lines, with median values shown above the plot. The p values were obtained by comparing the ind or mod class to the block class using Mann-Whitney U test.
Next we determined the number of genes in each class that required CHD9 or BRM for dexamethasone-regulated expression. Briefly, the set of genes in each gene class (block, ind, and mod) overlapped with two other gene sets derived from the samples depleted or not depleted of CHD9 or BRM, defined as illustrated in supplemental Fig. S3, B and C and explained in detail under “Experimental Procedures.” Although this procedure identified several subsets of genes (supplemental Fig. S3C, sectors i–iv), the genes of primary interest were genes in each class for which the dexamethasone-regulated expression was entirely dependent on CHD9 or BRM (supplemental Fig. S3C, sector iii), i.e. genes in the block, ind, or mod gene sets that were no longer dexamethasone-regulated after depletion of CHD9 or BRM.
Because block genes are only dexamethasone-regulated in cells depleted of Hic-5, we used data from cells that were depleted of Hic-5 alone or doubly depleted of Hic-5 and CHD9 or BRM to identify block genes that require CHD9 or BRM (supplemental Fig. S3, B and C). Using this algorithm (explained in detail under “Experimental Procedures”), we identified 296 of 534 block genes (55%) that require CHD9 for dexamethasone-regulated expression (Fig. 3B, left panel) and 292 of the 534 block genes (55%) that require BRM for dexamethasone-regulated expression (Fig. 3C, left panel). More than half of the block genes that required CHD9 were also dependent on BRM for dexamethasone-induced expression and vice versa (Fig. 3D, top left panel). Altogether, 78% (414 of 534 genes) of the block gene class were dependent on CHD9 and/or BRM for dexamethasone-induced expression, and 33% of block genes required both CHD9 and BRM (Fig. 3D, top left panel).
Similar analyses were conducted with the ind and mod gene classes to determine the genes that were dependent on CHD9 and/or BRM for dexamethasone-regulated expression. The effect of depleting each chromatin remodeler was determined in cells containing Hic-5 and in cells depleted of Hic-5. When the chromatin remodeler was depleted in cells containing Hic-5, 14 of 105 ind genes (13%) were dependent on CHD9, and 12 genes (11%) were dependent on BRM (Fig. 3, B and C, center panels). There were two genes (2%) regulated by both CHD9 and BRM, resulting in a total of 24 genes (23%) of the ind genes that required CHD9 or BRM or both for dexamethasone-induced expression (Fig. 3D, top right panel). When this analysis was performed in cells lacking Hic-5, 35% of the ind genes were dependent on CHD9 and/or BRM for dexamethasone-regulated expression (supplemental Fig. S3, D–F, left panels).
When mod genes were analyzed by single depletion of CHD9 or BRM in cells containing Hic-5, 80 of 364 mod genes (22%) were dependent on CHD9, and 63 (17%) were dependent on BRM for dexamethasone-regulated expression (Fig. 3, B and C, right panels). In total, 119 genes (33%) of the mod class were dependent on CHD9 or BRM or both, and only 7% required both CHD9 and BRM (Fig. 3D, bottom panel). Similar results were found when the effect of depleting CHD9 or BRM on mod gene regulation by dexamethasone was determined in cells lacking Hic-5 (supplemental Fig. S3, D–F, right panels). Because 138 of the 364 mod genes were no longer regulated by dexamethasone in cells depleted of Hic-5 (Fig. 3A), we only analyzed the remaining 226 mod genes that were still dexamethasone-regulated in cells depleted of Hic-5. In this analysis, 32% of the Hic-5 regulated mod genes were dependent on CHD9 and/or BRM for dexamethasone-regulated expression (supplemental Fig. S3, D–F, right panels).
Thus, the great majority of the block genes (78%) required CHD9, BRM, or both for their dexamethasone-regulated expression, whereas relatively minor fractions of the ind and mod genes (23–35%) required these chromatin remodelers; the comparison of gene classes was even more striking when considering the genes that required both CHD9 and BRM (33% for block and 2–9% for ind and mod) (Fig. 3D and supplemental Fig. S3F). Although these analyses were done with 1.3-fold change in expression and FDR adjusted p ≤ 0.05 as cutoff values, the large difference in the percentage of block genes that required CHD9 and/or BRM compared with the ind or mod genes was preserved when more stringent parameters were used to identify genes that were significantly affected by depletion of CHD9 or BRM (supplemental Fig. S4).
To assess the overall genome-wide magnitude of the effect of CHD9 and BRM depletion on the dexamethasone-regulated expression of all genes in each of the three classes, we compared the log2 -fold change in mRNA levels caused by dexamethasone treatment in cells containing (supplemental Fig. S5, A–C, red bars) or depleted (supplemental Fig. S5, A–C, blue bars) of each chromatin remodeler. The depletion of CHD9 or BRM caused a large decrease in the effect of dexamethasone on the expression of the great majority of the block genes, as indicated by the large amount of red visible in supplemental Fig. S5A. This trend was evident for block genes that were activated (supplemental Fig. S5A, left side) or repressed (supplemental Fig. S5A, right side) by dexamethasone. In contrast, depletion of CHD9 or BRM caused a much more modest decrease in dexamethasone regulation of the ind and mod genes, as indicated by the much lower amount of red visible in supplemental Fig. S5, B and C. In fact, depletion of CHD9 or BRM caused little or no change in dexamethasone regulation for a large percentage of the ind and mod genes (very little difference in the height of red and blue bars; supplemental Fig. S5, B and C). To analyze these genome-wide data quantitatively and statistically, we calculated the percent decrease in the dexamethasone regulation of each gene (see formula under “Experimental Procedures”) caused by depletion of CHD9 or BRM and displayed these data as a box plot showing the genes in quartiles for each gene class (Fig. 3E). For the block genes, the median decrease in dexamethasone regulation was 52.9% for CHD9 depletion and 59.6% for BRM depletion. In contrast, the median decrease in dexamethasone regulation for ind genes was 0.04% and 15.1% for CHD9 and BRM depletion, respectively; and the median decrease in dexamethasone regulation for mod genes was 12.5% and 18.9% for CHD9 and BRM depletion, respectively. Furthermore, the difference in the distribution of values between the block genes and each of the other two gene classes was highly significant (Fig. 3E).
Gene ontology analysis (38, 39) of block genes versus a combination of ind and mod genes indicated some common pathways (e.g. developmental and cell differentiation pathways), but also some distinct physiological pathways. The block class was enriched for genes involved in localization of cells and cell contents and for genes involved in phosphate metabolism. In contrast, the combined ind and mod classes were enriched for genes involved in angiogenesis, apoptosis, and pathways involved in oxygen and mitogen-activated protein kinase signaling (supplemental Fig. S5D).
CHD9 and BRM are required for GR occupancy at GBR associated with block genes
To explore the mechanism by which CHD9 and BRM contribute to dexamethasone-regulated transcription of the block genes and because we propose that efficient GR binding to DNA is a cooperative process between GR and chromatin remodelers (Fig. 1), we examined the effect of CHD9 and BRM on the binding of GR to GBR associated with the representative block genes RP1L1 and GRAMD4 discussed above. Because there is no data to indicate which GBR controls each dexamethasone -regulated gene, we focused on the GR binding site that was most closely associated with each GR target gene in terms of linear genomic distance (9, 40). Supporting this choice is our previous finding that Hic-5 blocks dexamethasone-induced expression of these block genes and also prevents robust dexamethasone-induced GR binding and chromatin remodeling at the GBRs closest to these genes (9); this phenotypic correlation supports the conclusion that the closest GBR is involved in regulation of the representative genes examined here. As stated earlier in our hypothesis, we propose that there is an initial weak interaction between GR and GBRs, but robust GR occupancy requires recruitment of chromatin remodelers by GR and the resulting opening of the chromatin conformation (e.g. by creating a nucleosome-free region at the GBR) (Fig. 1). We further propose that Hic-5 interferes with this chromatin remodeling process at GBR of block genes.
Consistent with our model, we observed weak dexamethasone-induced GR binding at the GBR of the two representative dexamethasone-induced block genes in cells containing Hic-5, and robust dexamethasone-induced GR occupancy depended on depletion of Hic-5, as reported previously (9). GR occupancy was mostly eliminated after the double depletion of CHD9 and Hic-5 or BRM and Hic-5 (Fig. 4A). The GR binding level for the double depletions was essentially the same as the low level of GR binding observed when none of the proteins were depleted, suggesting that both CHD9 and BRM are required for the robust GR binding observed when Hic-5 is depleted. In contrast to CHD9 and BRM, the double depletion of Hic-5 and BRG1 (Brahma-related gene 1, a product of the SMARCA4 gene), a chromatin remodeler that was not required for dexamethasone-induced expression of the representative block genes (Fig. 2A), had no significant effect on GR occupancy at the GBR associated with the two block genes (Fig. 4A). In a similar chromatin immunoprecipitation analysis of GBRs associated with two representative ind genes (MSX2 and IGFBP1) and two representative mod genes (SCNN1A and SLN), single depletion of CHD9, BRM, or BRG1 or double depletion of Hic-5 along with each chromatin remodeler had little or no effect on dexamethasone-induced GR binding (Fig. 4, B and C); there was a small but marginally significant decrease in GR binding to the MSX2 GBR after double depletion of Hic-5 and CHD9, but there were no other significant decreases in GR binding to the ind or mod genes when any of the three chromatin remodelers was depleted. A region where GR does not bind was examined as a negative control (Fig. 4D). Thus, CHD9 and BRM, but not BRG1, were selectively required for GR binding to GBR associated with block genes.
Figure 4.

CHD9 and BRM are required for GR occupancy at GBR of representative block genes. A–D, cells depleted of Hic-5 or a chromatin remodeler only (CHD9, BRM, or BRG1) or doubly depleted of both a chromatin remodeler and Hic-5 were treated with dexamethasone for 1 h. Chromatin immunoprecipitation with antibodies against GR or normal IgG followed by quantitative PCR at the associated GBRs was performed for the indicated block genes (A), ind genes (B), and mod genes (C); GR occupancy at a negative control region is also shown (D). Values shown are mean ± S.D. for n = 4 biological replicates performed on different days. *, p < 0.05; **, p < 0.01 from a paired t test. ns, not significant.
CHD9 and BRM are required for dexamethasone-induced chromatin remodeling at GBR of the block genes
To determine whether CHD9 and BRM are required for the dexamethasone-induced increase in chromatin accessibility at the GBR of the representative block genes, we performed formaldehyde-assisted isolation of regulatory elements (FAIRE) followed by quantitative PCR. As shown previously (9), dexamethasone treatment increased FAIRE signals at the GBR of the representative block genes RP1L1 and GRAMD4 after Hic-5 depletion but not in control cells transfected with nonspecific siRNA. Double depletion of CHD9 and Hic-5 or of BRM and Hic-5 eliminated most of the dexamethasone-induced FAIRE signal for these block genes (Fig. 5A). However, depletion of BRG1 along with Hic-5 did not significantly alter the dexamethasone-induced FAIRE signal at the GBR of the block genes compared with depletion of Hic-5 alone. In contrast to the block genes, a robust dexamethasone-induced FAIRE signal was observed for the representative ind and mod genes either in the presence or absence of Hic-5, and depletion of CHD9, BRM, or BRG1 did not significantly alter these FAIRE signals either in the presence of absence of Hic-5 (Fig. 5, B and C), except in the case of the SCNN1A GBR, where the double depletion of BRM and Hic-5 (but not the single depletion of BRM) caused a small but significant decrease in the dexamethasone-induced FAIRE signal. Thus, CHD9 and BRM, but not BRG1, are required for dexamethasone-induced chromatin remodeling activity at the GBR of the block genes, and the requirement of CHD9 and BRM was specific for the block gene class but not for ind and mod genes.
Figure 5.

CHD9 and BRM are selectively required for chromatin remodeling at GBR of representative block genes. A–C, cells depleted of Hic-5 or a chromatin remodeler only (CHD9, BRM, or BRG1) or doubly depleted of both a chromatin remodeler and Hic-5 were treated with ethanol (etoh) or dexamethasone (dex) for 1 h. Chromatin accessibility as measured by FAIRE followed by quantitative PCR at the associated GBRs was performed for the indicated block genes (A), ind genes (B), and mod genes (C). The FAIRE signal at a negative control region is also shown for comparison. Values shown are mean ± S.D. for n = 3 biological replicates performed on different days. *, p < 0.05; **, p < 0.01 from a paired t test. ns, not significant.
Hic-5 selectively inhibits the interaction between GR and block gene-specific chromatin remodelers
Because Hic-5 inhibited the chromatin remodeling actions of CHD9 and BRM, which are required for GR occupancy of block gene GBRs (Figs. 4 and 5), and because Hic-5 binds directly to the hinge region of GR located between the DNA-binding domain and the ligand-binding domain (14), we investigated whether or not Hic-5 inhibits the interaction between GR and CHD9 or BRM using a proximity ligation assay (PLA). PLA detects protein-protein interactions by using primary antibodies of differing species for each of the putative protein partners. When the proteins are in proximity of less than 40 nm, secondary antibodies attached with oligonucleotides guide the formation of circular DNA strands that serve as templates for rolling circle amplification. Fluorescent probes that bind to the amplified DNA generate a fluorescence signal for each bimolecular interaction that appears as a pinpoint signal against the background of the cell micrograph, which, in this case, includes DAPI-stained nuclei (Fig. 6A). The number of signals is quantifiable with image analysis software (Fig. 6B). Antibodies for GR, CHD9, BRM, and BRG1 were used to examine the interactions between GR and chromatin remodelers in cells containing or depleted of Hic-5 and treated with dexamethasone or ethanol. Signals above background were only detected in dexamethasone-treated cells (Fig. 6B and supplemental Fig. S6), presumably at least in part because GR is only localized to the nucleus after dexamethasone treatment. This result validated the specificity of the contribution of the GR antibody to the PLA signal. Validation of the antibodies for the chromatin remodelers was achieved by showing that the dexamethasone-induced signals for the GR-chromatin remodeler interactions were reduced to background by depletion of the relevant chromatin remodeler (Fig. 6, A and B). Robust dexamethasone-induced signals for the GR-CHD9 and GR-BRM interactions were observed only after depletion of Hic-5; in contrast, a robust GR-BRG1 interaction was observed either in the presence or absence of Hic-5. Hence, the PLA experiments demonstrate that Hic-5 inhibits the interaction between GR and the block gene-specific chromatin remodelers CHD9 and BRM but does not inhibit GR interaction with other chromatin remodelers such as BRG1.
Figure 6.

Hic-5 effect on the interaction between GR and chromatin remodelers. Cells were depleted of the specified protein(s) using siRNA and treated with ethanol (etoh) or dexamethasone (dex) for 1 h. A, PLA of dexamethasone-treated cells was performed to measure the indicated protein-protein interactions. Red fluorescent dots indicate individual bimolecular interactions between two molecules. DAPI (blue) staining specifies nuclei. PLA images of both ethanol- and dexamethasone-treated cells are shown in supplemental Fig. 6. B, quantification of PLA signals within the nuclei of ethanol- and dexamethasone-treated cells (the number of dots in the nuclei/number of nuclei). Values are mean ± S.D. for n = 3 biological replicates performed on different days. *, p < 0.05; **, p < 0.01 from a paired t test. ns, not significant. Scale bars = 20 μm.
Discussion
The dynamic co-dependence of GR occupancy and chromatin remodeling is regulated by Hic-5
Our results demonstrate that the block class of GR target genes is defined by its requirement for specific chromatin remodelers, CHD9 and BRM, which are not required for most of the genes in the ind and mod classes of GR target genes. 78% of block genes required at least one of these two chromatin remodelers, and 33% required both of them. In contrast, only 23–35% of ind and mod genes required BRM or CHD9, and only 2–9% required both (Fig. 3D and supplemental Fig. S3F). Of course, the specific number of genes assigned to each category depends on the stringency of the –fold change and statistical cutoffs chosen, but the dramatic difference in dependence on BRM and CHD9 for dexamethasone-regulated expression of the block versus ind and mod genes was consistent even when more stringent cutoff values were tested (supplemental Fig. S4). Several possible explanations can be offered for the 22% of the block genes that did not appear to require CHD9 or BRM: there may be other chromatin remodelers required for some block genes that were not tested in this study, the -fold change and statistical cutoffs chosen may have generated some false positive block genes, or the effects of CHD9 or BRM depletion on some block genes may have missed the statistical cutoff. Similarly, the fact that 23–35% of the ind and mod genes demonstrated a requirement for CHD9 or BRM could be due to the low 1.3-fold change cutoff chosen. This conclusion is supported by the generally small effect of CHD9 or BRM depletion on the ind and mod gene classes compared with the large effect on the block gene class (Fig. 3E).
The fact that 33% of the block genes require both CHD9 and BRM for dexamethasone-regulated expression suggests that each chromatin remodeler contributes some unique function to remodel the chromatin at the GBR associated with these genes and that both of these functions are required for dexamethasone-induced chromatin remodeling and for the resulting robust GR binding and transcriptional regulation. The requirement for multiple ATPases to maintain transcription factor binding sites has been demonstrated previously (26) and suggests that different types of chromatin remodeling activities cooperate to accomplish chromatin remodeling associated with transcriptional regulation. In fact, CHD9 and BRM belong to different families of ATPases, and in vitro assays indicate that they support different types of nucleosome and chromatin remodeling activities (28, 41).
Hic-5 binds to GR (14) and was not detectable on any of the block gene GBRs tested in the absence of dexamethasone but was recruited to the GBRs of ind and mod genes in a dexamethasone-induced manner (9). This indicates that Hic-5 is recruited to GBRs by GR. This raises the question of how Hic-5 can prevent robust GR occupancy on block gene GBRs. Our proposal that GR makes initial weak contact with the GBR but that robust occupancy and chromatin remodeling of GBRs are co-dependent processes (Fig. 1) resolves this conundrum. The co-dependence provides an opportunity for regulation, and, in this case, Hic-5 regulates this process selectively on block genes (Figs. 4 and 5) by interfering with the interaction between GR and the two block gene-specific chromatin remodelers CHD9 and BRM (Fig. 6). When Hic-5 is depleted, CHD9 and BRM are allowed to interact with GR, and these two chromatin remodelers are required for the robust binding of GR and chromatin remodeling at the GBR of the block genes (Fig. 7).
Figure 7.

Model illustrating the mechanism of Hic-5 action on dexamethasone-induced block genes. In the presence of Hic-5, interactions between GR and CHD9 and between GR and BRM are inhibited, preventing dexamethasone-induced chromatin remodeling and robust GR occupancy at the GBR. Hence, expression of the block gene is not dexamethasone-regulated. In the absence of Hic-5, GR is able to recruit block gene-specific chromatin remodelers, CHD9 and BRM, allowing the opening of chromatin, robust GR occupancy, recruitment of coregulators and RNA polymerase II (Pol II), and dexamethasone-induced expression of the block gene.
Our findings that different GR target genes require different chromatin remodelers is presumably related to a previous report that different DNase hypersensitive sites are maintained by different chromatin remodelers (26). It is well established that GBRs (and other transcription factor binding sites) are generally marked by DNase-hypersensitive sites, which are often present before GR binding but increase in accessibility after GR binding (42). Our results indicate that the increased accessibility of GBRs induced by dexamethasone requires gene-specific chromatin remodelers, and the dexamethasone dependence of the chromatin transition indicates a requirement for GR to recruit or activate the chromatin remodelers, thus supporting our conclusion that GR occupancy and chromatin remodeling are co-dependent processes. Also consistent with this is the previous demonstration that components of the Swi-Snf chromatin remodeling complex (for which BRG1 and BRM serve as alternative ATPase subunits) interact directly with GR and are recruited to many GBRs in a hormone-induced manner (22–24, 27). In addition, BRM and GR have been shown previously to regulate the occupancy of each other on GBRs in a gene-specific manner (27).
Another question suggested by our results is what drives gene-specific requirements for CHD9 and BRM. As with gene-specific coregulator actions in general, we propose that these come from inherent properties of the chromatinized genes. First, the specific DNA sequence to which GR binds is known to influence GR conformation and actions (43, 44). Second, the set of additional regulatory elements that bind additional transcription factors and coregulators is specific for each gene. Third, the chromatin conformation may also be gene-specific. Together, these factors establish a specific regulatory environment that determines the specific coregulators that are required for activation or repression of transcription. Protein-protein interactions among all transcription factors and coregulators at the site, as well as posttranslational modifications made by enzymatic coregulators present in this regulatory environment, should influence the coregulators that are required for transcription and the specific actions of the transcription factor and coregulators (e.g. whether they have positive or negative effects on transcription).
Physiological implications of the gene-specific requirements for and actions of Hic-5
There are two separate aspects of the gene-specific coregulator activity of Hic-5: first, it affects the GC-regulated expression of some but not all GR target genes; second, it acts by different mechanisms on different GR target genes, as evidenced by its distinctive mechanisms of action on mod and block genes. For mod genes that require Hic-5 for GC-induced expression, Hic-5 facilitates recruitment of the Mediator complex and RNA polymerase II; in contrast, Hic-5 prevents GC-induced GR binding and chromatin remodeling at GBR of block genes (9). There is accumulating evidence that the gene-specific actions of coregulators may correlate with specific physiological pathways. For example, GC regulates many different physiological pathways, including anti-inflammatory pathways, developmental pathways, and metabolism of glucose, lipids, and bone. If different coregulators are required for GC-regulated genes that control these different pathways, then regulation of the amounts or activities (e.g. by posttranslational modifications) of specific coregulators could modulate the specific physiological response to GC. It is thus interesting to note that gene ontology analysis indicated that the block gene class is enriched for biological processes that are different from those enriched in the combined mod and ind genes (supplemental Fig. S5D). The considerable number of block genes that require both CHD9 and BRM supports the notion that CHD9 and BRM affect similar biological processes. For example, previous studies indicate that BRM and CHD9 may both be involved in regulating osteogenic genes (35, 45). Because this study shows that the interaction between GR and these two chromatin remodelers is regulated by Hic-5, it would be interesting to explore how Hic-5 influences GR interaction with BRM and CHD9 to regulate osteogenic genes.
Because Hic-5 prevents GR binding to a specific subset of potential GBRs, Hic-5 actually alters the GR cistrome (the genome-wide set of sites occupied in a given cell type). The GR cistrome varies in a cell type-specific manner (42), but the factors that contribute to cell type-specific transcription factor binding are only partially understood. Cell type-specific heterochromatin domains certainly contribute (46), and chromatin remodelers have been shown to influence the locations of hypersensitive sites and transcription factor binding (26, 42). Hic-5 represents a new type of mechanism that contributes to determination of the GR cistrome.
Thus, studying the mechanism of Hic-5 action on GR provides a unique opportunity to advance our understanding of how transcription factor binding site selection is regulated and the role of coregulators in mediating transcription factor binding and activity. This study elucidates a mechanism that explains the block gene-specific actions of Hic-5 on a subset of GC-regulated genes. However, Hic-5 has been shown to block transcription factor occupancy and hormone-induced expression of genes by estrogen receptor (21) and actually promoted androgen receptor binding to some sites (20). Additionally, the coregulators cell cycle and apoptosis regulator 1 (CCAR1), cell cycle and apoptosis regulator 2 (CCAR2), calcium-binding and coiled-coil domain 1 (CALCOCO1), and zinc finger protein 282 (ZNF282) also block expression of a subset of GC-regulated genes in A549 cells, although not as robustly as Hic-5 (6). Therefore, although this study focuses on GR and Hic-5, it is likely that our model applies to other transcription factors and coregulators. Transcription factor binding site selection and gene-specific actions of coregulators are relevant to all transcription factors, and, hence, our study broadly contributes to our understanding of the role of coregulators in transcriptional regulation.
Because Hic-5 modulates (both positively and negatively) the GC-regulated expression of some GR target genes (mod genes) and blocks the activation or repression of others (block genes) in response to GC, it has the potential to dramatically influence the outcome of the response to GC, but only if the amount or activity of Hic-5 is regulated. It is thus interesting to note that Hic-5 expression is highly cell type- and tissue-specific (47, 48). In addition, phosphorylation of Hic-5 by multiple kinases on multiple residues of Hic-5 has been reported, with some of these modifications promoting and others inhibiting interaction between Hic-5 and androgen receptor (49–51). In addition, Hic-5 activity and changes in Hic-5 expression have been linked to various types of cancer, especially prostate cancer (17, 20, 48). In that light, it is also relevant to note that enhanced GR activity has been recently implicated in progression of castration-resistant prostate cancer (52). In future work, it will be very interesting to test whether posttranslational modifications of Hic-5 and the signaling pathways that control them alter Hic-5 effects on androgen receptor- and GR-regulated gene expression and emerge as novel therapeutic targets in cancer.
Experimental procedures
Cell culture and siRNA transfection
U2OS osteosarcoma cells stably expressing wild-type GRα (U2OS-GRα) were maintained as described previously (9). The actions of GC are well known in most cell types, including osteogenic lineages (53, 54). However, GR is expressed at extremely low levels in the parent U2OS cell line, and therefore the line used here was derived by introduction of a transgene (55). U2OS-GRα cells express levels of GR that are equivalent to levels expressed naturally in many cell lines and primary cells. Cells were grown in medium supplemented with 5% (v/v) FBS and transfected with siRNA using Lipofectamine RNAiMAX (Invitrogen). For double depletion of the chromatin remodeler and Hic-5, equivalent amounts siRNA for a chromatin remodeler and Hic-5 (siHic5) were added. For single-protein depletions, equivalent amounts of siRNA for the targeted protein and siNS were used so that the total volume and mass of siRNA was consistent. 48 h after siRNA transfection, the cells were either treated for the indicated length of time with 100 nm dexamethasone (Sigma) or an equivalent amount of ethanol as a control. siRNA sequences for siNS and siHic-5 were described previously (9). siRNAs for chromatin remodelers were designed and purchased through MISSION predesigned siRNA (Sigma). The sense sequences were as follows: siBRG1 (5′-GGAAUACCUCAAUAGCAUU-3′), siBRM (5′-CCAAAUGAUUGCUCGACGA-3′), siSNF2h (5′-CAACAGAUAUGCAUCUAGU-3′), siINO80 (5′-GCAUGAAUUGGUUGGCCAA-3′), siEP400 (5′-CUGAUGAGGAGUUUCAACA-3′), siCHD1 (5′-CUCAGUACCAUGAUCAUCA-3′), siCHD4 (5′-CAAACAGGAGCUUGAUGAU-3′), siCHD6 (5′-CAAACUUCUGGAGGGUCU-3′), siCHD7 (5′-GGACUUUGCACGUAGCACA-3′), siCHD8 (5′-CAGAAUCAUUCAGGUCUAU-3′), and siCHD9 (5′-CGAAUUGAUGGCAGAGUCA-3′). siRNA depletion was verified by immunoblot. Depletion of BRM and CHD9 was validated with a second siRNA with sense sequences as follows: siCHD9#2 (5′-AAGUAUUUGAUGGAGUU-3′) and siBRM#2 (5′-GGAACUUAGCCGAUGAAA-3′).
Immunoblot
48 h after transfection with siRNA, the cells were washed three times with PBS and lysed for 10 min with radioimmune precipitation assay lysis buffer containing protease inhibitor mixture (Roche) at 4 °C. Cell lysates were centrifuged, and the supernatant was resolved by electrophoresis in 4–15% Mini-Protean TGX precast polyacrylamide gradient protein gels (Bio-Rad). The following primary antibodies were used for immunoblotting: GAPDH (Sigma, G9545), Hic-5 (BD Transduction Laboratories, 611165), glucocorticoid receptor (Santa Cruz Biotechnology, SC8992), BRG1 (Abcam, EPNCIR11A), BRM (Abcam, ab15597), INO80 (Abcam, ab118787), SNF2H (Abcam, ab72499), EP400 (Abcam, ab5201), CHD1 (Bethyl Laboratories, A301218A), CHD4 (Abcam, ab72418), CHD6 (Bethyl Laboratories, A301221A), CHD7 (Cell Signaling Technology, 6505S), CHD8 (Cell Signaling Technology, 11891S), and CHD9 (Novus Biologicals, NB100-60419). The secondary antibodies used were goat anti-rabbit (Promega, W4011) and goat anti-mouse (Promega, W4021).
Quantitative RT-PCR
Total RNA was isolated from cells using TRIzol reagent (Thermo Scientific) and reverse-transcribed using the iScript cDNA synthesis kit (Bio-Rad). The resulting cDNA was analyzed by quantitative PCR amplification with the LightCycler 480 SYBR Green I Master (Roche) on the LightCycler 480 system (Roche). The following primers used to detect mRNA expression with quantitative PCR were described previously: RP1L1, HOXD1, IGFBP1, MSX2, and SCNN1A (9) and GRAMD4 (21). Other primers used were as follows: TIPARP (forward primer, 5′-TCCGCTCCTGTTTTATACTGC-3′; reverse primer, 5′-AGTTTGCTGAAGTGACCCC-3′) and SLN (forward primer, 5′-CAAGCCGCTGTGAAAATGG-3′; reverse primer, 5′-GAGCATCTCAGTCAATCCCAG-3′).
RNA sequencing analysis
U2OS-GRα cells transfected with combinations of siRNA were treated with either ethanol or 100 nm dexamethasone for 8 h. 12 different conditions were examined: control (siNS/siNS) with or without dexamethasone, Hic-5 only depletion (siHic5/siNS) with or without dexamethasone, CHD9-only depletion (siCHD9/siNS) with or without dexamethasone, CHD9 and Hic-5 double depletion (siCHD9/siHic5) with or without dexamethasone, BRM-only depletion (siBRM/siNS) with or without dexamethasone, and BRM and Hic-5 double depletion (siBRM/siHic5) with or without dexamethasone. Total RNA was extracted using TRIzol (Thermo Scientific), and three biological replicates were performed on different days. RNA samples were reverse-transcribed using the iScript cDNA synthesis kit (Bio-Rad). The quality of the cDNA was assessed using an Agilent Technologies 2100 bioanalyzer, and quantitative RT-PCR analysis was performed for selected GR target genes. A total of 36 high-quality samples (12 conditions for three replicates each) were submitted to the Next-Generation Sequencing Core at the University of Southern California Norris Comprehensive Cancer Center for library preparation and sequencing. Single-end 75-bp RNA sequencing data were generated for the samples using Illumina NEXTseq 500. The sequencing results produced 36–58 million raw reads per sample. After trimming the raw reads for quality and adapter sequence, the samples were mapped using TopHat 2.1.1 against the GRCh38/hg38 human reference genome (56). Mapped reads were quantified to known UCSC Genes using the GenomicAlignments R package (57). Gene expression levels were normalized with the upper quantile method, and low-expressing genes were excluded so that genes with more than one count per million in at least three samples were analyzed (58). We implemented the “remove unwanted variation” strategy to account for unknown nuisance technical effects between samples (59). Differentially expressed genes were identified with edgeR using a 1.3-fold change in expression (log2 = 0.4) and false discovery rate-adjusted p ≤ 0.05 as cutoffs (60). Gene Ontology was used for functional annotation of the gene classes (38, 39).
Algorithms for defining ind, mod, and block gene classes and genes that require CHD9 or BRM
Assignment of genes to the block, ind, and mod classes involved a strategy developed previously (6) using three different comparisons from the RNA sequencing data to define the following gene sets from the RNA sequencing data (supplemental Dataset S1): dexamethasone-regulated genes in control cells transfected with nonspecific siRNA (supplemental Fig. S3A, comparison I), dexamethasone-regulated genes in Hic-5-depleted cells (supplemental Fig. S3A, comparison II), and genes with mRNA levels that were significantly different between the control dexamethasone-treated cells and Hic-5-depleted dexamethasone-treated cells (supplemental Fig. S3A, comparison III). Except where otherwise indicated, significant differences in mRNA levels were defined with a 1.3-fold change cutoff and FDR-adjusted p ≤ 0.05. Specific regions of overlap among the gene sets derived from these three comparisons defined the block, ind, and mod gene sets. The ind genes (Fig. 3A, blue region) were dexamethasone-regulated in control cells (included in set I) and in Hic-5-depleted cells (included in set II) and had dexamethasone-treated mRNA levels that were not significantly different in control and Hic-5-depleted cells (excluded from set III). mod genes (Fig. 3A, green regions) were defined by the intersection of set I (dexamethasone-regulated in control cells) and set III (different dexamethasone-treated mRNA levels in control versus Hic-5-depleted cells). The block genes (Fig. 3A, red region) were included in sets II and III but not in set I, i.e. they were dexamethasone-regulated only after Hic-5 depletion and had different dexamethasone-treated mRNA levels in control versus Hic-5-depleted cells.
The number of genes in each of the three classes (block, ind, and mod) that required CHD9 or BRM for dexamethasone-regulated expression was defined by overlapping each of the three gene classes (block, ind, and mod) with two other comparisons from the RNA sequencing data (supplemental Datasets S2–S4). Specifically, each gene class (block, ind, and mod) was overlapped with sets a and b or with sets c and d (supplemental Fig. S3, B and C). Sets a and b were derived by comparing mRNA levels in cells depleted of Hic-5 alone versus cells doubly depleted of Hic-5 and CHD9 or Hic-5 and BRM (supplemental Fig. S3B, left panel). Sets c and d were derived by comparing mRNA levels in cells depleted of CHD9 or BRM alone versus control cells with no depletions (supplemental Fig. S3B, right panel). Although overlapping these comparisons identified several gene subsets of the block, ind, and mod gene classes (supplemental Fig. S3C, sectors i–iv), the genes of primary interest were those for which significant regulation by dexamethasone was entirely dependent on CHD9 or BRM, i.e. genes that were excluded from set a or c and included in set b or d (supplemental Fig. S3C, sector iii).
Because block genes are only dexamethasone-regulated when Hic-5 is depleted, genes in the block class were overlapped with sets a and b. Again, except where otherwise specified, we used 1.3-fold change in expression and FDR-adjusted p ≤ 0.05 as cutoffs for the analysis. Similar analyses were conducted with the ind and mod gene classes to determine the genes that were dependent on CHD9 and/or BRM for dexamethasone-regulated expression. Because the dexamethasone regulation of ind genes is independent of the presence or absence of Hic-5, the ind genes can be overlapped with results from either the double depletion of chromatin remodeler and Hic-5 (supplemental Fig. S3B, a and b) or chromatin remodeler depletion only (supplemental Fig. S3B, c and d). Because many mod genes (138 of 364 genes) were no longer dexamethasone-regulated upon Hic-5 depletion (Fig. 3B, green region excluded from set II), genes in this class were best analyzed by single depletion of CHD9 or BRM (without depleting Hic-5) so that all mod genes were included in the analysis. Therefore, mod genes were overlapped with sets c and d (supplemental Fig. S3C). However, to test reproducibility, we analyzed the dependence of ind and mod genes on CHD9 and BRM both in cells containing Hic-5 (Fig. 3, B–D) and in cells lacking Hic-5 (supplemental Fig. S3, D–F).
For the box plot showing the quantitative effects of depleting CHD9 and BRM on the expression of block, ind, and mod genes (Fig. 3E), the following formula was used,
| (Eq. 1) |
where Y = % decrease in log2 -fold change after CHD9 or BRM depletion, X = log2 -fold change cause by dexamethasone in cells containing CHD9 or BRM, and Z = log2 -fold change caused by dexamethasone in cells depleted of CHD9 or BRM.
Chromatin immunoprecipitation
These experiments were performed as described previously (9) with slight modifications. Briefly, U2OS-GRα cells grown on 15-cm dishes were transfected with the appropriate siRNAs. After 48 h, the cells were treated with 100 nm dexamethasone or equivalent amounts of ethanol for 1 h before cross-linking with 1% (v/v) formaldehyde for 10 min at room temperature and extracting chromatin from the harvested cells. Chromatin was sonicated for 20–30 min (30-s on/off cycles) with a Biorupter (Diagenode) at 4 °C to produce a DNA fragment size of 400–600 bp. Immunoprecipitation of the sonicated chromatin samples was conducted with a mixture of GR antibodies: H300 (6 μg, Santa Cruz Biotechnology), PA1-511A (2 μg, Thermo Scientific), and D6H2L (2 μg, Cell Signaling Technology). Protein G-Sepharose magnetic beads (GE Healthcare) were used to isolate the immune complexes with the cross-linked DNA. When the DNA was purified, quantitative PCR amplification with the LightCycler 480 SYBR Green I Master (Roche) on the LightCycler 480 system (Roche) was performed with primers for the GBRs at the genes of interest. The following are the primer sequences for the GBRs used in this study: RP1L1, IGFBP1, MSX2, and SCNN1A (9); GRAMD4 (21); and SLN (forward primer, 5′-CAGGCTACCCATCACACTTCTTT-3′; reverse primer, 5′-TCAAGGTCACCATTAAAGTGCAAGA-3′).
FAIRE
The protocol used for the FAIRE experiment was described previously (61). Cells were treated as in the chromatin immunoprecipitation experiments. When free DNA was purified from cross-linked, sonicated chromatin by phenol extraction, quantitative PCR primers for GBRs at the genes of interest as mentioned for chromatin immunoprecipitation were used to assess chromatin accessibility.
Proximity ligation assay
The PLA technology developed by Olink Bioscience allows the visualization of protein-protein interactions in situ (62). U2OS-GRα cells were grown on coverslips in 12-well plates and transfected with siRNA for 48 h, followed by 1-h dexamethasone or ethanol treatment. The cells were fixed with methanol for 2 min and treated according to the PLA probe protocol in the instructions of the manufacturer (Olink Bioscience). Samples were first saturated with blocking solution and then incubated with two primary antibodies of differing species that bind to their respective, potentially interacting proteins for 1 h in a 37 °C humidified chamber. Next, secondary antibodies conjugated with complementary oligonucleotides serving as PLA probes were added for 1 h in a 37 °C humidified chamber. Ligation and rolling circle amplification were performed for 100 min at 37 °C. During the amplification step, fluorescently labeled oligonucleotides hybridized to the amplified product. The coverslips were then dried and mounted using Duolink II mounting medium with DAPI (Sigma) for nucleus staining. Slides were analyzed using a fluorescence microscope. Each PLA fluorescent dot represents one bimolecular protein interaction. ImageJ version 1.49 (https://imagej.nih.gov/ij/) was used to quantify the fluorescent dots in the nucleus. For each sample, interactions were counted for at least 400 cells.
Statistical analysis
Statistical analyses of quantitative RT-PCR, chromatin immunoprecipitation with quantitative PCR, FAIRE with quantitative PCR, and quantification of PLA interactions were performed using paired t test. The number of biological replicates (n) and p values for each experiment are indicated in the figure legends. Box plots were generated using R. The horizontal center lines indicate the median, with the upper and lower box limits denoting the 25th and 75th percentile, respectively. The whiskers extend 1.5 times the interquartile range from the 25th and 75th percentiles, and outliers are represented by dots. The p values indicating statistical significance between the datasets in the box plots were obtained using Mann-Whitney U test. RNA sequencing data were analyzed using edgeR. Differentially expressed genes were obtained using 1.3-fold change (log2 = 0.4) and 0.05 FDR-adjusted p value cutoff. For higher stringency analysis, 1.5- or 2.0-fold change and 0.01 FDR-adjusted p values were used for the cutoff.
Author contributions
B. H. L. performed all experiments and analyzed the data; he also participated equally in conception of the project and writing the paper. M. R. S. participated equally in conception of the project and writing the paper.
Supplementary Material
Acknowledgments
We thank Rajas Chodankar and Coralie Poulard for experimental advice, Dan Gerke for technical assistance, and Ruchi Bajpai for providing antibodies against CHD6–9 and experimental advice. We also thank Yibu Chen, Meng Li, Suhn Rhie, and Kim Siegmund for advice on bioinformatics analysis.
This work was supported by National Institutes of Health grants R37DK055274 and R01DK043093 (to M. R. S.) and Cancer Center Support Grant P30CA014089 (to the USC Norris Comprehensive Cancer Center, which supported the Molecular Genomics, Bioreagent, Biostatistics, and Molecular and Cell Biology Core Facilites used in this project). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Figs. S1–S6 and Datasets S1–S4.
The RNA sequencing data were submitted to the Gene Expression Omnibus (GEO) under accession number GSE93871.
- GR
- glucocorticoid receptor
- GC
- glucocorticoid
- GBR
- glucocorticoid receptor-binding region
- FDR
- false discovery rate
- FAIRE
- formaldehyde-assisted isolation of regulatory elements
- PLA
- proximity ligation assay
- BRM
- Brahma homologue, product of the SMARCA2 gene.
References
- 1. Glass C. K., and Rosenfeld M. G. (2000) The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev. 14, 121–141 [PubMed] [Google Scholar]
- 2. Lonard D. M., and O'Malley B. W. (2012) Nuclear receptor coregulators: modulators of pathology and therapeutic targets. Nat. Rev. Endocrinol. 8, 598–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rosenfeld M. G., Lunyak V. V., and Glass C. K. (2006) Sensors and signals: a coactivator/corepressor/epigenetic code for integrating signal-dependent programs of transcriptional response. Genes Dev. 20, 1405–1428 [DOI] [PubMed] [Google Scholar]
- 4. Won Jeong K., Chodankar R., Purcell D. J., Bittencourt D., and Stallcup M. R. (2012) Gene-specific patterns of coregulator requirements by estrogen receptor-α in breast cancer cells. Mol. Endocrinol. 26, 955–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bittencourt D., Wu D. Y., Jeong K. W., Gerke D. S., Herviou L., Ianculescu I., Chodankar R., Siegmund K. D., and Stallcup M. R. (2012) G9a functions as a molecular scaffold for assembly of transcriptional coactivators on a subset of glucocorticoid receptor target genes. Proc. Natl. Acad. Sci. U.S.A. 109, 19673–19678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wu D. Y., Ou C. Y., Chodankar R., Siegmund K. D., and Stallcup M. R. (2014) Distinct, genome-wide, gene-specific selectivity patterns of four glucocorticoid receptor coregulators. Nucl. Recept. Signal. 12, e002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yang C. K., Kim J. H., and Stallcup M. R. (2006) Role of the N-terminal activation domain of the coiled-coil coactivator in mediating transcriptional activation by β-catenin. Mol. Endocrinol. 20, 3251–3262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rogatsky I., Luecke H. F., Leitman D. C., and Yamamoto K. R. (2002) Alternate surfaces of transcriptional coregulator GRIP1 function in different glucocorticoid receptor activation and repression contexts. Proc. Natl. Acad. Sci. U.S.A. 99, 16701–16706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chodankar R., Wu D. Y., Schiller B. J., Yamamoto K. R., and Stallcup M. R. (2014) Hic-5 is a transcription coregulator that acts before and/or after glucocorticoid receptor genome occupancy in a gene-selective manner. Proc. Natl. Acad. Sci. U.S.A. 111, 4007–4012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Biddie S. C., Conway-Campbell B. L., and Lightman S. L. (2012) Dynamic regulation of glucocorticoid signalling in health and disease. Rheumatology 51, 403–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brown M. C., Curtis M. S., and Turner C. E. (1998) Paxillin LD motifs may define a new family of protein recognition domains. Nat. Struct. Biol. 5, 677–678 [DOI] [PubMed] [Google Scholar]
- 12. Bach I. (2000) The LIM domain: regulation by association. Mech. Dev. 91, 5–17 [DOI] [PubMed] [Google Scholar]
- 13. Kim-Kaneyama J. R., Lei X. F., Arita S., Miyauchi A., Miyazaki T., and Miyazaki A. (2012) Hydrogen peroxide-inducible clone 5 (Hic-5) as a potential therapeutic target for vascular and other disorders. J. Atheroscler. Thromb. 19, 601–607 [DOI] [PubMed] [Google Scholar]
- 14. Yang L., Guerrero J., Hong H., DeFranco D. B., and Stallcup M. R. (2000) Interaction of the tau2 transcriptional activation domain of glucocorticoid receptor with a novel steroid receptor coactivator, Hic-5, which localizes to both focal adhesions and the nuclear matrix. Mol. Biol. Cell 11, 2007–2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Drori S., Girnun G. D., Tou L., Szwaya J. D., Mueller E., Xia K., Kia X., Shivdasani R. A., and Spiegelman B. M. (2005) Hic-5 regulates an epithelial program mediated by PPARγ. Genes Dev. 19, 362–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Aghajanova L., Velarde M. C., and Giudice L. C. (2009) The progesterone receptor coactivator Hic-5 is involved in the pathophysiology of endometriosis. Endocrinology 150, 3863–3870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li X., Martinez-Ferrer M., Botta V., Uwamariya C., Banerjee J., and Bhowmick N. A. (2011) Epithelial Hic-5/ARA55 expression contributes to prostate tumorigenesis and castrate responsiveness. Oncogene 30, 167–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang H., Song K., Krebs T. L., Yang J., and Danielpour D. (2008) Smad7 is inactivated through a direct physical interaction with the LIM protein Hic-5/ARA55. Oncogene 27, 6791–6805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shibanuma M., Kim-Kaneyama J. R., Sato S., and Nose K. (2004) A LIM protein, Hic-5, functions as a potential coactivator for Sp1. J. Cell. Biochem. 91, 633–645 [DOI] [PubMed] [Google Scholar]
- 20. Leach D. A., Need E. F., Trotta A. P., Grubisha M. J., DeFranco D. B., and Buchanan G. (2014) Hic-5 influences genomic and non-genomic actions of the androgen receptor in prostate myofibroblasts. Mol. Cell. Endocrinol. 384, 185–199 [DOI] [PubMed] [Google Scholar]
- 21. Chodankar R., Wu D. Y., Gerke D. S., and Stallcup M. R. (2015) Selective coregulator function and restriction of steroid receptor chromatin occupancy by Hic-5. Mol. Endocrinol. 29, 716–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nagaich A. K., Walker D. A., Wolford R., and Hager G. L. (2004) Rapid periodic binding and displacement of the glucocorticoid receptor during chromatin remodeling. Mol. Cell 14, 163–174 [DOI] [PubMed] [Google Scholar]
- 23. Fryer C. J., and Archer T. K. (1998) Chromatin remodelling by the glucocorticoid receptor requires the BRG1 complex. Nature 393, 88–91 [DOI] [PubMed] [Google Scholar]
- 24. Burd C. J., and Archer T. K. (2013) Chromatin architecture defines the glucocorticoid response. Mol. Cell. Endocrinol. 380, 25–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McNally J. G., Müller W. G., Walker D., Wolford R., and Hager G. L. (2000) The glucocorticoid receptor: rapid exchange with regulatory sites in living cells. Science 287, 1262–1265 [DOI] [PubMed] [Google Scholar]
- 26. Morris S. A., Baek S., Sung M. H., John S., Wiench M., Johnson T. A., Schiltz R. L., and Hager G. L. (2014) Overlapping chromatin-remodeling systems collaborate genome wide at dynamic chromatin transitions. Nat. Struct. Mol. Biol. 21, 73–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Engel K. B., and Yamamoto K. R. (2011) The glucocorticoid receptor and the coregulator Brm selectively modulate each other's occupancy and activity in a gene-specific manner. Mol. Cell. Biol. 31, 3267–3276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Clapier C. R., and Cairns B. R. (2009) The biology of chromatin remodeling complexes. Annu. Rev. Biochem. 78, 273–304 [DOI] [PubMed] [Google Scholar]
- 29. Wiechens N., Singh V., Gkikopoulos T., Schofield P., Rocha S., and Owen-Hughes T. (2016) The chromatin remodelling enzymes SNF2H and SNF2L position nucleosomes adjacent to CTCF and other transcription factors. PLoS Genet. 12, e1005940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bao Y., and Shen X. (2007) INO80 subfamily of chromatin remodeling complexes. Mutat. Res. 618, 18–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pradhan S. K., Su T., Yen L., Jacquet K., Huang C., Côté J., Kurdistani S. K., and Carey M. F. (2016) EP400 deposits H3.3 into promoters and enhancers during gene activation. Mol. Cell 61, 27–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schnetz M. P., Bartels C. F., Shastri K., Balasubramanian D., Zentner G. E., Balaji R., Zhang X., Song L., Wang Z., Laframboise T., Crawford G. E., and Scacheri P. C. (2009) Genomic distribution of CHD7 on chromatin tracks H3K4 methylation patterns. Genome Res. 19, 590–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Menon T., Yates J. A., and Bochar D. A. (2010) Regulation of androgen-responsive transcription by the chromatin remodeling factor CHD8. Mol. Endocrinol. 24, 1165–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lutz T., Stöger R., and Nieto A. (2006) CHD6 is a DNA-dependent ATPase and localizes at nuclear sites of mRNA synthesis. FEBS Lett. 580, 5851–5857 [DOI] [PubMed] [Google Scholar]
- 35. Marom R., Shur I., Hager G. L., and Benayahu D. (2006) Expression and regulation of CReMM, a chromodomain helicase-DNA-binding (CHD), in marrow stroma derived osteoprogenitors. J. Cell. Physiol. 207, 628–635 [DOI] [PubMed] [Google Scholar]
- 36. Gaspar-Maia A., Alajem A., Polesso F., Sridharan R., Mason M. J., Heidersbach A., Ramalho-Santos J., McManus M. T., Plath K., Meshorer E., and Ramalho-Santos M. (2009) Chd1 regulates open chromatin and pluripotency of embryonic stem cells. Nature 460, 863–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sparmann A., Xie Y., Verhoeven E., Vermeulen M., Lancini C., Gargiulo G., Hulsman D., Mann M., Knoblich J. A., and van Lohuizen M. (2013) The chromodomain helicase Chd4 is required for Polycomb-mediated inhibition of astroglial differentiation. EMBO J. 32, 1598–1612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ashburner M., Ball C. A., Blake J. A., Botstein D., Butler H., Cherry J. M., Davis A. P., Dolinski K., Dwight S. S., Eppig J. T., Harris M. A., Hill D. P., Issel-Tarver L., Kasarskis A., Lewis S., et al. (2000) Gene ontology: tool for the unification of biology: the Gene Ontology Consortium. Nat. Genet. 25, 25–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. The Gene Ontology Consortium (2017) Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Res. 45, D331–D338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schiller B. J., Chodankar R., Watson L. C., Stallcup M. R., and Yamamoto K. R. (2014) Glucocorticoid receptor binds half sites as a monomer and regulates specific target genes. Genome Biol. 15, 418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bartholomew B. (2014) Regulating the chromatin landscape: structural and mechanistic perspectives. Annu. Rev. Biochem. 83, 671–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. John S., Sabo P. J., Thurman R. E., Sung M. H., Biddie S. C., Johnson T. A., Hager G. L., and Stamatoyannopoulos J. A. (2011) Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nat. Genet. 43, 264–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lefstin J. A., and Yamamoto K. R. (1998) Allosteric effects of DNA on transcriptional regulators. Nature 392, 885–888 [DOI] [PubMed] [Google Scholar]
- 44. Meijsing S. H., Pufall M. A., So A. Y., Bates D. L., Chen L., and Yamamoto K. R. (2009) DNA binding site sequence directs glucocorticoid receptor structure and activity. Science 324, 407–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nguyen K. H., Xu F., Flowers S., Williams E. A., Fritton J. C., and Moran E. (2015) SWI/SNF-mediated lineage determination in mesenchymal stem cells confers resistance to osteoporosis. Stem Cells 33, 3028–3038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Becker J. S., Nicetto D., and Zaret K. S. (2016) H3K9me3-dependent heterochromatin: barrier to cell fate changes. Trends Genet. 32, 29–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yuminamochi T., Yatomi Y., Osada M., Ohmori T., Ishii Y., Nakazawa K., Hosogaya S., and Ozaki Y. (2003) Expression of the LIM proteins paxillin and Hic-5 in human tissues. J. Histochem. Cytochem. 51, 513–521 [DOI] [PubMed] [Google Scholar]
- 48. Deakin N. O., Pignatelli J., and Turner C. E. (2012) Diverse roles for the paxillin family of proteins in cancer. Genes Cancer 3, 362–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ishino M., Aoto H., Sasaski H., Suzuki R., and Sasaki T. (2000) Phosphorylation of Hic-5 at tyrosine 60 by CAKβ and Fyn. FEBS Lett. 474, 179–183 [DOI] [PubMed] [Google Scholar]
- 50. Wang X., Yang Y., Guo X., Sampson E. R., Hsu C. L., Tsai M. Y., Yeh S., Wu G., Guo Y., and Chang C. (2002) Suppression of androgen receptor transactivation by Pyk2 via interaction and phosphorylation of the ARA55 coregulator. J. Biol. Chem. 277, 15426–15431 [DOI] [PubMed] [Google Scholar]
- 51. Maudsley S., Davidson L., Pawson A. J., Freestone S. H., López de Maturana R., Thomson A. A., and Millar R. P. (2006) Gonadotropin-releasing hormone functionally antagonizes testosterone activation of the human androgen receptor in prostate cells through focal adhesion complexes involving Hic-5. Neuroendocrinology 84, 285–300 [DOI] [PubMed] [Google Scholar]
- 52. Arora V. K., Schenkein E., Murali R., Subudhi S. K., Wongvipat J., Balbas M. D., Shah N., Cai L., Efstathiou E., Logothetis C., Zheng D., and Sawyers C. L. (2013) Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell 155, 1309–1322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pockwinse S. M., Stein J. L., Lian J. B., and Stein G. S. (1995) Developmental stage-specific cellular responses to vitamin D and glucocorticoids during differentiation of the osteoblast phenotype: interrelationship of morphology and gene expression by in situ hybridization. Exp. Cell Res. 216, 244–260 [DOI] [PubMed] [Google Scholar]
- 54. Shalhoub V., Conlon D., Tassinari M., Quinn C., Partridge N., Stein G. S., and Lian J. B. (1992) Glucocorticoids promote development of the osteoblast phenotype by selectively modulating expression of cell growth and differentiation associated genes. J. Cell. Biochem. 50, 425–440 [DOI] [PubMed] [Google Scholar]
- 55. Rogatsky I., Trowbridge J. M., and Garabedian M. J. (1997) Glucocorticoid receptor-mediated cell cycle arrest is achieved through distinct cell-specific transcriptional regulatory mechanisms. Mol. Cell. Biol. 17, 3181–3193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Trapnell C., Williams B. A., Pertea G., Mortazavi A., Kwan G., van Baren M. J., Salzberg S. L., Wold B. J., and Pachter L. (2010) Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lawrence M., Huber W., Pagès H., Aboyoun P., Carlson M., Gentleman R., Morgan M. T., and Carey V. J. (2013) Software for computing and annotating genomic ranges. PLoS Comput. Biol. 9, e1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bullard J. H., Purdom E., Hansen K. D., and Dudoit S. (2010) Evaluation of statistical methods for normalization and differential expression in mRNA-seq experiments. BMC Bioinformatics 11, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Risso D., Ngai J., Speed T. P., and Dudoit S. (2014) Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechnol 32, 896–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Robinson M. D., McCarthy D. J., and Smyth G. K. (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Simon J. M., Giresi P. G., Davis I. J., and Lieb J. D. (2012) Using formaldehyde-assisted isolation of regulatory elements (FAIRE) to isolate active regulatory DNA. Nat. Protoc. 7, 256–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Söderberg O., Gullberg M., Jarvius M., Ridderstråle K., Leuchowius K. J., Jarvius J., Wester K., Hydbring P., Bahram F., Larsson L. G., and Landegren U. (2006) Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods 3, 995–1000 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.