

Abstract
Despite certain quantum concepts, such as superposition states, entanglement, ‘spooky action at a distance’ and tunnelling through insulating walls, being somewhat counterintuitive, they are no doubt extremely useful constructs in theoretical and experimental physics. More uncertain, however, is whether or not these concepts are fundamental to biology and living processes. Of course, at the fundamental level all things are quantum, because all things are built from the quantized states and rules that govern atoms. But when does the quantum mechanical toolkit become the best tool for the job? This review looks at four areas of ‘quantum effects in biology’. These are biosystems that are very diverse in detail but possess some commonality. They are all (i) effects in biology: rates of a signal (or information) that can be calculated from a form of the ‘golden rule’ and (ii) they are all protein–pigment (or ligand) complex systems. It is shown, beginning with the rate equation, that all these systems may contain some degree of quantumeffect, and where experimental evidence is available, it is explored to determine how the quantum analysis aids in understanding of the process.
Keywords: quantum, enzymes, olfaction, photosynthesis, magnetodetection
1. Introduction
Four living processes are considered in this review: a reaction mechanism (enzyme catalysis), a sensory signal (olfaction), the transfer of energy (photosynthetic energy capture) and the encoding of information (magnetodetection). Though diverse areas, they have at least two common threads (i) they are biosystems that may be understood in terms of a rate and (ii) they are all biosystems consisting of a pigment (or small, non-protein, molecule such as a ligand/odorant/flavin) in a protein environment. Examining the rate equation, based on Fermi's golden rule, will allow us to consider ‘quantum effects’ and, surprisingly, it will be seen that the protein environment of these systems does not camouflage or collapse any state, but conversely seems not only to enable the said rate but also to further- to enhance- it. The ways in which the full system is treated involve various degrees of ‘quantumness’ and we shall see briefly how this is done in the four systems that follow. Firstly, it is shown that the remarkably fast reaction rates of enzymes are accelerated by tunnelling phenomena. Secondly, it is conjectured that the signalling in olfaction is also a tunnelling effect, enabled by the presence of an odorant with a signature quantum mechanical vibration. Thirdly, it is seen that the superposition of excited states upon light excitation in photosynthesis supports long-lived coherent states, conjectured to explain the efficiency of energy transfer. Fourthly, magnetoreception is briefly examined, where it may be seen that entangled states encode the angle of magnetic field lines, perceived in animals as differences in reaction rates. Where available, this review is written around experimental evidence and theory is described in order to explain/predict the quantum effects, depicted when possible in the figures. Whether any quantum theorizing illuminates and explains a biological effect is then briefly discussed in the conclusions with a final mention of the role of the environment and what remains as future work and challenges in the field.
(a). Rate equation
Be it a charge or energy transfer (or both), the systems discussed here share one commonality: a transition from one state to another, and the rate of the transition can be analysed with the rate equation, or the ‘golden rule’ [1]:
| 1.1 |
where the rate describes a transition from D (donor, initial state or reactant) to A (acceptor, final state or product), where ρ is the density of states that can accept the transferring particle (or Franck–Condon (FC) factors), is the Planck constant (quantum of action) and HDA are Hamiltonian transition matrix, where the transition may be tunnelling. In all cases discussed here, transfer occurs between an initial (donor, D) and final (acceptor, A) state, and always within a protein environment that may be in a complex with a small molecule be it a substrate, odorant or chromophore. The transit can be treated classically, or with various degrees of quantum definition (e.g. of the electronic states and/or nuclear states). One tool simple and effective for describing any transfer event is a configuration coordinate diagram, as in figure 1, where a classical picture is illustrated. In its simplest form, the rate is classical; through examining the four topics in this article, it will be shown that in some cases semi-classical approximations may explain well experimental observations, but in others (particularly §4) where coherence is proved to be an important effect the ‘quantum part’ is more complex. In tunnelling quantum mechanics enters via HDA, to determine the probability of transfer of the moving particle, equation (1.1), but it emerges in some calculation of rates, such as photosynthetic energy transfer, that vibronic effects (coupled electronic and nuclear motion) must be included which reflects the importance of treating the whole (protein) environment quantum mechanically.
Figure 1.

(a) A configuration coordinate diagram showing the activation energy barrier E‡, separating the reactant states (donor D) from the product states (acceptor A) which are shown as diabatic Born–Oppenheimer surfaces, with κ=1, as in equation (1.2). Shown also, ΔE the energy difference at minimum between the D and A, at XD and XA, respectively, λ the reorganization energy and the reaction coordinate Q [2,1]. Xc indicates a crossing point for the transitioning particle at the asterisk. (b) a classical tunnelling (or insulating) barrier that separates the donor and acceptor positions, barrier height is E‡, barrier width is distance r.
In classical absolute reaction rate theory, Eyring [3], the rate constant of a transitioning particle from D to A, as in figure 1, would be given by the following:
| 1.2 |
and
| 1.3 |
where B∼1011M−1s−1 is the collision frequency of the reacting particles depending on the nature and phase of the mixture. The above relates to the Arrhenius equation [1]:
| 1.4 |
where ΔE‡ is the energy of activation, ΔG‡ is the free energy, ΔH‡ is the enthalpy and ΔS‡ is the entropy. Within this classical regime, the factor κ↦|HDA|2, see equation (1.1), and its value determine adiabacity/non-adiabacity, with κ=1 or κ<1, respectively, i.e. the nature of the Born–Oppenheimer surfaces (as depicted in figure 1. The configuration coordinate diagram shows these terms, and by the parabola geometry it can be found that E‡=(λ+ΔE)2/4λ, where λ is the reorganization (or relaxation) energy coupled to the particle transfer. The total reorganization energy is usually considered as a summation of inner shell atoms (i) and solvent molecules (o) re-arranging, respectively, λ=λi+λo where
| 1.5 |
and the Qs indicate harmonic oscillators, i.e. the displacement from equilibrium position upon charge transfer, as governed by Hooke's Law, where kij is a spring constant, assuming normal mode vibrations. The outer molecular environment re-arrangement is given by
| 1.6 |
where the charged particle in this case is an electron e and r are the radii of two reactants, r12 of the two reactants in contact, DS is the static dielectric constant and Dop is the square of refractive index of reaction [1]. The reorganization energy can be generalized to λ=μω2Q2/2, where Q is a normal mode, μ is the reduced mass and ω=2πv is the angular frequency of oscillation. Examining figure 1, it can be seen that this reorganization energy is the energy required to displace Q without charge transfer, Q=XD−XA.
In the purely classical picture, it is the case that for a transition D↦A there is a probability of occurrence determined mainly by overcoming the activation energy barrier E‡, and the ability of the environment to arrange such that there is crossing at Xc, and there is energy conservation ED=EA. That is, if the particle is considered a ball as in figure 1. If the particle is considered a wave, the probabilities change is determined by the overlap of wave functions. The energy barrier depicted by the wall ΔE‡ becomes penetrable: tunnelling occurs. In the quantum mechanical penetration of a square barrier like that shown in figure 1 [1], the probability of a particle penetrating is given by [4]
| 1.7 |
such that the probability decays exponentially with increasing barrier width r and the decay constant varies with the square root of the barrier height ΔE‡ and mass m of the particle. Thus in classical activation, the height of the barrier is the main hurdle, whereas in quantum activation the width of the barrier is also an important obstacle. This review highlights the use of this equation (1.1) for tunnelling phenomena, as Oppenheimer first showed in 1928 [5], but applied to biology. Furthermore, it will be seen that various other effects in biological systems beyond tunnelling, such as coherence (§4) and entanglement1 (§5) arise from the analysis of the rate equation and the determination of the Hamiltonian that defines the transition from D to A, HDA, and in some cases the quantum effects of the environment.
(b). The environment helps or hinders?
In the rate equation, the Hamiltonian matrix HDA may simply describe two eigenstates, D and A. In some cases, this is enough to describe rate observations, in others a fuller definition of the Hamiltonian matrix is required, which may be limited by what is definitively known of the system. Quantum systems are not isolated from the environment, thus the total Hamiltonian representing a system may be written as follows:
| 1.8 |
Here, HS is the Hamiltonian for the system, HE describes the free evolution of the environment and HI the interaction between the environment and the system [6]. If we are describing electron transfer for example, HS may involve electronic states on D and A. In fact in equation (1.8), it most often is the case that HS represents electronic states, HE the nuclear states and HI the nuclear–electronic interactions. The Hamiltonian is often written so that electronic states are coupled to a ‘bath’ of nuclear motion that may be represented by a collection of (bosonic) oscillators as in equation (1.5) or as a collection of spin states see §5. The system is considered ‘closed’ if the system-environment evolves under a Hamiltonian; the evolution is unitary (see §4a) but ‘open’ if the Hamiltonian, as in equation (1.8), is insufficient to describe the dynamics. The open quantum system thus accounts fully for the quantum effects of the protein dynamics on the rate (e.g. of charge/energy transfer), typically expected to dissipate and ‘leak’ information in a non-classical way, in other words, decoherence [7].
Usually, environmental variables are controlled where possible in physical tests: one variable is isolated and the effect of changes on the rest of the system monitored (or vice versa). Where quantum effects are typically observed, the system is usually a relatively simple, perhaps a single molecule. By contrast, biological systems of many molecules (and atoms), degrees of freedom and so, many variables, are not simple. In their simplest forms, the following examples constitute a particle (e.g. an electron and a photon) a ligand (or pigment) and a protein which hosts the interaction. Proteins have many degrees of freedom and so potential energy landscape, and are dynamical beasts which move within this multidimensional landscape. This environment is often referred to in the literature as a ‘wet, noisy and warm system’ in contrast the isolated, single molecule view that a quantum optics physicist may prefer. This wet warm noise is thought to ‘drown’ out any fine-tuned quantum effects. Whatever an electron or a photon may be doing would be washed away by the large amplitude, governed by kT, of nuclear vibrations insensitive to the transitions in and around them. Fascinatingly, and contrary to expectations, it seems that, far from being insensitive to, or drowning out, these small effects, it is becoming clear that the protein environment is not only integral to, but also assists these rate changes contrary to expectations. Such ‘protein–pigment complexes’ or PPCs will be described through four examples. Notably, only one of the PPCs is depicted here, in figure 7, with protein data bank information (pdb code 3BSD).2 While structure data for PPCs are ubiquitous for enzymatic systems, they are unknown in the case of olfaction and magnetodetection. Unsurprisingly, much more testing of ‘quantum effects’ has ensued in the (much larger, and growing) field of photosynthesis. One reason being the photosynthetic materials present less challenges experimentally, but also they are more water soluble (more than olfactory receptors, for example), available and cheap (e.g. spinach can be used to extract a light harvesting system for testing relatively easily).
Figure 7.

(a) A PPC from pdb code 3BSD showing the seven pigments in the Fenna–Matthews–Olson (FMO) complex in Chlorobaculum tepidum (formerly Chlorobium tepidum) from [55]. The structure of the FMO is shown in blue ribbon and the chromophores: bacteriochlorophyll a (BChl a) is in green. The two-dimensional molecular structure of the seven BChl a is also shown next to the protein [55]. (b) Born–Oppenheimer surfaces to show the shift from ground to excited state on a BChl a pigment in FMO [56].
2. Enzymes
Enzymatic processes are catalytic mechanisms in the body for essential biochemical processes, for example hydrolysis of the neurotransmitter acetylcholine by the enzyme acetylcholinesterase. The paradigm for enzymatic reactions is the ‘lock and key’ concept (see also §3) derived from the fact that the enzyme substrate binds into an ‘active site’ on the enzyme structure, making particular intermolecular contacts with amino acid residues on the enzyme. These residues typically act as nucleophiles or acid/base catalysts in the reaction, which occurs only if these point contacts are made: as a key (substrate) fitting into a lock (enzyme). The process of obtaining this particular arrangement, like a ‘lock and key’ or a ‘hand in glove’ between the enzyme and substrate, may involve orbital steering, and ‘pushes’ the substrate towards a transition state (TS), along the reaction trajectory from the initial (reactant) to the final (product) state (or from D to A as in figure 1). This fit then drives a transition along a reaction pathway, on a configuration coordinate diagram and the activation energy of the reaction is lowered (see equation (1.4)). The phrase for this process, first coined by Pauling [2,8], is the ‘enhanced transition state theory’, ‘enhanced’ because of this preferential binding between enzyme and substrate that cannot be easily mimicked or substituted. However, a confounding puzzle exists: the efficiency (or speed) with which this transition occurs (i.e. there can be a 1025-fold difference in rate, experimentally observed, when comparing the molecules reacting in solution versus enzymatic environments [2]) cannot solely be explained by classical mechanics or any purely classical transition state theory (TST). Not only are these fast rates observed, but enzymatic processes exhibit weird behaviour with respect to temperature. How are these strange results and faster rates achieved?
Key experiments in 1966 by DeVault & Chance first showed that a tunnelling effect was present in enzymes, see figure 2 [9]. In Chromatium vinsosum, a photosynthetic bacterium, light-induced oxidation of cytochrome initiates electron transfer from cytochrome (donor, D) to bacteriochlorophyll, BChl (acceptor, A; figure 2). Unless the two molecules are in close proximity (within Van der Waals contact), electron transfer from cytochrome to BChl is divided by a barrier, a classically forbidden/insulating barrier. The experiments showed that, as expected, the oxidation at high temperatures is temperature dependent; the rates are faster at higher temperatures indicating there is an activation barrier to surmount as in figure 1. But, surprisingly, at lower temperatures (4–100 K) the temperature dependence is lost, and the reaction still occurs without the ‘required’ energy to overcome the TST activation barrier. This implies that without the requirement of kinetic energy to surmount the barrier the electrons may be quantum mechanically tunnelling through. This finding thus first highlighted the inability of TST in its classical regime to explain the temperature independence at low temperature and the high rates of electron transfer. While increasing the temperature does increase the reaction rate, the classical theory still does not account for the fastest rates. Quantum tunnelling is one way to penetrate barrier crossing, avoiding the barrier height issue and so to enabling and enhancing a rate.
Figure 2.

DeVault & Chance's pictorial description of the tunnelling region between cytochrome and bacteriochlorophyll (BChl) from [9] with permission from Elsevier. Energetic levels occupied by valence electrons are shown. The BChl is excited by light which leaves a hole quickly filled by an electron tunnelling from the cytochrome. DeVault & Chance also indicate the possibility of an ‘activated’ path versus the tunnelling path, where the activated path may be enabled, for example by a vibration in the cytochrome, bringing together the energetic states to the crossing point (asterisk, see figure 1) and/or the tunnelling path also may be ‘stabilized by a phonon emission’ [9].
The DeVault and Chance experiments measure the temperature dependence of light-activated electron tunnelling. The enzyme protochlorphyllide oxidoreductase (POR) synthesizes chlorophyll by a light-activated hydride and proton transfer. Measurement of this reaction rate is also possible at a range of temperatures (physiological and cryogenic) and probes a nuclear tunnelling event enhanced by motion along the H-transfer coordinate [10]. The phenomenon of nuclear tunnelling in bio-catalysis offers an opportunity to test rates with the kinetic isotope effect (KIE). Increasing the mass of an atom (where to within a suitable degree, all else remains the same) by isotopic substitution will alter the rate, in a mass (frequency-dependent) manner given , where k is the force constant, μ the reduced mass and ω is the bond frequency, and ω=2πv. Heavier isotopes have lower frequencies v, a higher activation energy ΔE‡ and so a lower rate, if determined by equation (1.4). Changing the mass in the rate equation determined by TST thus yields an expected rate change, e.g. for deuterium versus hydrogen (D/H) and tritium over hydrogen (T/H). So isotopic substitution allows a test for how well the observed rates can be predicted in a KIE [11–13]:
| 2.1 |
where l denotes the light particle and h denotes the heavy particle, A is an Arrhenius prefactor, R is a gas constant and E‡ is an activation energy (figure 1) [2]. This model calculates rates and provides clear experimental tests of the predictions; however, deviations from the expected rate have been consistently observed, indicating the classical model is insufficient. Considering a hydrogen transfer event (figure 3) [14]. It can be expected that as hydrogen would have a lower activation energy, it will have a faster rate. Not considered in the classical regime is that it is more probable not just because of a reduction in the barrier height (activation enthalpy ΔE‡) but also a reduction in the distance of transition (width of barrier) r, see equation (1.7) and figure 1. Lighter particles can tunnel over larger distances. Heavier particles have comparably shorter de Broglie wavelengths: i.e. they are more sensitive to distance fluctuations and so do not tunnel as far. In 1989, Klinman was the first to show proton tunnelling in enzyme reactions using the KIE with consideration of full quantum mechanical vibrations to narrow the D–A gap (r) and facilitate tunnelling, providing less width for more efficient tunnelling which explains the increased rates better [15].
Figure 3.

Showing the internal hydrogen transfer in malonaldehyde from Hay & Scrutton [14]. (a,b) The transition towards product from reactant, via the transition state (TS). (c) Donor (D) and acceptor (A) moving along a reaction coordinate, Q, the dashed line shows where tunnelling is most probable at the TS. The TRC denotes ‘tunnelling ready coordinate’. (d) The potential energy surface along a reaction coordinate for hydrogen and deuterium transfer at the same D–A distances. The idea of a ‘promoting mode’: to enhance the hydrogen transition is depicted, if motion in c d(Q) is conducive to promotion, then the tunnelling distance dtun is reduced and in combination with a lowering of the activation enthalpy a rate enhancement results.
Using equations (1.1) and (1.4), and so treating the nuclear motion classically but the charge transition quantum mechanically,
| 2.2 |
we acquire the non-adiabatic semi-classical Marcus theory expression which can be used in the high-temperature limit (when all the vibrations are excited) [1,16]. So transitions at high temperature can be measured and calculated for their rates; however, a fully quantum picture is required to explain the temperature independence at low temperatures as observed in the experiments by DeVault and Chance. At thermal equilibrium, oscillations have large energy kBT with the square of standard deviation given by σ2=kBT/ki, and a probability of displacement from equilibrium given by
| 2.3 |
accounting for quantum mechanical vibrations instead of classical, the average energy is given by
| 2.4 |
such that, for the equally spaced vibrational quanta (phonons) of difference , even at n=0, the lowest energy level has and a probability of displacement:
| 2.5 |
which converges to kBT at high temperature, and at low temperature , where independence of T is achieved [1]. So, now treating the nuclear vibrations as quantum, we arrive at the semi-classical regime [17]:
| 2.6 |
The fully quantum regime can be obtained by using equation (1.1) with FC factors. Using FC factors that assume the Condon approximation, that the transition HDA is constant over the cross-over region, see the asterisk in figure 1, and the nuclear and the electronic parts of the Hamiltonian can be separated and factored out. As nuclear vibrations are on the scale of 0.1 Åversus 1–10 Åtransit of a charge tunnelling (for example), it is reasonable to suppose that charge feels a fixed field with respect to the nuclear environment. The full quantum transition rate was first determined by Huang & Rhys [18], for F-centres in diamonds. It takes into account not just the probability of charge transfer due to the charge's ability to tunnel, but also to the ability of oscillators to make quantum transitions that facilitate the processes. This is measured by the nuclear part, an overlap integral:
| 2.7 |
Where C2(n,n′)=FC, is a Franck–Condon factor, n′ is a vibrational state on A and n is a vibrational state on D, χ is a vibrational wave function [1]. Evidence provided by the C. vinosum data [9] is best fitted by the full quantum mechanical (QM) model:
| 2.8 |
where is a Huang–Rhys factor, is the average quantum number of ensemble oscillators, Ip is a Bessel function [19]. The full quantum consideration, equation (2.8), converges to the classical Arrhenius activation barrier, equation (1.4), in the limit of high temperature. Thus, equation (2.8) explains the results at the higher temperatures but also the temperature independence at low temperatures. Modelling the process with a full quantum mechanical treatment fits the experimental results well [2,20]. For example, for the process of ascorbate oxidation by ferricyanide, the observed KIE at room temperature is larger than the semi-classical limit by a magnitude approximately 7, indicating that nuclear quantum mechanical tunnelling of the proton occurred (and it is perhaps vibrationally assisted) [21]. Thus, calculating the rates of such charge transfer, with full quantum analysis not only converges to the classical result (the regimes are not mutually exclusive), but also explains the empirical tests provided via isotopic substitutions.
(a). Challenges and future prospects
Although it is very well established that quantum mechanical tunnelling plays an important role in enzymatic processes that sustain life, it is still the case that this information is not fully applied in the development of catalysts, for example, which scientists still fail to make as efficient as biocatalysis by enzymes. Partly, this may be because it is not fully clear exactly what role the protein environment takes. As in photosynthetic energy capture see §4 (and potentially olfaction, see §3), it is becoming clear that the ‘hot and messy’ environment (the protein) may actually aid rates and transitions rather than hinder, in ways that have not yet been fully quantified. Protein conformations from equilibrium in a tunnelling pathway may cause decoherence of states, they may also modulate the energy gap between donor and acceptor sites in the protein electron transfer reactions, i.e. thermal control can be helpful. An interesting notion of ‘promoting modes’ versus ‘accepting modes’ has been defined with respect to tunnelling in enzymes [14] (figures 3 and 4). The latter meaning a non-vanishing overlap integral accounts for charge transfer, as in equation (2.7), i.e. the transition is enabled,versus the former which leads to active compression of the activation barrier, by reducing the height or the width, i.e. the transition may be enhanced. As in figure 3, a vibrational motion d(Q) coupled to the reaction coordinate compresses the reaction barrier [14]. Identifying such promoting and/or demoting modes present in the protein's vibrational environment may be key to engineering better rates. The development of terahertz spectroscopy that is able to determine single molecule vibrations may lead to a promising future in this area [22,23]. One difficulty in this field, evaded in POR, is that it is not always possible to measure rates at low temperatures and to initiate the catalysis within the ultrafast time scales. Photosynthetic systems, for example (see §4), are much more amenable to the testing of quantum effects because the reaction is photoinitiated and does not require mixing of the reactants (as in enzyme and substrate) [10].
Figure 4.
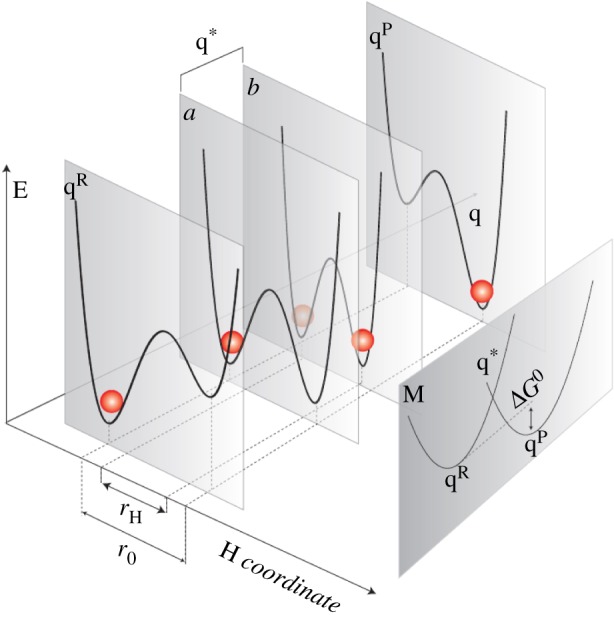
Depiction of vibrationally assisted models of hydrogen transfer from [14]. E denotes energy on the potential energy surface, q is the environmental reaction coordinate, r is the hydrogen coordinate where transfer is indicated by the red sphere, occurring at crossing point q*. qR denotes hydrogen in the initial/D state, and qP the hydrogen on the final/ A state. ‘M’ denotes this Marcus-like depiction, see equation (2.2), of the Born–Oppenheimer states on the environmental reaction coordinate. In a promoting mode, a vibrational motion must be coupled to the itinerant charge. (Online version in colour.)
3. Olfaction
In olfaction, the process by which we detect odour, it is not fully understood how an odorant molecule initiates the odorant response. It does so by ‘turning on’ an olfactory protein, a G-protein coupled receptor (GPCR) [24], but the physical mechanism by which it does this has not as yet been fully determined. One theory is that the odorant fits the receptor like a ‘lock and key’, similarly to the previous §2. However, designing odorants based on such shape complementarity between receptor and odorant (analgous to the substrate and enzyme) proves that this theory alone is not sufficient to explain olfactory response [25]. For one thing, very diversely shaped molecules can initiate the same receptor and conversely some same-shaped molecules initiate non-mutual receptors, such that the receptors are often referred to as simultaneously ‘discriminatory’ and ‘promiscuous’. Furthermore, olfaction does not alter the chemical composition of the odorant; there is no driving towards a product state from a TS (in the odorant at least) as there is in TST. There are around 390 types of functional human olfactory receptors [26], all of which are somehow ‘tuned’ to respond to the different molecular stimuli offered by odorants in order to span a ‘smell space’ of potentially thousands of odorants [27]. And they do so in a combinatorial process: many receptors initiated constitute a ‘code’ that determines a smell character [24]. The receptors do this consistently (there is no subjectivity at the receptor level), and are shown to be exquisitely sensitive to ‘something’ of the odorant (in approx. 390 ways), but that ‘something’ is currently unknown. This is not a problem unique to smell: many ligand–protein recognition events are not well understood and, notoriously, rational design of drugs based on ‘structure activity’ relationships fails to come up with successful lead candidates. Since GPCRs are an important drug targeting class, approximately 50% of pharmaceuticals designed are targeted at this class of protein [28], advancement in this area is of great importance to global health. Determining a completed theoretical basis for molecular recognition, when the ligand is not chemically altered, that is truly explanatory and predictive is an ongoing process.
It was first proposed by Dyson and then Wright [29,30], that the ‘something’ described above could be the odorant's molecular vibrations: that the receptor is somehow transducing the thermal fluctuations of an odorant. However, as everything in biological processes is thermally fluctuating, how would the receptor extract this information from the noise? Later, Turin [31] proposed that the receptor is detecting the odorant's quantum mechanical vibrations. This process had already been established as a signaling transduction mechanism by Jaklevik & Lambe [32] in 1968, where they showed inelastic electron tunnelling (IET) through a small molecule that bridges an electrode junction encodes information of the molecular energetics (quantized vibrations or phonons) in the resulting current (figure 5).
Figure 5.

Lambe & Jacklevic demonstrate inelastic electron tunnelling through a metal junction, from [32]. There are two possible ways for the electron to cross the tunnelling layer (insulating barrier) via (a) an elastic or (b) an inelastic transition. The inelastic transition occurs when there is a molecule bridging the tunnelling layer with a mode of vibration, , that the tunnelling electrons excites, and so loses energy too.
In electron tunnelling, there are two possible ways for the electron to cross the tunnelling layer via (a) an elastic or (b) an inelastic transition. In the elastic case, the electrons tunnelling across the insulating barrier (in figure 5 see ‘tunnelling layer’) do so without losing energy, transitioning from one Fermi level to the other in crossing from metal 1 to metal 2. In the inelastic case, the electron loses energy that directly matches whatever may be filling the insulating barrier, such as a molecular ‘bridge’ with an excitable mode of vibration. Typically, the elastic current is dominant and can be extracted from I/V plots, whereas the inelastic channel is extracted from the second derivative, d2I/dV 2, as peaks in the spectra that correspond to increases in conductance due to vibrational modes thus identifying what molecule fills the tunnelling layer by its spectrum. Turin's proposal was that the GPCR olfactory receptors implement a similar mechanism [31]. Since it has been established that a mechanism for biological IET is feasible as a detection mechanism in the nose if you consider Fermi's ‘golden rule’ for electron transfer where the odorant is treated as a small perturbing presence [33]. Fermi's ‘golden rule’, see equation (1.1), is a non-adiabatic rate equation that allows calculation of the times of a transition event (such as an inelastically or an elastically tunnelling electron).3 The rule holds provided there is only weak electronic coupling between two electronic states φD and φA, e.g. an electron in an initial state on a donor and in the final state on an acceptor (both fixed) and there is a separation of D and A energy levels, for example, as in a binding site of a GPCR (or in the contacts between enzyme-substrate). Similar to the metal junction in figure 5, the hypothesis is that an odorant docks at the GPCR binding site and fills an insulating barrier. A donor and acceptor state for electrons must exist somewhere in the odorant–receptor system such as a residue that hydrogen-bonds to the odorant at the binding site (where the protein replaces the metal junction in this instance). The binding event and the presence of the weakly coupled states at reasonable electron tunnelling distances [4] enable the transfer of an electron due to the perturbing effect of the odorant's presence. The key question, however, is whether the electron transfer is elastic (not discriminatory—no energy loss) or inelastic (discriminatory— is lost) and so if the odorant molecule can be identified by its phonon excitation, or whether the non-identifying elastic route dominates (as in the metal junction IETS) disguising the odorant's effect?
Using Fermi's golden rule in this way results in a non-adiabatic semi-classical Marcus theory expression for olfaction:
| 3.1 |
cf. equation (2.2), except the particular parameters for olfaction, where , εD is the energy state of the donor, εA the acceptor and is the vibrational mode of the odorant, n=1 corresponds to one phonon excitation, n=0 corresponds to zero phonon excitation on the odorant. , is a Poisson expression for the dependence on Huang–Rhys factors S, is the relaxation (or reorganization) energy of the environment, Sq is the Huang–Rhys factor for all the oscillations in the environment and S is the Huang–Rhys factor similar as in equation (2.8), but explicitly for the odorant. In the olfactory theory, it is the ratio of the change in energy to , where the change in energy is due to the change in force that occurs to initiate the electron transfer due to the odorants perturbing presence, i.e. the ‘kick’ to cause the transition. Notice that again the Born–Oppenheimer principle is used: the electronic and nuclear wave functions are separated as usual in Marcus theory [34]. There is an electronic coupling matrix element contained in t that determines the strength between D and A at the nuclear configuration of the TS and is sensitive exponentially to distance [4]. In the case of olfaction, a hopping integral between D and A is estimated, resulting from an admixture of these states with the odorant. The rest of the terms are FC factors that determine the nuclear rearrangement upon electron transfer, and can be portrayed in a configuration coordinate similar to figure 1 [35]. The crucial result is that the inelastic tunnelling rate is higher (preferable) than the elastic rate. In other words, when an odorant with a phonon of docks at the receptor with a donor–acceptor energy gap matching this energy, then vibrationally assisted tunnelling occurs the electron is aided in its transition by the presence of the odorant. With no odorant present (or one with an ‘incorrect’ ), the tunnelling event is less likely, thus quantum mechanics enhances the rate in order for discrimination: smell! This result has been replicated [36], and extended further by Nazir et al. [37], from the original hypothesis beyond the semi-classical Marcus theory of [33] to solve a polaron master equation and find that accounting for dissipation increases the frequency resolution of the receptor and the sensitivity to the presence of the odorant. It can be seen that, against intuition, dissipation assists the population of acceptor over donor states and the quantum dynamics of the system are more thoroughly accounted for in this model. The dissipative model also explains the phenomena of chiral recognition in olfaction [38,39].
Any vibrational based theory of olfaction offers a simple test (similar again to §2), provided by isotopic substitutions. Substituting atoms in the odorant enable a change in mass (and so frequency) that will shift the mode of vibration (e.g. by 700 cm−1 [40], 86 meV) , while keeping other physical properties the same, binding to the receptor, for example, should not be significantly disturbed. Changing the vibration should alter the smell (which would be hard to explain classically). However evidence in this area is ambiguous. Haffenden et al. [40] in 1996 showed that human subjects could distinguish benzaldehyde from benzaldehyde-d6. Then in 2004 Keller & Voshall reported that human subjects could notdistinguish acetophenone and d-8 acetophenone [41] and then more recently in 2013 [42] this was contested and it was shown that in fact these particular isotopes could be differentiated when expert smellers and gas chromatography (for purity) were used. Note also behavioural studies using insects have suggested that Drosophila melanogaster (fruit flies [43]) and Apis mellifera (honeybees [44]) are able to distinguish isotopically substituted odorants. Such behavioural studies of insects evade the subjective elements unavoidable in human tests, but still do not isolate the effects at the receptor level, where there is no room for contention. One recent study by Zhuang and colleagues [45], does just this, one of the approximate 390 receptor types they screened (‘OR5AN1’ identified as a human ‘musk-recognizing’ receptor) is tested in vitro against deuterated and non-deuterated muscone. This test eliminates any process in perireception and post reception and directly probes the odorant–receptor activation process. They show for a number of musk odorants that dose–response curves do not show any differentiation between isotopic forms of the odorants with any statistical significance (e.g. figure 6 [45]).
Figure 6.

IR spectra for ‘4-d30’ (red line) and undeuterated muscone ‘4’ (black line) indicating differing vibrational modes (particularly in the 1380–1550 cm−1 region) from [45] copyright Proceedings of National Academy of Sciences, USA. Also shown are dose-response curves of OR5AN1 plots for (1) cyclopentadecanone and (4) muscone and the scatter plots with 96% confidence interval log EC50 values, showing there is no differentiation between isotopes that is statistically significant [45].
This study shows definitively that the muscone receptor is not sensitive to deuterated muscone odorants and may seem fairly damning for any vibrations based theory of olfaction. Note, however, that it concentrates on one particular ‘musk-sensitive receptor’ and further it tests the hypothesis that C−H vibrations alone are signatures in smell spectra. According to the theory developed [33], it would a better test to probe vibrational modes such as C=O, for example, (substitute with isotopic oxygen), which are more likely responsible for the large changes in forces (there are larger moving partial charges upon phonon excitation) that would give rise to an enhanced electron tunnelling event (or promoted, as in §2). It is likely in all these (C–H targeted) deuteration experiments that the wrong mode of vibration is being tested one that would not typically stand out against others in the odorant–receptor environment. This concept is explored by Lin & Besohn [46], as in §2, showing that ‘promoting modes’ in napthalene and actephenone correspond to C–C stretching vibrations which give appreciable derivatives of electronic wave functions with respect to the nuclear coordinates, as opposed to the C–H stretching vibrations which provide ‘accepting modes’ (non-vanishing overlap integrals) [46]. Both can occur, but the promoting state is better at enhancing the rate, or perhaps, in the case of olfaction, being the signature vibration that turns ‘on’ the receptor.
(a). Challenges and future prospects
Though at first sight the recent study by Zhuang and colleagues [45] appears to refute a quantum-based olfaction theory, it is put here and elsewhere that this is not definitive [47]. Testing further various isotopic substitutions in this manner at the receptor level would be very informative. Recent results indicate that isotopic substitutions are differentiated in drosophila by the measurements of electroantennogram amplitudes suggesting receptor-level discrimination [48]. Also odorant response maps in honeybees show discrimination of similar-shaped odorant with different vibrational frequencies [49]. Any detailed quantum mechanical analysis of the binding site in olfaction is limited by the lack of crystal structures available for the olfactory receptors. Energy states εD and εA are assumed to be molecular orbital states (highest occupied molecular orbitals). Is it even possible for an energetic donor–acceptor splitting to match such a small (typically 200 meV)? What donors and acceptors of electrons might there be available in the olfactory receptor? Calculating the electronic coupling for initial and final wave functions without any structural information would constitute very involved guess-work, given structural fluctuations on the picosecond time scale and sub-ångström distances may substantially affect any quantum process. However, recent studies have advanced knowledge of the binding site [50] and new research is emerging to show that biological IETS may play a role in neuroreceptors [51]. So let the notion of phonon-assisted electron tunnelling (as a signalling mechanism) be emphasized and not ignored, if not a role in olfaction, but perhaps in other systems in biology that rely upon ligand–receptor activation.
4. Photosynthetic energy capture
Photosynthesis, abundant in plants, algae and bacteria, is arguably one of the most important fundamental interactions on Earth: the conversion of sunlight energy into the chemical energy forms the basis of life. In brief, a photosynthetic system uses an ‘antenna’ (100 000s of light-absorbing molecules) to capture sunlight energy which initiates the process by creating an ‘exciton’, an electron-hole pair, that travels to the reaction centre (RC) and ultimately results in the production of ATP, the ‘molecule of life’. For maximum efficiency, all photons absorbed have to make it to the RC, but there are so many random paths and the photon does not ‘know’ the most efficient route. So the suggestion is that, rather than taking one path, quantum coherence is used to travel all paths simultaneously. Given the photosynthetic molecule's raisôn de ẽtre, often if surviving in hostile (low-light level) conditions, is to gather as many photons of light at the RC as possible, what benefits are provided by quantum physics? One possibility is that coherence and interference exhibited by the absorbed photons may enable a quantum ‘random walk’—most efficiently getting the energy to where it needs to be [52,53]. This process is excitation energy transfer (EET), rather than energy transfer, but equation (1.1) can again be used to calculate transfer rates.
These first photosynthetic steps, energy harnessing and transfer to the reaction centre for charge separation, all take place within femtoseconds–picoseconds [54]. The PPCs that constitute the antennae that funnel the excitation energy to where it needs to be are exquisitely manufactured such that these systems may operate at low light levels and still gather enough excited states but are also able to operate in excess sunlight, and therefore contain ‘quenching’ molecules that protect against light damage. The key chromophoric pigments in photosynthesis (those that absorb photons to make an ‘exciton’) are chlorophylls, for example as in figure 2, the primary molecules in photosynthesis. However, carotenoids are often in close proximity in order to ‘tune’ energetic states, like the protein may do in olfaction and/or enzymatic processes. Carotenoids actually most often serve as regulatory molecules, they photoprotect chlorophylls from population of triplet states in excess light conditions so that damaging free radicals are not generated.
Figure 7 illustrates the structure of the Fenna–Matthews–Olson (FMO protein) complex and seven bacteriochlorophyll a (pigment) molecules from Chlorobaculum tepidum,a photosynthetic green sulfur bacteria.4 The FMO shown here is a monomer unit that exists in trimeric form in nature and links the chlorosome to the RC. The BChl a pigments absorb light at 800 nm, which is referred to as the Qy transition. Passing this excitation energy on to other pigments en route to the reaction centre without fluorescing or being lost to any other route is integral to the efficiency of the overall process.
For FMO, the Hamiltonian is given by [56]
| 4.1 |
where and are nuclear Hamiltonians for ground and excited states, respectively and Q and P are mass scaled coordinates [56] (e.g. ). These Born–Oppenheimer surfaces with a shift along the reaction coordinate can be pictured, again using a configuration coordinate diagram, as in figure 7b, to describe the excitation. The ground state can be considered D, and the excited state A. It is assumed there is no orbital overlap between these pigments and that the assignment of electrons is unambiguous. An electronic energy gap correlator, , operator quantifies the energy difference between the excited and the ground state at a given nuclear configuration, where
| 4.2 |
Following the notation by [56], the V denotes the ground or the excited state potential energy as a function of the coordinate Q. Equation (4.2) can be understood in terms of parabola geometry, corresponds to the energy gap between the transition, and is the reorganization energy (see §3, Huang–Rhys factors also emerge in analogous ways). Examination of a reduced energy gap operator allows comparison to the open quantum system model [56]:
| 4.3 |
where is the Hamiltonian of the system, in this case electronic excited states, is the Hamiltonian of the ‘bath’, typically nuclear vibrations in the environment (protein), and represents the system–bath interaction. and are coordinates for the system and the bath, indices j and k are over the system and bath degrees of freedom respectively, and the creation and annihiliation operators are used where . Examining bath correlation functions is important with respect to accurately determining the system–bath interaction, ways of doing so is considered by Valleau et al. [56]. Most protein systems involve many degrees of freedom so that it is difficult to find a full quantum solution. The energy gap correlator allows one to take account of environmental motion modelled as random fluctuations if the operator is assumed to evolve with time (t), , for n=m correlators, where the choice is determined by fluctuations of normal modes at temperature T. At high T and very large amplitude oscillations, the quantum superposition state is collapsed and a classical solution is recovered. It is also possible to determine an electronic energy gap correlator which finds a quantum solution beyond simple shifting harmonic surfaces.
When the inter-pigment coupling is small with respect to the relaxation in the environment, this is called the small coupling limit and the electron–nuclear coupling can be treated perturbatively. Nuclear reorganization occurs before energy transfer (EET) and the motion can be described as incoherent hopping. This gives Förster dynamics which shares similar characteristics to the rates derived in the §§2 and 3; FC factors are used to separate out the vibrational overlap and spectral features indicating that vibrations modulate EET, again similar to the previous topics. It is not always the case that the pigment coupling is small. In the non-Förster regime, the pigment excitation may be delocalized over several pigment sites and so the exciton is robust with respect to dissipation of the reorganization energy. In this case, coherent transfer is observed where the exciton wave packet travels with phase coherence; this may be referred to as the very strong coupling (or Redfield) regime [57] (figure 8). Förster and Redfield theory are second-order perturbative methods used for small and very large electronic couplings between pigments, respectively. Note that both approximations have limitations and much more sophisticated analysis is now required, which is beyond the scope of this article. EET in photosynthesis may also be solved using numerically exact hierarchical equations of motion (HEOM) [59] and density matrix renormalization techniques [60,61]; the reader is referred to the references for comprehensive analysis.
Figure 8.

Figure to show the incoherent Förster (very weak coupling), intermediate coherent (large coupling) and Redfield (very large coupling) regime for EET in photosynthesis, from [58]. The electronic coupling refers to an electrostatic (Coloumb) dipole–dipole interaction between pigment BChl sites.
(a). Two-dimensional electronic spectroscopy
The FMO example, as in the above section, is so important because it was the first photosynthetic system to display long-lived coherent excited states in experiment [62]. Ultrafast (femtosecond) laser pulses are able to populate excitonic states within the BChl a Qy band. Using , the energy gaps of 70–400 cm−1 correspond to time periods of 80–500 fs [63]. A third-order coherence response is generated by three pump pulses from a laser. Results are frequency and phase resolved using frequency domain heterodyne detection, obtaining a magnitude and phase of the signal. When the three laser pulses are directed at the sample, the time period between the first two pulses allows a ‘one-quantum coherence’ τ (the coherence time) between the ground and resonant excited states. The second pulse promotes population of more excited states, ground states and coherences between ‘zero-quantum coherences’. The time period between the second and third pulse is the ‘waiting time’ T. The third pulse causes a radiative coherence (the third-order coherence response). The period between the final pulse and the emitted signal is the rephasing time t. Two-dimensional spectra are taken at specific waiting times T (figure 9). The two-dimensional spectra are generated by fixing T and taking a two-dimensional Fourier transform over the t and τ times [63].
Figure 9.

Two-dimensional electronic spectroscopy (2DES), in (a) showing the time evolution of excitation between 2 pigments, ‘cross peaks’ amplitudes oscillate with v2−v1, Fourier transform gives the oscillation frequency. (b) shows the two-dimensional spectra at T=55 fs (real part of total signal) for PC645, a marine cryptophyte algae that contains 8 billion (chromophore) pigments, coloured bars are peak absorptions for the chromophores. The green cross indicates a cross peak with coherent oscillation as a function of waiting time T (at 570 nm, 600 nm). (c) plots the oscillation's amplitude with waiting time (d) plots the rephasing (red) and non-rephasing (purple) amplitudes of this cross peak and (e) is a Fourier transform of the traces in (d) showing that peaks occur at 26 THz in both signals but 21 THz only in the rephasing spectrum, from [58].
The important point to note from these experiments, shown clearly in figure 9 for a marine cryptophyte algae (PC645) and in figure 10, where it was discovered for the first time in FMO, [62], is the fact that the amplitude of the signal is oscillating with time—quantum coherent ‘beats’. Long-lived quantum mechanical evolution of electronic coherences is observed.
Figure 10.

The first evidence of electronic coherence between single states in FMO, a non-rephasing signal appears on the main diagonal in (a) and evolved in time, see (b). Plotting the amplitude of the lowest exciton peak beating with time, upon Fourier transform (d) reproduces frequencies that agree well with the predicted beat times and intensities, from [62].
To understand the nature of these quantum beats, we return to the system Hamiltonian, equation (1.8), which can be evolved with time as in , making a unitary transform operator ρ(t)=u(t)ρ′(0)u†(t) for a density matrix in any basis set [63]. The time evolution of the density matrix, of an isolated (closed) quantum system, is given by the Liouville–von-Neumann equation [54]:
| 4.4 |
giving a first-order differential equation, with operator L, the quantum master equation (QME) [6]
| 4.5 |
when expressed in the Hamiltonian eigenbasis with εi as the energy of the ith eigenstate:
| 4.6 |
and
| 4.7 |
It can be seen that under time evolution of ρij(t) [63], that there are oscillations or ‘beats’, that are coherent for the off-diagonal elements of the density matrix that represents the ensemble i≠j (the transfer terms), and diagonal elements are the populations of energetic (spectroscopic) states i=j which remain constant (energies of individual sites). For an observable to be measured (in spectroscopy) , the observable operator is the dipole moment change and is determined by the amplitude intensity, see vertical axes in figures 9–11. The dipole moment operator does not commute with the Hamiltonian and so off-diagonal elements (the coherent beats) will be observed in measurements of the amplitude with respect to time, as is seen in the experiments.
Figure 11.

Quantum beats at room temperature in FMO from [64], copyright Proceedings of National Academy of Sciences, USA. Data shown are at waiting time T=400 fs and (a) is 77 K (b) 125 K (c) 150 K and (d) 277 K. At higher temperature, peaks are broader (dephasing between g and e states). Theoretical analysis [65], extracts the quantum beat at the cross peak (white circle) and plotted in (e) as a function of waiting time T to observe the amplitude oscillating. The beat occurs around 200 fs even at room temperature.
Note some criticisms in the past have targeted these initial experiments, mainly that the laser excitation conditions do not match the incoherent light actually received by plants at ambient temperatures. At least with respect to addressing the latter concern, it has been shown that quantum coherence at higher temperatures (>77 K) occurs in phycobiliproteins in cryptophyte algae [55], and further in FMO also [64] where even at room temperature quantum beats are observed (figure 11).
Work is ongoing with respect to the understanding completely the results of 2DES [66–68]. Bloch–Redfield has limitations [69,57]. Indeed all approaches such as those outlined above, that are perturbative with weak coupling, Markovian, and lead to the standard Master equation are not sufficient. HEOM interpolate between the Bloch–Redfield and Förster regimes [58] and provide numerically exact treatments for describing the energy transfer process [70].
(b). Challenges and future prospects
The development of femtosecond (ultra-fast) spectroscopy, see §4a, in probing photosynthetic complexes has been a major breakthrough in the field ‘quantum effects in biology’. It shows the first instance of observing quantum superposition and coherence dynamics in vital biological systems. Note, however, that this is just the beginning of the story of energy transfer in photosynthesis. Section 4a refers to the (unitary) time evolution of a closed system and shows how an initial superposition of energy eigenstates will exhibit oscillatory coherences, equation (4.7) (figure 10). However, it is not straightforward interpreting the spectra. Coherences may not just be electronic, but vibrational and/or both (vibronic). Vibrations are not necessarily merely responsible for providing the thermal relaxation of excitons. Given relevant vibrational modes and excitonic energy gaps, it is entirely likely that vibronic coherences are prevalent. This raises questions as to whether the excitonic quantum ‘beats’ will be washed out by coupling to the environment or whether the environment involves vibronic coherences which is actually accountable for the efficient energy transfer (rather than simply excitonic states). The latter interpretation would require analysis of non-unitary evolution. Theory along these lines is beyond the scope of this article and the interested reader is pointed to the phenomenally exciting and fast developing work of the following references [61,71–74]. Experimental work that supports the importance of vibronic coherence makes use of the efficient energy transfer observed in photosynthesis, photovoltaics and artifical light harvesting [75–79].
5. Bird magnetoreception
Ornithologists Wolfgang and Roswitha Wiltschko were the first to observe and carefully document evidence of the phenomenon of bird migration which appeared to be directed by sensory clues sensitive to the Earth's magnetic field. They suggested that the garden warblers migrating from Northern Scandinavia to Southern Africa every year are directed by their detection of the Earth's magnetic field. This has since been shown in birds such as European Robins and also more recently it was found in domestic chickens, and also turtles and spiny lobsters [80]. The original tests simply placed the birds (European robins) in cages with no sky view, within which they would scratch at the southwesterly direction, the direction in which they would be migrating if free. The concept of magnetodetection was deduced from the main finding that when the birds were exposed to an external magnetic field that interfered with the inclination (angle) of the Earths magnetic field, they become lost and scratched at all directions. Furthermore, when their eyes were completely covered, they become similarly disorientated, suggesting the process is photoinitiated. Not only is the process light-dependent, but magnetodetection in European Robins has shown to be wavelength-dependent (figure 12) [81].
Figure 12.
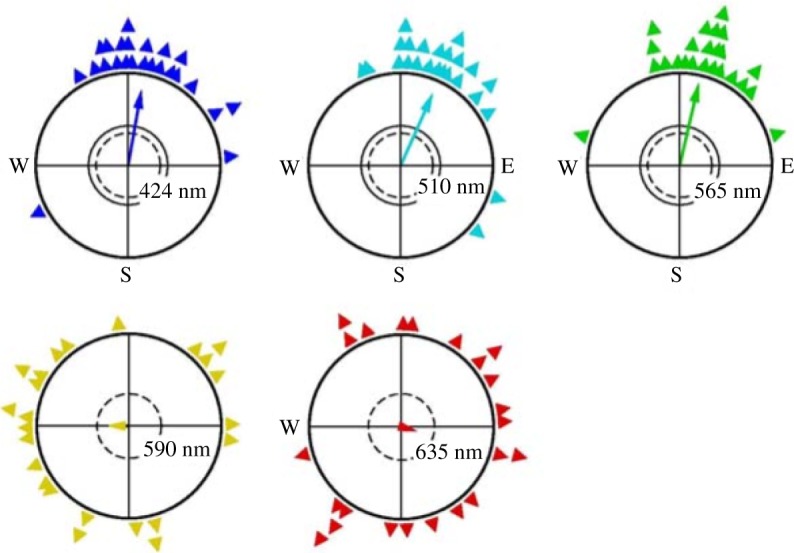
Experiments show birds exposed to monochromatic light exhibit differing orientation behaviour—orientation is better in ‘blue’ light, from [81].
As explanation, there are two dominant theories currently in the field. One is essentially classical, based on ferrimagnetic iron oxide particles in the bird's body. The classical picture describes a magnetically sensitive protein that acts analogously to a physically moving compass. A navigational ‘Magnetoreceptor’ of this kind has recently been discovered [82]. However, for the purposes of this review, we concentrate on the alternative hypothesis which conjectures quantum processes in photoinitiated radical reactions: a radical pair mechanism (RPM). This is because the classical picture of a moving compass does not explain key observations that are clearly light (and wavelength dependent), as in figure 12. The RPM was first introduced by Schulten and colleagues [83] and developed further by Ritz and colleagues [84–88], his idea is that the ‘chemical compass’ in the bird is a light-initiated RPM, where a disruption to the pair recombination due to magnetic field effects results in interpretation and recognition in the birds nervous system. Such a proposed RPM can be described as in figure 13 [89].
Figure 13.

RPM mechanism adapted from [89]. The left shows the photo-initiated radical pair AB (FH and W here), the spin Hamiltonian interconverts the pair from singlet to triplet states, with differing resulting singlet or triplet products as a consequence of the entanglement in the middle: structures in the box show the tryptophan radical (W) and the flavin radical (FH) at the proposed orientation in vivo, (the possible AB pair). On the left are the two-dimensional structures for FH and W. The orientation of this possible RP (from pdb code 1DNP) was used to calculate anisotropic magnetic field effects in [89].
Light first excites the donor (D) and acceptor (A) molecules from the electronic ground state, causing electron transfer from D→A, resulting in unpaired electrons on each: radical pairs (RPs).
| 5.1 |
It is proposed (figure 13) that the donor and acceptor may be FH and W, respectively, as in (5.1). The RPs are initially singlet states (anti-parallel electron spins), but an interaction due to hyperfine coupling leads to conversion into triplet states (parallel electron spins). This singlet–triplet mixing (S-T) is a coherent oscillation of the total electron spin state, determined by the hyperfine constant (which arises due to the interaction between the spin states and the magnetic nuclei in the environment) but also the Zeeman interaction with the external magnetic field B. The rate of this transition back to a recombination of singlet states depends on the orientation of the (Earth's) magnetic field [6] and the interconversion is oscillatory coherent: another quantum beat. For more detail of the coherent oscillations, Kominis [90] shows that different approaches to solve equation (4.4) versus the full QME can be used to show the time evolution of coherent states 〈QS〉T. What is interesting to calculate are the rates, respectively, of kT triplet product formation versus kS singlet product formation since this encodes information about the magnetic field. What are the rates for product SP or ST formation based on S/T populations?
For this problem, the QME (cf. equation (4.5)) is given by the Haberkorn approach [6]:
| 5.2 |
where QT projects on the triplet subspace and similarly for the singlet subspace. The Hamiltonian of the RP in the protein (nuclear) environment is the sum of the hyperfine coupling (left term) and the Zeeman contribution (right term) [6]:
| 5.3 |
which introduces the effect of the Earth's magnetic field B, as discussed above. The Hamiltonian of the RP interaction is given by Iij and si which are nuclear and electron spin operators and Tij which is the hyperfine coupling tensor. g is the gyromagnetic ratio and μ0 is the Bohr magneton, and B the external magnetic field, the strength and direction of which defines the population of triplet versus singlet states. It is found that the longer lived the pair, the more sensitive to B the system [89]; if the magnetic Hamiltonian has no time to mix the electron spin, all reaction products will remain singlet. The singlet yield is then determined from [6]:
| 5.4 |
The orientation information is derived from the anisotropic hyperfine interaction, the interconversion of S↔T states is dependent on the RP (fixed in a protein in the birds anatomy) relative to the direction of the magnetic field. The hyperfine interactions are anisotropic, so that kS≠kT depending on the orientation of the chromophoric molecules with respect to the field, see figure 13, which shows the variation of ΦS with respect to orientation (angles Phi and Theta) of the RP at B=50 μT and kS=2×105 s−1 (European robins magneto-detect within 43–54 μT [89]). The changes in rate, according to the orientation dependence, suggest that the bird responds somehow to the information of the Earth's field with respect to these D/A molecules in its anatomy (in its head movement; figure 14).
Figure 14.

The orientation dependence of the singlet recombination probability ΦS of the radical pair [FH•++W•−] as in figure 13, from [89]. Results are for a weak field of B=50 μT and kS=2×105 s−1.
(a). Challenges and future prospects
Though the theory is fascinating, an analogous reaction mechanism is shown (figure 15) it is unknown where anatomically the RPM takes place. The proof of principle is shown, in the case of a synthetic triad of carotenoid (C)–porphyrin (P)–fullerene molecules [92] that RPs do indeed encode magnetic fields through S/T reaction rates. However, this is a synthetic construct and experimental verification and identification of the biological system that may host in actuality this mechanism in animals has not yet been determined. There are various suggestions as to which protein is responsible, and cryptochrome is the popular contender [93]. But this is not certain, and certainly exactly which molecules are the donor and acceptors in the RP is not ascertained either. Figure 13, for example, is based on a D/A pair FH and W which is modelled on FADH and Trp-306 in the E. coli DNA photolyase. This photolyase repairs UV damage to DNA and involves electron transfer, energy transfer and enzymatic processes (all of the above!), but it is not clear this is the protein used in animals for magnetodetection (it is a model construct like figure 15). The RPM is also known to occur in photosynthetic reaction centres, and has been probed with EPR; zero quantum coherences and quantum beats have been observed [94]. Furthermore, if the protein conjectured to house the RPM iscryptochrome (which has not been established via EPR and ENDOR experiments to show spin-correlated RPs of certain lifetimes), it is still not known how the field information is then passed on to the bird. Similar to the limitations in §3, more needs to be known about the receptor structure responsible, and the possible D/A candidates, especially as in this case: it has been seen that there is an orientation dependence, but how is the singlet recombination rate then transduced to the bird's nervous system?
Figure 15.

Reaction mechanism (b) and two-dimensional structures of the carotenoid (C)–porphyrin (P)–fullerene (F) system (a) that demonstrates the ‘chemical compass’ principle, from [91]. Interconversion between singlet and triplet pair is shown [C•+−P−F•−] as moderated by weak magnetic fields. The rates at which they recombine to C–P–F is spin-selective accounting for rates kS and kT. C•+ shows the anisotropic hyperfine couplings calculated on C.
6. Conclusion
Four systems have been discussed. All describe rates with differing degrees of possible ‘quantum effects’ hosted in a type of PPC. These quantum effects range from tunnelling, as observed in enzyme processes and conjectured in olfaction, to the observations of quantum coherent superpositions in photosynthetic energy capture, and finally to entanglement—Einstein's ‘spooky action at a distance’—in a possible mechanism for magneto-detection in animals. Arguably, one of the most astonishing, and common, inferences from this examination is, counterintuitively, that the environment (the protein) in the PPC does not hinder any of these processes, but actually that it may help.
(a). Environment helps
This review particularly focuses on an environment modelled using normal modes (bosonic baths). This assumes anharmonic, large amplitude, and long time-scale motions are on irrelevant time scales (with respect to the rates described here). If it were otherwise any quantum superposition state carrying information would collapse. It is found that, actually, normal mode vibrations, at least, do not decohere, but rather support and/or accelerate rates. In §2 Enzymes, the possibility of tunnelling being ‘stabilized by phonon emission’ is first introduced. In §3 Olfaction, tunnelling is conjectured to be assisted by phonon emission by an odorant. In both, it is possible that a vibrational mode may accept and/or promote the rate. Of course, there may also be ‘demoting’ modes. In §4, it is shown that a weak perturbative regime can model the excitonic energy transfer observed, but it is likely that the protein motions and so the mixing of electronic and vibrational wave functions is important [95,96]. Thus, the analysis goes well beyond that of vibrational modes and any semi-classical version of the golden rule. There is much exciting work in this area [97] and it is fascinating to consider whether these more ‘quantum’ models may be useful in the analysis of olfaction and enzyme reaction rates. It is less obvious how environmental vibrations contribute in magnetoreception; however, it has been experimentally shown that the environment does not disguise any effect [91], and it is likely that the D/A pair that encode the field are held at relative orientations optimal for the effect transduction (figure 13) which is of course determined by the host protein. As is the nuclear environment (the nuclear spin) which is key to coupling to the D/A states (equation (13)) for the asymmetry in reaction rates.
Intriguingly, although protein environments (e.g. enzymes) are more often thought of as insulating barriers (e.g. figure 2) and as ‘wet and noisy’ environments though not to be any way conducive to the survival of any ‘quantum effect’, it has been seen that protein motion may serve to promote key quantized events such as charge and energy transfer. Typically, proteins can facilitate transfer by: (i) reducing the effective tunnelling mass by solvent exclusion, (ii) enabling crossing by equalization of energy states reactants and products (i.e. moving the Born–Oppenheimer surfaces closer together; figure 1), and (iii) by reducing barrier widths. Perhaps, the most exciting is the idea that protein motion may support the persistence of coherent oscillations, seen in figure 11, for example, at ambient temperatures. The phenomena of vibronic coupling emerging in photosynthesis suggests the protein nuclear motion supports the efficient energy transfer in photosynthesis, these coherent effects have been observed in charge separation in natural systems [75,76]. Of course, this is exciting with respect to understanding the fundamental nature of one of the most essential processes in life, but beyond this, knowing the importance of vibrations in proteins allows for the manipulation of the effect: this can be tested and exploited in artificial systems [77–79].
(b). Challenges in general and future outlook
As mentioned above, it is clear the field of ‘quantum effects in biology’ is the most well tested in photosynthesis—where quantum processes have been directly observed. Structures of photosynthetic apparatus provide more reliable information on the relevant states involved. This is not true exactly for magnetodetection and olfaction, for example. The analysis can only be as good as the representative Hamiltonian, which defines all interactions and forces driving the system, and not knowing the likely D/A states, let alone with the precision required, is a huge bottleneck in these fields. Designing experiments then that directly test hypotheses here, at the quantum level, must thus be a priority for advancement. Until then, though interesting, quantum effects remain conjecture, and any analysis is really redundant without experimental proof. It is hoped, however, that the theory and some concepts mentioned here highlight promise, and that the developments will continue. In these fields and beyond, there is still much more to discuss: neglected here is work in quantum computing, or quantum simulators as in Mostame et al. [98], where biological mimetics may suggest promise. In fact, ‘quantum effects’ and the exploration thereof have indicated to be useful in various fields: DNA mutations [99], vision [100], to name a couple, which may also exhibit surprising quantum effects and provide fascinating avenues for future examination.
Acknowledgements
This work has benefitted enormously by input from Gavin Vinson, Dorothy Duffy, Joonsuk Huh, JohnJoe McFadden and Alex Connor, much gratitude to them for their time and assistance. With thanks also to the support I have received over years from Andrew Horsfield, Andrew Fisher and Rachel McKendry and the late Marshall Stoneham.
Footnotes
Electron spins in the magnetodetection system are assumed to be entangled (versus just spin coherence) by virtue of the correlation between spins being long-lived enough to be observed: the effect on the singlet–triplet conversion rate encodes the information of the magnetic field.
Note the abbreviation PPC is typical within the field of photosynthetic energy transfer, the author uses it here to umbrella all protein complexes for these four topics, though technically in the other systems the equivalent of the ‘pigment’ is simply a non-protein molecule.
‘Non-adiabatic’, in particular, the ‘non-adiabatic’ operator in this context corresponds to the derivatives of electronic and nuclear wave functions with respect to nuclear coordinates (electron–phonon mixing) which determine crossing at the configuration coordinate surfaces.
Note eight BChl pigments are now known to exist [56].
Competing interests
I declare I have no competing interests.
Funding
I received no funding for this study.
Disclaimer
I am the sole author of this review and am responsible for the context thereof.
References
- 1.De Vault D. 1984. Quantum-mechanical tunneling in biological systems, vol. 2 Cambridge, UK: Cambridge University Press. [Google Scholar]
- 2.Klinman JP, Kohen A. 2013. Hydrogen tunneling links protein dynamics to enzyme catalysis. Annu. Rev. Biochem. 82, 471–496. (doi:10.1146/annurev-biochem-051710-133623) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eyring H. 1935. The activated complex and the absolute rate of chemical reactions. Chem. Rev. 17, 65–77. (doi:10.1021/cr60056a006) [Google Scholar]
- 4.Gray HB, Winkler JR. 2005. Long-range electron transfer. Proc. Natl Acad. Sci. USA 102, 3534–3539. (doi:10.1073/pnas.0408029102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oppenheimer JR. 1928. Three notes on the quantum theory of aperiodic effects. Phys. Rev. 31, 66–81. (doi:10.1103/PhysRev.31.66) [Google Scholar]
- 6.Huelga SF, Plenio MB. 2013. Vibrations, quanta and biology. Contemp. Phys. 54, 181–207. (doi:10.1080/00405000.2013.829687) [Google Scholar]
- 7.Schlosshauer M. 2014. The quantum-to-classical transition and decoherence. Handbook of Quantum Information. Berlin, Germany: Springer.
- 8.Pauling L. 1948. Chemical achievement and hope for the future. Am. Sci. 36, 51–58. [PubMed] [Google Scholar]
- 9.DeVault D, Chance B. 1966. Studies of photosynthesis using a pulsed laser. I. Temperature dependence of cytochrome oxidation rate in chromatium. Evidence for tunneling. Biophys. J. 6, 825–847. (doi:10.1016/S0006-3495(66)86698-5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heyes DJ, Sakuma M, de Visser SP, Scrutton NS. 2009. Nuclear quantum tunneling in the light-activated enzyme protochlorophyllide oxidoreductase. J. Biol. Chem. 284, 3762–3767. (doi:10.1074/jbc.M808548200) [DOI] [PubMed] [Google Scholar]
- 11.Kihara T, McCray JA. 1973. Water and cytochrome oxidation-reduction reactions. Biochim. Biophys. Acta 292, 297–309. (doi:10.1016/0005-2728(73)90037-6) [DOI] [PubMed] [Google Scholar]
- 12.Northrop DB. 1975. Steady-state analysis of kinetic isotope effects in enzymic reactions. Biochemistry 14, 2644–2651. (doi:10.1021/bi00683a013) [DOI] [PubMed] [Google Scholar]
- 13.Okamura MY, Feher G. 1986. Isotope effect on electron transfer in reaction centers from Rhodopseudomonas sphaeroides. Biophysics 83, 8152–8156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hay S, Scrutton NS. 2012. Good vibrations in enzyme-catalysed reactions. Nat. Chem. 4, 161–168. (doi:10.1038/nchem.1223) [DOI] [PubMed] [Google Scholar]
- 15.Grant KL, Klinman JP. 1989. Evidence that both protium and deuterium undergo significant tunneling in the reaction catalyzed by bovine serum amine oxidase. Biochemistry 28, 6597–6605. (doi:10.1021/bi00442a010) [DOI] [PubMed] [Google Scholar]
- 16.Bendall DS. 1996. Protein electron transfer. Oxford, UK: BIOS Scientific Publishers. [Google Scholar]
- 17.Hopfield JJ. 1974. Kinetic proofreading: a new mechanism for reducing errors in biosynthetic processes requiring high specificity. Proc. Natl Acad. Sci. USA 71, 4135–4139. (doi:10.1073/pnas.71.10.4135) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang K, Rhys A. 1950. Theory of light absorption and non-radiative transitions in F-centres. Proc. R. Soc. Lond. A 204, 406–423. (doi:10.1098/rspa.1950.0184) [Google Scholar]
- 19.Jortner J, Ben-Reuven A. 1976. Intermolecular electronic energy trasnfer induced by an intense optical radiation field. Chem. Phys. Lett. 41, 401–406. (doi:10.1016/0009-2614(76)85382-1) [Google Scholar]
- 20.Pudney CR, Lane RSK, Fielding AJ, Magennis SW, Hay S, Scrutton NS. 2013. Enzymatic single-molecule kinetic isotope effects. J. Am. Chem. Soc. 135, 3855–3864. (doi:10.1021/ja309286r) [DOI] [PubMed] [Google Scholar]
- 21.Kandathil SM, Driscoll MD, Dunn RV, Scrutton NS, Hay S. 2014. Proton tunnelling and promoting vibrations during the oxidation of ascorbate by ferricyanide? Phys. Chem. Chem. Phys. 16, 2256–2259. (doi:10.1039/c3cp55131h) [DOI] [PubMed] [Google Scholar]
- 22.Vlček A, Kvapilová H, Towrie M, Záliš S. 2015. Electron-transfer acceleration investigated by time resolved infrared spectroscopy. Acc. Chem. Res. 48, 868–876. (doi:10.1021/ar5004048) [DOI] [PubMed] [Google Scholar]
- 23.Vinh NQ, Redlich B, van der Meer AFG, Pidgeon CR, Greenland PT, Lynch SA, Aeppli G, Murdin BN. 2013. Time-resolved dynamics of shallow acceptor transitions in silicon. Phys. Rev. X 3, 011019 (doi:10.1103/PhysRevX.3.011019) [Google Scholar]
- 24.Buck L. 1991. A novel multigene family may encode odorant receptors: a molecular basis for odor recognition. Cells 65, 175–187. [DOI] [PubMed] [Google Scholar]
- 25.Sell CS. 2006. On the unpredictability of odor. Angew. Chem. 45, 6254–6261. (doi:10.1002/anie.200600782) [DOI] [PubMed] [Google Scholar]
- 26.Tobar HF, Moreno PA, Vélez PE. 2009. Highly conserved regions in the 5’ region of human olfactory receptor genes. Genet. Mol. Res. 8, 117–128. (doi:10.4238/vol8-1gmr550) [DOI] [PubMed] [Google Scholar]
- 27.Harini K, Sowdhamini R. 2015. Computational approaches for decoding select odorant-olfactory receptor interactions using mini-virtual screening. PLoS ONE 10, e0131077 (doi:10.1371/journal.pone.0131077) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Salom D, Lodowski DT, Stenkamp RE, Le rong I, Golczak M, Jastrzebska B, Harris T, Ballesteros JA, Palczewski K. 2006. Crystal structure of a photoactivated deprotonated intermediate of rhodopsin. Proc. Natl Acad. Sci. USA 103, 16 123–16 128. (doi:10.1073/pnas.0608022103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Malcolm Dyson G. 1938. The scientific basis of odour. J. Soc. Chem. Industry 57, 647–651. (doi:10.1002/jctb.5000572802) [Google Scholar]
- 30.Wright RHH. 1977. Odor and molecular vibration: neural coding of olfactory information. J. Theoret. Biol. 64, 473–502. (doi:10.1016/0022-5193(77)90283-1) [DOI] [PubMed] [Google Scholar]
- 31.Turin L. 1996. A spectroscopic mechanism for primary olfactory reception. Chem. Senses 21, 773–791. (doi:10.1093/chemse/21.6.773) [DOI] [PubMed] [Google Scholar]
- 32.Lambe J, Jaklevic RC. 1968. Molecular vibration spectra by inelastic electron tunneling. Phys. Rev. 165, 821–832. (doi:10.1103/PhysRev.165.821) [Google Scholar]
- 33.Brookes JC, Hartoutsiou F, Horsfield AP, Stoneham AM. 2007. Could humans recognize odor by phonon assisted tunneling? Phys. Rev. Lett. 98, 38101 (doi:10.1103/PhysRevLett.98.038101) [DOI] [PubMed] [Google Scholar]
- 34.Narth C, Gillet N, Cailliez F, Lévy B, de la Lande A. 2015. Electron transfer, decoherence, and protein dynamics: insights from atomistic simulations. Acc. Chem. Res. 48, 1090–1097. (doi:10.1021/ar5002796) [DOI] [PubMed] [Google Scholar]
- 35.Brookes JC. 2011. Olfaction: the physics of how smell works? Contemp. Phys. 52, 385–402. (doi:10.1080/00107514.2011.597565) [Google Scholar]
- 36.Solov’yov IA, Chang PY, Schulten K. 2012. Vibrationally assisted electron transfer mechanism of olfaction: myth or reality? Phys. Chem. Chem. Phys. 14, 13 861–13 871. (doi:10.1039/c2cp41436h) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Checinska A, Pollock FA, Heaney L, Nazir A, Chȩcińska A, Pollock FA. 2014. Dissipation enhanced vibrational sensing in an olfactory molecular switch. J. Chem. Phys. 142, 025102 (doi:10.1063/1.4905377) [DOI] [PubMed] [Google Scholar]
- 38.Tirandaz A, Taher Ghahramani F, Shafiee A. 2015. Dissipative vibrational model for chiral recognition in olfaction. Phys. Rev. E 92, 032724 (doi:10.1103/PhysRevE.92.032724) [DOI] [PubMed] [Google Scholar]
- 39.Takai Y, Touhara K. 2015. Enantioselective recognition of menthol by mouse odorant receptors. Biosci. Biotechnol. Biochem. 79, 1980–1986. (doi:10.1080/09168451.2015.1069697) [DOI] [PubMed] [Google Scholar]
- 40.Haffenden LJW, Yaylayan Va, Fortin J. 2001. Investigation of vibrational theory of olfaction with variously labelled benzaldehydes. Food Chem. 73, 67–72. (doi:10.1016/S0308-8146(00)00287-9) [Google Scholar]
- 41.Keller A, Vosshall LB. 2004. A psychophysical test of the vibration theory of olfaction. Nat. Neurosci. 7, 337–338. (doi:10.1038/nn1215) [DOI] [PubMed] [Google Scholar]
- 42.Gane S, Georganakis D, Maniati K, Vamvakias M, Ragoussis N, Skoulakis EMC, Turin L. 2013. Molecular vibration-sensing component in human olfaction. PLoS ONE 8, e55780 (doi:10.1371/journal.pone.0055780) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Franco MI, Turin L, Mershin A, Skoulakis EMC. 2011. Molecular vibration-sensing component in Drosophila melanogaster olfaction. Proc. Natl Acad. Sci. USA 108, 3797–3802. (doi:10.1073/pnas.1012293108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gronenberg W, Raikhelkar A, Abshire E, Stevens J, Epstein E, Loyola K, Rauscher M, Buchmann S. 2014. Honeybees (Apis mellifera) learn to discriminate the smell of organic compounds from their respective deuterated isotopomers. Proc. R. Soc. B 281, 20133089 (doi:10.1098/rspb.2013.3089) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Block E. et al. 2015. Implausibility of the vibrational theory of olfaction. Proc. Natl Acad. Sci. USA 112, E2766–E2774. (doi:10.1073/pnas.1503054112) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lin SH, Besohn R. 1968. Effect of partial deuteration and temperature on triplet-state lifetimes. J. Chem. Phys. 48, 2732 (doi:10.1063/1.1669507) [Google Scholar]
- 47.Turin L, Gane S, Georganakis D, Maniati K, Skoulakis EMC. 2015. Plausibility of the vibrational theory of olfaction: fig. 1. Proc. Natl Acad. Sci. USA 112, E3154 (doi:10.1073/pnas.1508035112) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Drimyli E, Gaitanidis A, Maniati K, Turin L, Skoulakis EMC. 2016. Differential electrophysiological responses to odorant isotopologues in drosophilid antennae. eNeuro 3, ENEURO.0152-15.2016 (doi:10.1523/ENEURO.0152-15.2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Paoli M, Anesi A, Antolini R, Guella G, Vallortigara G, Haase A. 2016. Differential odour coding of isotopomers in the honeybee brain. Sci. Rep. 6, 21893 (doi:10.1038/srep21893) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barwich AS. 2015. What is so special about smell? Olfaction as a model system in neurobiology. Postgrad. Med. J. 92, 27–33. (doi:10.1136/postgradmedj-2015-133249) [DOI] [PubMed] [Google Scholar]
- 51.Hoehn RD, Nichols D, Neven H, Kais S. 2015. Neuroreceptor activation by vibration-assisted tunneling. Sci. Rep. 5, 9990 (doi:10.1038/srep09990) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Plenio MB, Huelga SF. 2008. Dephasing-assisted transport: quantum networks and biomolecules. New J. Phys. 10, 113019 (doi:10.1088/1367-2630/10/11/113019) [Google Scholar]
- 53.Mohseni M, Rebentrost P, Lloyd S, Aspuru-Guzik A. 2008. Environment-assisted quantum walks in photosynthetic energy transfer. J. Chem. Phys. 129, 174106 (doi:10.1063/1.3002335) [DOI] [PubMed] [Google Scholar]
- 54.Fassioli F, Dinshaw R, Arpin PC, Scholes GD. 2014. Photosynthetic light harvesting: excitons and coherence. J. R. Soc. Interface 11, 20130901 (doi:10.1098/rsif.2013.0901) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hayes D, Wen J, Panitchayangkoon G, Blankenship RE, Engel GS. 2011. Robustness of electronic coherence in the Fenna-Matthews-Olson complex to vibronic and structural modifications. Faraday Discuss. 150, 459–469. (doi:10.1039/c0fd00030b) [DOI] [PubMed] [Google Scholar]
- 56.Valleau S, Eisfeld A, Aspuru-Guzik A. 2012. On the alternatives for bath correlators and spectral densities from mixed quantum-classical simulations. J. Chem. Phys. 137, 224103 (doi:10.1063/1.4769079) [DOI] [PubMed] [Google Scholar]
- 57.Ishizaki A, Fleming GR. 2009. On the adequacy of the Redfield equation and related approaches to the study of quantum dynamics in electronic energy transfer. J. Chem. Phys. 130, 234110 (doi:10.1063/1.3155214) [DOI] [PubMed] [Google Scholar]
- 58.Chenu A, Scholes GD. 2015. Coherence in energy transfer and photosynthesis. Annu. Rev. Phys. Chem. 66, 69–96. (doi:10.1146/annurev-physchem-040214-121713) [DOI] [PubMed] [Google Scholar]
- 59.Tanimura Y, Kubo R. 1989. Time evolution of a quantum system in contact with a nearly Gaussian-Markoffian Noise Bath. J. Phys. Soc. Jpn 58, 101–114. (doi:10.1143/JPSJ.58.101) [Google Scholar]
- 60.Prior J, Chin AW, Huelga SF, Plenio MB. 2010. Efficient simulation of strong system-environment interactions. Phys. Rev. Lett. 105, 1–4. (doi:10.1103/PhysRevLett.105.050404) [DOI] [PubMed] [Google Scholar]
- 61.Chin AW, Prior J, Rosenbach R, Caycedo-Soler F, Huelga SF, Plenio MB. 2013. The role of non-equilibrium vibrational structures in electronic coherence and recoherence in pigment-protein complexes. Nat. Phys. 9, 113–118. (doi:10.1038/nphys2515) [Google Scholar]
- 62.Engel GS, Calhoun TR, Read EL, Ahn TK, Mancal T, Cheng YC, Blankenship RE, Fleming GR. 2007. Evidence for wavelike energy transfer through quantum coherence in photosynthetic systems. Nature 446, 782–786. (doi:10.1038/nature05678) [DOI] [PubMed] [Google Scholar]
- 63.Mohseni M, Omar Y, Engel GS, Plenio MB. 2014. Quantum effects in biology. Cambridge, UK: Cambridge University Press. [Google Scholar]
- 64.Panitchayangkoon G, Hayes D, Fransted KA, Caram JR, Harel E, Wen J, Blankenship RE, Engela GS. 2010. Long-lived quantum coherence in photosynthetic complexes at physiological temperature. Proc. Natl Acad. Sci. USA 107, 12 766–12 770. (doi:10.1073/pnas.1005484107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Adolphs J, Renger T. 2006. How proteins trigger excitation energy transfer in the FMO complex of green sulfur bacteria. Biophys. J. 91, 2778–2797. (doi:10.1529/biophysj.105.079483) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Roden JJJ, Bennett DIG, Whaley KB. 2016. Long-range energy transport in photosystem II. J. Chem. Phys. 144, 245101 (doi:10.1063/1.4953243) [DOI] [PubMed] [Google Scholar]
- 67.Allegra M, Giorda P, Lloyd S. 2015. Global coherence of quantum evolutions based on decoherent histories: theory and application to photosynthetic quantum energy transport. Phys. Rev. A 93, 042312 (doi:10.1103/PhysRevA.93.042312) [Google Scholar]
- 68.Shim S, Rebentrost P, Valleau S, Aspuru-Guzik A. 2012. Atomistic study of the long-lived quantum coherences in the Fenna-Matthews-Olson complex. Biophys. J. 102, 649–660. (doi:10.1016/j.bpj.2011.12.021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jeske J, Ing DJ, Plenio MB, Huelga SF, Cole JH. 2015. Bloch-Redfield equations for modeling light-harvesting complexes. J. Chem. Phys. 142, 064104 (doi:10.1063/1.4907370) [DOI] [PubMed] [Google Scholar]
- 70.Hein B, Kreisbeck C, Kramer T, Rodríguez M. 2012. Modelling of oscillations in two-dimensional echo-spectra of the Fenna-Matthews-Olson complex. New J. Phys. 14, 023018 (doi:10.1088/1367-2630/14/2/023018) [Google Scholar]
- 71.Womick JM, Moran AM. 2009. Exciton coherence and energy transport in the light-harvesting dimers of allophycocyanin. J. Phys. Chem. B 113, 15 747–15 759. (doi:10.1021/jp907644h) [DOI] [PubMed] [Google Scholar]
- 72.Christensson N, Kauffmann HF, Pullerits T, Mančal T. 2012. Origin of long-lived coherences in light-harvesting complexes. J. Phys. Chem. B 116, 7449–7454. (doi:10.1021/jp304649c) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tiwari V, Peters WK, Jonas DM. 2013. Electronic resonance with anticorrelated pigment vibrations drives photosynthetic energy transfer outside the adiabatic framework. Proc. Natl Acad. Sci. USA 110, 1203–1208. (doi:10.1073/pnas.1211157110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chenu A, Christensson N, Kauffmann HF, Mančal T. 2013. Enhancement of vibronic and ground-state vibrational coherences in 2D spectra of photosynthetic complexes. Sci. Rep. 3, 2029 (doi:10.1038/srep02029) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fuller FD. et al. 2014. Vibronic coherence in oxygenic photosynthesis. Nat. Chem. 6, 706–711. (doi:10.1038/nchem.2005) [DOI] [PubMed] [Google Scholar]
- 76.Romero E, Augulis R, Novoderezhkin VI, Ferretti M, Thieme J, Zigmantas D, van Grondelle R. 2014. Quantum coherence in photosynthesis for efficient solar-energy conversion. Nat. Phys. 10, 676–682. (doi:10.1038/nphys3017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lim J. et al. 2015. Vibronic origin of long-lived coherence in an artificial molecular light harvester. Nat. Commun. 6, 7755 (doi:10.1038/ncomms8755) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.De Sio A. et al. 2016. Tracking the coherent generation of polaron pairs in conjugated polymers. Nat. Commun. 7, 13742 (doi:10.1038/ncomms13742) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang L, Griffin GB, Zhang A, Zhai F, Williams NE, Jordan RF, Engel GS. 2017. Controlling quantum-beating signals in 2D electronic spectra by packing synthetic heterodimers on single-walled carbon nanotubes. Nat. Chem. 9, 219–225. (doi:10.1038/nchem.2729) [DOI] [PubMed] [Google Scholar]
- 80.Wiltschko W, Wiltschko R. 2005. Magnetic orientation and magnetoreception in birds and other animals. J. Comp. Physiol. A Neuroethol. Sensory Neural Behav. Physiol. 191, 675–693. (doi:10.1007/s00359-005-0627-7) [DOI] [PubMed] [Google Scholar]
- 81.Wolfgang Wiltschko RW, Wiltschko W, Wiltschko R. 2007. Magnetoreception in birds: two receptors for two different tasks. J. Ornithol. 148, 61–76. (doi:10.1007/s10336-007-0233-2) [Google Scholar]
- 82.Qin S. et al. 2015. A magnetic protein biocompass. Nat. Mater. 15, 217–226. (doi:10.1038/nmat4484) [DOI] [PubMed] [Google Scholar]
- 83.Ritz T, Adem S, Schulten K. 2000. A model for photoreceptor-based magnetoreception in birds. Biophys. J. 78, 707–718. (doi:10.1016/S0006-3495(00)76629-X) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ritz T, Thalau P, Phillips JB, Wiltschko R, Wiltschko W. 2004. Resonance effects indicate a radical-pair mechanism for avian magnetic compass. Nature 429, 177–180. (doi:10.1038/nature02534) [DOI] [PubMed] [Google Scholar]
- 85.Ritz T, Wiltschko R, Hore PJ, Rodgers CT, Stapput K, Thalau P, Timmel CR, Wiltschko W. 2009. Magnetic compass of birds is based on a molecule with optimal directional sensitivity. Biophys. J. 96, 3451–3457. (doi:10.1016/j.bpj.2008.11.072) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ritz T. 2011. Quantum effects in biology: bird navigation. Procedia Chem. 3, 262–275. (doi:10.1016/j.proche.2011.08.034) [Google Scholar]
- 87.Wang K, Mattern E, Ritz T. 2006. On the use of magnets to disrupt the physiological compass of birds. Phys. Biol. 3, 220–231. (doi:10.1088/1478-3975/3/3/007) [DOI] [PubMed] [Google Scholar]
- 88.Wiltschko R, Thalau P, Gehring D, Nießner C, Ritz T, Wiltschko W. 2015. Magnetoreception in birds: the effect of radio-frequency fields. J. R. Soc. Interface 12, 20141103 (doi:10.1098/rsif.2014.1103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cintolesi F, Ritz T, Kay CWM, Timmel CR, Hore PJ. 2003. Anisotropic recombination of an immobilized photoinduced radical pair in a 50-μT magnetic field: a model avian photomagnetoreceptor. Chem. Phys. 294, 385–399. [Google Scholar]
- 90.Kominis IK. 2012. Magnetic sensitivity and entanglement dynamics of the chemical compass. Chem. Phys. Lett. 542, 143–146. (doi:10.1016/j.cplett.2012.06.014) [Google Scholar]
- 91.Maeda K, Liddell P, Gust D, Hore PJ. 2013. Spin-selective recombination reactions of radical pairs: experimental test of validity of reaction operators. J. Chem. Phys. 139, 234309 (doi:10.1063/1.4844355) [DOI] [PubMed] [Google Scholar]
- 92.Maeda K, Henbest KB, Cintolesi F, Kuprov I, Rodgers CT, Liddell PA, Gust D, Timmel CR, Hore PJ. 2008. Chemical compass model of avian magnetoreception. Nature 453, 387–390. (doi:10.1038/nature06834) [DOI] [PubMed] [Google Scholar]
- 93.Foley LE, Gegear RJ, Reppert SM. 2011. Human cryptochrome exhibits light-dependent magnetosensitivity. Nat. Commun. 2, 356 (doi:10.1038/ncomms1364) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bittl R, Kothe G. 1991. Transient EPR of radical pairs in photosynthetic reaction centers: prediction of quantum beats. Chem. Phys. Lett. 177, 547–553. (doi:10.1016/0009-2614(91)90082-K) [Google Scholar]
- 95.Romero E, Novoderezhkin VI, van Grondelle R. 2017. Quantum design of photosynthesis for bio-inspired solar-energy conversion. Nature 543, 355–365. (doi:10.1038/nature22012) [DOI] [PubMed] [Google Scholar]
- 96.Scholes GD. et al. 2017. Using coherence to enhance function in chemical and biophysical systems. Nature 543, 647–656. (doi:10.1038/nature21425) [DOI] [PubMed] [Google Scholar]
- 97.O’Reilly EJ, Olaya-Castro A. 2014. Non-classicality of the molecular vibrations assisting exciton energy transfer at room temperature. Nat. Commun. 5, 3012 (doi:10.1038/ncomms4012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mostame S, Rebentrost P, Eisfeld A, Kerman AJ, Tsomokos DI, Aspuru-Guzik A. 2012. Quantum simulator of an open quantum system using superconducting qubits: exciton transport in photosynthetic complexes. New J. Phys. 14, 105013 (doi:10.1088/1367-2630/14/10/105013) [Google Scholar]
- 99.McFadden J, Al-Khalili J. 1999. A quantum mechanical model of adaptive mutation. Biosystems 50, 203–211. (doi:10.1016/S0303-2647(99)00004-0) [DOI] [PubMed] [Google Scholar]
- 100.Kukura P, McCamant DW, Yoon S, Wandschneider DB, Mathies RA. 2005. Structural observation of the primary isomerization in vision with femtosecond-stimulated Raman. Science 310, 1006–1009. (doi:10.1126/science.1118379) [DOI] [PubMed] [Google Scholar]