

Abstract
Minifascicular neuropathy (MN) is an extremely rare developmental malformation in which peripheral nerves are composed of many small fascicles. Only one patient with MN with 46XY gonadal dysgenesis (GD) was found to carry a mutation affecting the start codon in desert hedgehog (DHH). We identified an identical novel rearrangement mutation of DHH in two consanguineous families with MN, confirming mutations in DHH cause MN with 46XY GD. The patients with the 46XY karyotype developed GD, whereas a patient with the 46XX karyotype did not. These findings further support that DHH has important roles in perineural formation and male gonadal differentiation.
Introduction
Minifascicular neuropathy (MN) is an extremely rare developmental malformation of peripheral nerves and is characterized by the presence of many small fascicles with glove and stocking‐type sensory impairment.1 Two unrelated Japanese patients with 46XY gonadal dysgenesis (GD) have been reported.2 MN was also reported in an Italian patient, who had the 46XX karyotype and whose gynecological ultrasound examinations showed normal findings.3 On the basis of the finding that DHH knockout mice show MN, Umehara et al. performed mutational analysis of DHH and found a homozygous mutation involving the translation initiation codon in the first Japanese family found to be affected by MN.4 However, no DHH mutations were detected in the second Japanese family reported by Sugie et al.2 or in the Italian family, and genes and mutations responsible for MN have remained to be elucidated. We have recently identified Japanese MN patients with or without GD born to consanguineous parents. Employing linkage analysis and detailed mutational analyses, we identified a novel rearrangement mutation in DHH. We further confirmed the same mutation in the second Japanese family.2 Our findings provide further evidence that DHH is the gene responsible for MN in humans.
Subjects and Methods
Written informed consent was obtained from all the participants. The present study was approved by the institutional review board of the University of Tokyo. Genomic DNA was prepared from peripheral blood leukocytes of the participants in accordance with the standard procedure.
Subjects
MN‐1 family
Two patients with MN born to consanguineous parents were investigated in the study (Fig. 1A). The 55‐year‐old patient (patient 1) was born by normal delivery and raised as a female. She started to suffer from muscle cramps on exercise since childhood. Gynecological examination for amenorrhea revealed the absence of ovaries and uterus at the age of 16, when chromosomal analysis showed 46XY. At the age of 43, pigmentation of the nails and hyperkeratosis of the sole skin appeared. She underwent amputation of some of her toes because of ulceration. On examination at the age of 55, skin pigmentations in the four extremities were observed. Pelvic magnetic resonance imaging (MRI) confirmed the absence of ovaries and uterus with a blind‐ended vagina and an undescended testicle in the left inguinal canal. Neurological examinations showed sensory disturbance and the absence of tendon reflexes. The Wechsler Adult Intelligence Scale (WAIS‐III) test revealed high IQ score (131; normal 90–109). Nerve conduction study revealed sensory motor dysmyelinating polyneuropathy (Table S1). The left sural nerve was biopsied. Light microscopy examination revealed numerous nerve fascicles, ranging in diameter from 20 to 200 μm subdivided into several minifascicles with connective tissue. The density of myelinated fibers was markedly reduced in each fascicle (Fig. 1B and C).
Figure 1.
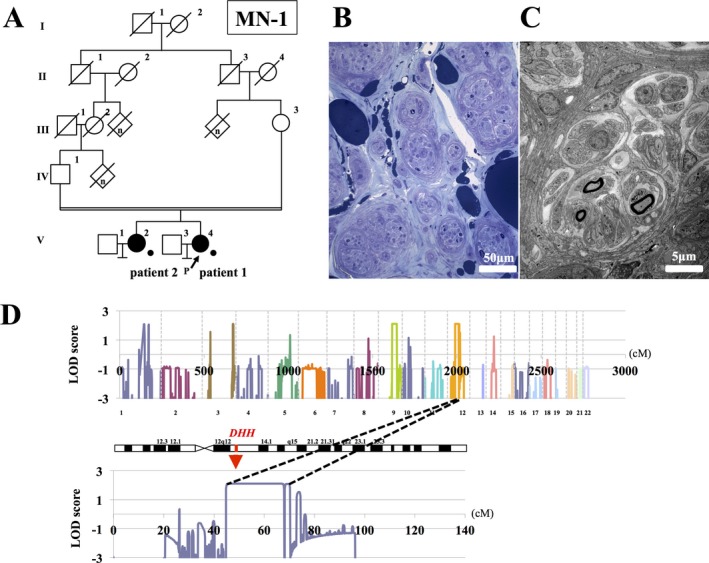
(A) Pedigree chart of MN‐1. Squares and circles indicate males and females, respectively. Affected persons are indicated by filled symbols. A diagonal line through a symbol represents a deceased person. The person with the arrow is the index patient. Persons with available genomic DNAs are indicated by dots. (B) Light microscopy findings of sural nerve specimens stained with toluidine blue. Sural nerve contains many small fascicles. Many fascicles have smaller fascicles inside them. (C) Electron microscopy findings of sural nerve specimens. There are few myelinated fibers. Minifascicles contain several myelinated and unmyelinated fibers. Endoneurial cells carry on processes and encircle the minifascicles. (D) Multipoint parametric linkage analysis (autosomal recessive model) of MN‐1 family. Multipoint parametric LOD scores spanning all the chromosomes are shown above. The horizontal axis is the cumulative genetic distance (centimorgan) starting at the short arm of chromosome 1. The vertical axis represents cumulative LOD scores. Regions on chromosome 12 give the highest multipoint parametric LOD score of 2.1. Multipoint parametric LOD scores of chromosome 12 is shown below. The horizontal axis is the genetic distance (centimorgan) starting at the arm of chromosome 12. The vertical axis shows multipoint parametric LOD scores.
Her elder sister (patient 2) also complained of muscle cramps on exercise since childhood. She showed pigmentation of the nails and hyperkeratosis of the sole skin, and she had some of her toes amputated. Nerve conduction studies showed sensory motor dysmyelinating polyneuropathy (Table S1). Although she did not have a child, her menstrual cycle was regular until the menopause and her karyotype was 46XX (Table 1).
Table 1.
Clinical and genetic findings in four neuropathologically confirmed MN families
| Patient | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| Age | 55 | 60 | 47 | 27 | 28 |
| Age at onset | Childhood | Childhood | 39 | 26 | 2 |
| Sex (phenotype) | F | F | F | F | F |
| Family history | + | + | − | − | + |
| Consanguinity | + | + | + | + | − |
| Neuropathy | Sensory dominant | Sensory dominant | Sensory dominant | Sensory dominant | Motor dominant |
| Weakness | − | − | + | − | + |
| Muscle cramp | + | + | − | − | − |
| Cutaneous ulcer | + | + | − | + | − |
| NCS (median nerve) | |||||
| CMAP (mV) | 12 | 5.5 | ND | 6.3 | 2 |
| MCV (m/sec) | 39.6 | 38.1 | 37.2 | 48 | 38 |
| Minifascicular formation | + | NE | + | + | + |
| Karyotype | 46XY | 46XX | 46XY | 46XY | 46XX |
| Gonads/Internal genitalia |
Bilateral streak gonads Hypoplastic uterus and ovaries |
No abnormality was pointed out | Hypoplastic uterus |
Right‐sided streak gonads Hypoplastic uterus and ovaries |
Normal |
| Mutation | c.304‐572_492dup | c.304‐572_492dup | c.304‐572_492dup | c.2T>C | ND |
| Source | Patient 1 | Patient 2 | Patient 3 (2) | 4 | 3 |
CMAP, compound muscle action potential; MCV, motor conduction velocity; NCS, nerve conduction studies; ND, not detected, NE, not examined.
MN‐2 family
The clinical presentations of patient 3 (Fig. S1) were described previously.2 Briefly, she noticed distal numbness and weakness in the four extremities at the age of 39, which gradually progressed. At the age of 47, a nerve conduction study revealed sensory dominant neuropathy. Sural nerve biopsy revealed minifascicle formation. She had amenorrhea and gynecological examinations revealed normal female external genitalia and a blind‐ended vagina. Chromosomal analysis revealed 46XY. Direct nucleotide sequence analysis revealed no point mutations in the sex‐determining region Y gene (SRY) and DHH.4
Whole exome sequence analysis
Exonic sequences were enriched using a SureSelect Human All Exon V4+UTRs kit (Agilent Technology, Santa Clara, CA, USA), followed by massively parallel sequence analysis (2 × 100 bp paired end reads) using Hiseq 2000 (Illumina, San Diego, CA, USA). The sequences were aligned against the reference genome (hg19) using Burrows‐Wheeler Aligner (BWA), and SAMtools5, 6 was used for extracting single‐nucleotide variants (SNVs) and small insertions and deletions (indels) with default parameter settings.
Linkage analysis and haplotype analysis
Three affected family members of the two families (MN‐1 and MN‐2) were genotyped using Genome‐wide Human SNP array 6.0 (Affymetrix, Santa Clara, CA)7 following the manufacturer's instructions. The genotype of each family member was determined using Genotyping Console 4.0 (Affymetrix). Parametric multipoint linkage analysis (autosomal recessive model with complete penetrance) was conducted using the pipeline software SNP‐HiTLink8 and Allegro version 29 employing single‐nucleotide polymorphisms (SNPs) satisfying a P > 0.001 in the Hardy–Weinberg test, a call rate of >0.98, a genotyping confidence score of <0.02, a minor allele frequency in the controls >0, and intermarker distances of 80–120 kb. Haplotypes were reconstructed manually.
Long‐range PCR analysis of DHH
We amplified the entire DHH region by long‐range PCR using the primer set: F1 (5′‐GCAGCTTCCAACTGAGAAGTCA‐3′) and R1 (5′‐GCTGATATGCCCTTGTTTAGGG‐3′). To search for the breakpoint junctions, we performed genomic PCR using the primer set: F2 (5′‐CTACCATCGACTCAGATTCT‐3′) and R2 (5′‐GCTCCCCTCCCTCCGCCTGA‐3′) (Fig. 2C), followed by direct nucleotide sequence analysis using a Big Dye Terminator version 3.1 and an ABI 3130xl genetic analyzer (Applied Biosystems, Carlsbad, CA).
Figure 2.
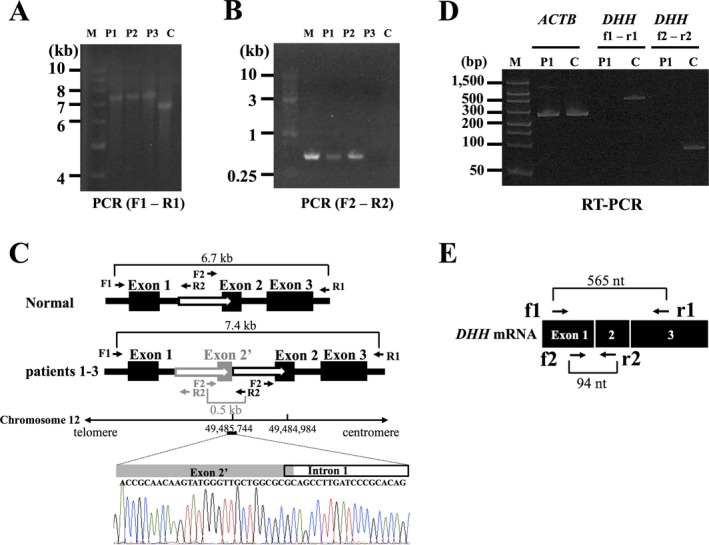
(A) Long‐range PCR amplification of the whole region of DHH in the three MN patients (left). We amplified the entire DHH region including all three exons using the following primer set: F1 (5′‐GCAGCTTCCAACTGAGAAGTCA‐3′) and R1 (5′‐GCTGATATGCCCTTGTTTAGGG‐3′). DHH is approximately 700 bp longer in the three cases than in the normal control. M, size standard marker (1 kb DNA ladder); C, normal control; P1, patient 1; P2, patient 2; P3, patient 3. (B): Detection of breakpoint junction by PCR. We performed genomic PCR using the following primer set: F2 (5′‐CTACCATCGACTCAGATTCT‐3′) and R2 (5′‐GCTCCCCTCCCTCCGCCTGA‐3′). M, size standard marker (1 kb DNA ladder); C, normal control; P1, patient 1; P2, patient 2; P3, patient 3. Only the patients’ genomic DNAs carrying the duplication can be PCR amplified. (C) Structure of mutation in DHH. Direct nucleotide sequence analysis of the PCR products amplified using a primer set (F2 and R2) showed the breakpoint junctions. One of them was found in exon 2 and the other in intron 1 (chromosome 12: 49,484,984‐49,485,744). The black boxes represent exons and the white arrows duplicated regions. (D) RT‐PCR analysis of DHH mRNA expression in the sural nerves. In the RT‐PCR analyses of DHH using primers located on exons 1 and 3 (f1 and r1 in Fig 2E) and primers located on exons 1 and 2 (f2 and r2 in Fig 2E), the PCR products (565 bp and 94 bp bands) were revealed only in a control subject, confirming the absence of DHH mRNA expression in the sural nerve of the patient 1. RT‐PCR of ACTB revealed bands corresponding to 285 bp in both the patient 1 and a control subject. M, size standard marker (FlashGel DNA marker); C, normal control; P1, patient 1; ACTB, actin beta. (E) A schematic presentation of normal DHH mRNA along with primers used in the RT‐PCR analysis.
RT‐PCR analysis of DHH mRNA expression in sural nerves
Total RNAs were extracted from the sural nerves of patient 1 and a control individual using TRIzol reagent (Invitrogen, Carlsbad, CA). cDNA synthesis was performed using the ReverTra Ace‐α‐™ (Toyobo, Osaka, Japan) with an oligo dT primer. RT‐PCR was performed using DHH‐specific primer sets: f1 (5′‐TCGTGCCGCTACTCTACAAG‐3′), r1 (5′‐GCCGCCAAAACCCAGTCTC‐3′), f2 (5′‐TCGTGCCCAACTACAACCC‐3′), and r2 (5′‐TTCACCCGCTCCTTACAACG‐3′) (Fig. 2E). As an internal control, we performed RT‐PCR of ACTB using the primer set: (5′‐AGCGAGCATCCCCCAAAGTT‐3′) and (5′‐GGGCACGAAGGCTCATCATT‐3′).
Results
Mutation discovery
Multipoint parametric linkage analysis of the MN‐1 family with the assumption of autosomal recessive inheritance revealed five regions in four chromosomes with the highest multipoint LOD score of 2.1. Notably, the candidate regions overlapped with the locus of DHH on chromosome 12 (Fig. 1D). In whole exome sequencing analyses, no single‐nucleotide variants or small insertion–deletions were determined by SAMtools in the region of DHH. We next tried to amplify the entire DHH region employing a long‐range PCR method using the primers F1 and R1. The PCR products obtained from patients 1 and 2 were larger than those from a control (Fig. 2A), raising the possibility of a rearrangement such as insertion and/or multiplication. Direct nucleotide sequence analysis of the PCR products revealed that a 761‐bp genomic segment was tandemly duplicated with the 5′ end located in intron 1 (chr12:49,485,744 in hg19) and the 3′ end in exon 2 (chr12:49,484,984) (Figs. 2B and C). We also conducted a mutational analysis of DHH of the patient 3 in MN‐2 family employing the same strategy. The same mutation was found in the patient (Figs. 2A and B).
To investigate whether mutant DHH mRNA is expressed in sural nerves, we extracted total RNAs from the sural nerves of patient 1 and a control individual. RT‐PCR analysis revealed that DHH mRNA was not expressed in the patient's sural nerve (Fig. 2D).
Haplotype analysis of the two families revealed that three individuals shared a substantially large region of 4.34 Mb in their disease‐associated haplotypes with the centromeric and telomeric boundaries defined by rs10735810 and rs11170044, respectively (Fig. S2). The result suggested the existence of a common founder.
Discussion
We identified a novel rearrangement mutation as the cause of MN with 46XY GD in two Japanese families. The duplication spans 761 bp and one end of the breakpoints includes exon 2. Given the structure of the duplication and the absence of DHH mRNA expression in the sural nerve in patient 1 assessed by RT‐PCR analysis, we suppose that the mutation would result in the loss of function of DHH.
DHH located on 12q12‐q13.1 comprises three exons, encoding a polypeptide of 396 amino acids.10 DHH is a member of the hedgehog gene family which also includes sonic hedgehog and Indian hedgehog.11 Paramantier et al.12 found that the peripheral nerves of dhh null mutant mice showed minifascicular formation. Analysis of null mutant mice demonstrated that dhh signaling plays an essential role in the regulation of mammalian spermatogenesis.13 Because mammalian hedgehog genes share a striking homology to the Drosophila segment polarity hedgehog gene, these findings suggest that DHH plays crucial roles in both peripheral nerve sheath development and spermatogenetic regulation in humans.
Umehara et al.4 discovered a homozygous mutation involving the start codon, which presumably leads to the failure of the transcription of DHH, in an MN patient with 46XY GD. In this study, we revealed that the patients in MN‐1 and MN‐2 had an identical rearrangement mutation in DHH. This finding further confirms that mutations in DHH cause MN.
The rearrangement mutation of our subjects results in the absence of DHH expression. To date, seven DHH mutations have been reported.14, 15, 16 Three of the seven mutations, including the mutation found in the study, cause 46XY GD with neuropathy, whereas some mutations did not cause neuropathy. (Table S2). Although nerve conduction studies, nerve biopsy, and/or functional study of the mutations were performed in a limited number of patients, mutations presumably leading to complete loss of function of DHH (c.304–572dup, c.2T>C, c.485T>C,17 and c.57–60dupAGCC) does not always cause both neuropathy and GD. It is still uncertain whether there is a phenotype correlation with complete loss versus partial loss of function of DHH gene.
One of our patients (patient 2) in MN‐1 and the patient reported by Malandrini et al.4, both of whom karyotype was 46XX, showed MN and normal sex characteristics. We suggest that DHH may be involved in different manners in peripheral nerve development or gonadal genesis.
To detect medium‐sized structural variants (100–1000 bp) as found in the study is still a challenge. Indeed, the rearrangement mutation which we detected was not revealed by standard whole exome sequencing. The finding in this study indicates that we should always give attention to structural variants, even if standard exome sequencing did not reveal mutations. The causative mutations of a patient with minifascicular neuropathy3 and a patient showing 46XY GD and neuropathy without minifascicular formation18 have not been identified. There may be clinical and genetic heterogeneity of this rare clinical entity, but our finding suggests that mutations that may have been overlooked by Sanger sequencing or standard exome sequencing should be assessed in these patients.
In conclusion, we identified a novel mutation in human DHH. The patients with the 46XY karyotype exhibited MN with 46XY GD, whereas the patient with the 46XX karyotype showed MN, but apparently normal sexual development. These findings further confirm that DHH is an important gene in both perineural formation in peripheral nerves and male gonadal differentiation.
Authors' Contribution
N.S.S. and R.M. contributed equally to this study.
Conflicts of Interest
None declared.
Supporting information
Figure S1. Pedigree chart of MN‐2.
Figure S2. Haplotypes around DHH locus constructed using SNPs of two families are shown.
Table S1. Nerve conduction study findings of patients 1 and 2 in MN‐1.
Table S2. Clinical and genetic findings in 11 patients who have mutations in DHH.
Acknowledgments
We gratefully acknowledge Dr. Mana Higashihara (Department of Neurology, Tokyo Metropolitan Geriatric Hospital and Institute of Gerontology) and Prof. Masahiro Sonoo (Department of Neurology, Teikyo University School of Medicine) for providing normal values of nerve conduction study according to the facility criteria.
References
- 1. Umehara F, Yamaguchi N, Kodama D, et al. Polyneuropathy with minifascicle formation in a patient with 46XY mixed gonadal dysgenesis. Acta Neuropathol 1999;98:309–312. [DOI] [PubMed] [Google Scholar]
- 2. Sugie K, Futamura N, Tate G, Umehara F. Hereditary motor and sensory neuropathy with minifascicle formation in a patient with 46XY pure gonadal dysgenesis: a new clinical entity. Ann Neurol 2002;51:385–388. [DOI] [PubMed] [Google Scholar]
- 3. Malandrini A, Gambelli S, Muglia M, et al. Motor‐sensory neuropathy with minifascicle formation in a woman with normal karyotype. Neurology 2005;65:776. [DOI] [PubMed] [Google Scholar]
- 4. Umehara F, Tate G, Itoh K, et al. A novel mutation of desert hedgehog in a patient with 46XY partial gonadal dysgenesis accompanied by minifascicular neuropathy. Am J Hum Genet 2000;67:1302–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li H, Durbin R. Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics 2009;25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Li H, Handsaker B, Wysoker A, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009;25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Taira M, Ishiura H, Mitsui J, et al. Clinical features and haplotype analysis of newly identified Japanese patients with gelsolin‐related familial amyloidosis of Finnish type. Neurogenetics 2012;13:237–243. [DOI] [PubMed] [Google Scholar]
- 8. Fukuda Y, Nakahara Y, Date H, et al. SNP HiTLink: a high‐throughput linkage analysis system employing dense SNP data. BMC Bioinformatics 2009;10:121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gudbjartsson DF, Thorvaldsson T, Kong A, et al. Allegro version 2. Nat Genet 2005;37:1015–1016. [DOI] [PubMed] [Google Scholar]
- 10. Tate G, Satoh H, Endo Y, et al. Assignment of desert hedgehog (DHH) to human chromosome bands 12q12 3 q13.1 by in situ hybridization. Cytogenet Cell Genet 2000;88:93–94. [DOI] [PubMed] [Google Scholar]
- 11. Ingham PW. Transducing Hedgehog: the story so far. EMBO J 1998;17:3505–3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Parmantier E, Lynn B, Lawson D, et al. Schwann cell‐derived Desert hedgehog controls the development of peripheral nerve sheaths. Neuron 1999;23:713–724. [DOI] [PubMed] [Google Scholar]
- 13. Bitgood MJ, Shen L, McMahon AP. Sertoli cell signaling by Desert hedgehog regulates the male germline. Curr Biol 1996;6:298–304. [DOI] [PubMed] [Google Scholar]
- 14. Canto P, Soderlund D, Reyes E, Mendez JP. Mutations in the Desert hedgehog (DHH) gene in patients with 46XY complete pure gonadal dysgenesis. J Clin Endocr Metab 2004;89:4480–4483. [DOI] [PubMed] [Google Scholar]
- 15. Das DK, Sanghavi D, Gawde H, et al. Novel homozygous mutations in Desert hedgehog gene in patients with 46XY complete gonadal dysgenesis and prediction of its structural and functional implications by computational methods. Eur J Med Genet 2011;54:e529–e534. [DOI] [PubMed] [Google Scholar]
- 16. Werner R, Mertz H, Birnbaum W, et al. 46, XY gonadal dysgenesis due to a homozygous mutation in desert hedgehog (DHH) identified by exome sequencing. J Clin Endocrinol Metab 2015;100:E1022–E1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Castro JJ, Méndez JP, Coral‐Vázquez RM, et al. In vitro and molecular modeling analysis of two mutant desert hedgehog proteins associated with 46XY gonadal dysgenesis. DNA Cell Biol 2013;32:524–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Baets J, Dierick I, Groote CC, et al. Peripheral neuropathy and 46XY gonadal dysgenesis: a heterogeneous entity. Neuromuscul Disord 2009;19:172–175. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Pedigree chart of MN‐2.
Figure S2. Haplotypes around DHH locus constructed using SNPs of two families are shown.
Table S1. Nerve conduction study findings of patients 1 and 2 in MN‐1.
Table S2. Clinical and genetic findings in 11 patients who have mutations in DHH.