

Abstract
Objective
Clinical neurological assessment is challenging for severe traumatic brain injury (TBI) patients in the acute setting. Waves of neurochemical abnormalities that follow TBI may serve as fluid biomarkers of neurological status. We assessed the cerebrospinal fluid (CSF) levels of glutamate, lactate, BDNF, and GDNF, to identify potential prognostic biomarkers of neurological outcome.
Methods
This cross‐sectional study was carried out in a total of 20 consecutive patients (mean [SD] age, 29 [13] years; M/F, 9:1) with severe TBI Glasgow Coma Scale ≤ 8 and abnormal computed tomography scan on admission. Patients were submitted to ventricular drainage and had CSF collected between 2 and 4 h after hospital admission. Patients were then stratified according to two clinical outcomes: deterioration to brain death (nonsurvival, n = 6) or survival (survival, n = 14), within 3 days after hospital admission. CSF levels of brain‐derived substances were compared between nonsurvival and survival groups. Clinical and neurological parameters were also assessed.
Results
Glutamate and lactate are significantly increased in nonsurvival relative to survival patients. We tested the accuracy of both biomarkers to discriminate patient outcome. Setting a cutoff of >57.75, glutamate provides 80.0% of sensitivity and 84.62% of specificity (AUC: 0.8214, 95% CL: 54.55–98.08%; and a cutoff of >4.65, lactate has 100% of sensitivity and 85.71% of specificity (AUC: 0.8810, 95% CL: 54.55–98.08%). BDNF and GDNF did not discriminate poor outcome.
Interpretation
This early study suggests that glutamate and lactate concentrations at hospital admission accurately predict death within 3 days after severe TBI.
Keywords: glutamate, lactate, Traumatic brain injury
Introduction
Traumatic brain injury (TBI) is a leading cause of mortality and morbidity among young individuals in low‐ and middle‐income countries. In Brazil, the estimated incidence of TBI is 400 cases per 100.000 inhabitants.1 TBI survivors, commonly suffer persisting cognitive dysfunction, and have a heightened risk of premature death, progressive neurodegeneration, and Alzheimer's disease.2, 3 The pathophysiological mechanisms governing short‐ and long‐term TBI outcomes include axonal injury3, blood‐brain barrier disruption4, glutamate neurotoxicity5, neuroenergetic deficits6, and lack of neurotrophic support.7 These neurochemical signatures also provide an opportunity for identifying molecules in body fluids that may serve as candidate biomarkers with putative clinical relevance.8, 9
Glutamate, the predominant excitatory neurotransmitter in the mammalian brain, may become excitotoxic at high extracellular concentrations through overstimulation of glutamate receptors.5, 10 Remarkably, high concentrations of extracellular glutamate are commonly observed after severe TBI, the magnitude of which correlates with increased intracranial pressure (ICP) and poor clinical outcomes.11, 12, 13, 14 Immediately after TBI, there is an exacerbated glutamate‐induced neuronal calcium influx and ionic imbalance, which evokes membrane depolarization and further glutamate release. As consequence, the membrane pumps work to rapidly restore the membrane gradients thereby increasing the glucose flux through glycolytic pathway and lactate production.8, 10 However, extracellular lactate may arise not only from neuronal glycolysis. The astrocyte‐neuron lactate shuttle model proposes a metabolic coupling between astrocytes and neurons based on the glutamate‐stimulated glycolysis in astrocytes, and the extrusion of lactate to support the energy demands associated with increased neuronal activity.15 Based on this concept, one could argue that increased extracellular brain lactate concentrations after TBI may reflect increased astrocytic metabolic responsiveness to high glutamate concentrations. This mechanism seems to be important for the survival of severe TBI patients since hypertonic sodium lactate infusion improves ICP, hemodynamic, and energetic parameters16, 17, whereas disturbances in this metabolic coupling are associated with worse prognosis.18
Moreover, experimental studies have demonstrated that in response to increased extracellular glutamate levels, astrocytes and neurons release brain‐derived neurotrophic factor (BDNF) and the glial cell line‐derived neurotrophic factor (GDNF). Notably, both of these neurotrophins modulate the activity of glutamatergic neurotransmission, which includes neuronal synaptic release and astrocytic glutamate uptake transporters activity/expression19, 20, while BDNF specifically modulates the expression of the high‐affinity monocarboxylate transporters 2 (MCT2) in neurons.21 The MCT2 controls the influx of energetic substrates including lactate to the oxidative metabolism in neurons.21 Based on the aforementioned, increased concentration of these molecules could likely indicate trophic responses intending to halt glutamatergic toxicity and improve metabolic support.
Although not yet shown in TBI patients, demonstration of a mechanistic interaction between neurotrophins, lactate, and glutamate neurotransmission may shed light on the high incidence of death in the initial days after a severe TBI. In addition, this interaction may provide an opportunity to evaluate relative concentrations of neurotrophins and glutamate levels as potential biomarkers with prognostic value. Indeed, since clinical neurological assessment is challenging for severe TBI patients in the acute setting, due to the common comatose state, etc., fluid biomarkers are being vigorously pursued.
Therefore, in this study, we assessed the cerebrospinal fluid (CSF) concentrations of glutamate, lactate, BDNF, and GDNF at hospital admission, to identify prognostic biomarkers of clinical outcomes.
Subjects and Methods
Study population and clinical management
The cohort used in this study was previously established in Böhmer et al., 20112. Briefly, a cross‐sectional study was carried out between 2006 and 2009 with a total of 20 consecutive patients with severe TBI, presenting score 8 or less in Glasgow Coma Scale (GCS) and abnormal computed tomography (CT) scan on their admission to the Emergency Unit of Cristo Redentor Hospital (Porto Alegre, RS, Brazil). CT classification was performed according to Eisenberg et al., 199022. Further details regarding patient's CT on admission were available on Böhmer et al., 20112. The TBI patients were predominantly young male (mean [SD] age, 29 [13] years; M/F, 9:1) and were admitted to emergency mainly because of motor vehicle accidents. The inclusion criteria for participating in this study was patients with isolated severe TBI, with no previous history of inflammatory, metabolic, and neuropsychiatric disorders that could influence the biomarkers’ concentrations and clinical outcomes. The exclusion criteria were diabetes, drug abuse, and neuropsychiatric disorders. The characteristics of patients are shown in Table 1. Severe TBI patients were submitted to Hospital Cristo Redentor emergency clinical protocol as previously reported by our group2. Briefly, after intubation, patients were supported with continuous mild hyperventilation induced with arterial carbon dioxide tension (PaCO2 between 30 and 35 mmHg). Mean arterial pressure was kept at satisfactory levels to allow cerebral perfusion pressure (mean arterial blood pressure minus intracerebral pressure) above 60 mmHg. Immediately after initial resuscitation, one intracranial pressure monitor (Codman® microsensor) was introduced into the brain parenchyma through a right frontal trepanation. Ventriculostomy was performed to monitor ICP and to allow drainage of CSF when the ICP exceeded 20 mmHg, which was the proposed protocol of Brain Trauma Foundation for clinical management of severe TBI patients with GCS of 8 or less and abnormalities on the computed tomography. The clinical management followed the same standard protocol for ICP management in all patients. CSF drained from the intraventricular catheter was collected via an extraventricular drainage system. The intraventricular catheter was removed when patients died, or when survival patients presented stable ICP for 24 h. The primary ventricular CSF samples collected between 2 and 4 h after hospitalization were used to determine biomarkers’ concentrations. For an overview about advantages and disadvantages of CSF as a matrix to measure candidate biomarkers of brain injury, please see Zetteberg et al., 2016.23
Table 1.
Demographics and clinical characteristics of patients
| Variables | Survival (n = 14) (median ± range) | NonSurvival (n = 6) (median ± range) | P ‐value |
|---|---|---|---|
| Age | 29 (17–53) | 21.5 (16–38) | P = 0.140 |
| ICP | 8.5 (3–20) | 10 (2–45) | P = 0.095 |
| MAP | 93 (70–124) | 92.5 (63–113) | P = 0.534 |
| CPP | 87.5 (57–114) | 82 (21–88) | P = 0.149 |
| GCS | 6 (4–8) | 4 (4–7) | P = 0.085 |
| CSF glucose (mg/dL)a | 108.2 (75.9–129.4) | 118.3 (80.7–191.9) | P = 0.341 |
ICP, intracranial pressure; MAP, mean arterial pressure; CPP, cerebral perfusion pressure; GCS, Glasgow coma scale.
CSF glucose levels (controls): median: 55.29 range: 29.8–94.1mg/dL.
Intracranial mass lesions associated with midline displacement greater than 5 mm were surgically removed when necessary. No undesirable complications associated with intraventricular catheter placement or ICP monitoring including intracranial hemorrhage or infection were observed. Intracranial hypertension was managed initially in all patients by conventional ICP reduction therapy, such as CSF drainage, mannitol bolus, and mild hyperventilation (PaCO2 up to 30 mmHg). In patients with sustained high ICP (from 20 mmHg) and unresponsive to conventional therapy, other alternatives were employed, including barbiturate coma, hyperventilation (PaCO2 < 30 mmHg), control with jugular bulb monitoring, and decompress craniotomy.
Moreover, 20 healthy subjects (ASA I status) scheduled for elective urological, gynecological, general, or vascular procedures were selected as age and sex‐matched controls. Surgeries essentially included inguinal or umbilical herniorrhaphy, perineoplasty, abdominal hysterectomy, vaginal hysterectomy, myomectomy, transurethral prostate or bladder resection, prostatectomy, and saphenectomy. These subjects were interviewed for the absence of cognitive or neurological disorders.2 Control subjects received a spinal anesthesia technique. Experienced anesthesiologists collected the CSF. The first 0.5 mL of CSF aspirated was discarded to reduce cells contamination. The CSF samples were inspected visually and discarded if blood contamination was present. A total of 0.5 mL of CSF was collected from the patients after successful subarachnoid puncture and before the intrathecal injection of anesthetics or analgesics. The CSF samples from patients and controls were immediately centrifuged at 10,000g in an Eppendorf centrifuge during 5 min to obtain cell‐free supernatants and stored at −70°C within 30 min of collection.
Informed consent for participating in this study was obtained from patients’ family members and directly from healthy individuals, according to the Declaration of Helsinki. At the moment when the consent form was applied, family members were also questioned about patient's lifestyle and preexistent diseases. The local institutional Ethics Committee approved this protocol (project number 0038.0.164.165‐05).
Outcome measures
Considering that patients died up to 3 days from hospital admission, we decided to use 3 days as an arbitrary cutoff point. Patients were then stratified according to two clinical outcomes: deterioration to brain death (nonsurvival n = 6) or survival (survival, n = 14), within 3 days after hospital admission. The ICP, hemodynamic, and metabolic variables including mean arterial blood pressure and cerebral perfusion pressure were daily assessed.22 Two years after discharge, phone calls were placed to confirm that survival patients remained alive, and to investigate the grade of long‐term functional disability. TBI patients were then assessed for the modified Rankin Scale (mRS) and scored by an experienced neurologist (R.M). The mRS score is the commonly used outcome classification scale for disabilities and handicaps after cerebral stroke or other causes of neurological disability. The scale has six grades ranging from 0 (fully independent) to grade 6 (dead), and is considered a reliable endpoint for clinical neurological studies.24
CSF Biomarkers analysis
Glutamate assay
Glutamate concentration in TBI patients and controls were analyzed in 25 μL of the CSF cell‐free supernatant samples through high‐performance liquid chromatography (HPLC). Briefly, samples were filtered and derivatized with o‐phthalaldehyde and mercaptoethanol. CSF samples were separated by reverse phase column (Supelcosil LC‐18, 250 mm × 4.6 mm, Supelco) in a Shimadzu Instruments liquid chromatograph. The mobile phase flowed at a rate of 1.4 mL/min at 24°C. Buffer composition is A: 0.04 mol/L sodium dihydrogen phosphate monohydrate buffer, pH 5.5, containing 20% of methanol; and B: 0.01 mol/L sodium dihydrogen phosphate monohydrate buffer, pH 5.5, containing 80% of methanol. The gradient profile was modified according to the content of buffer B in the mobile phase: 0% at 0.00 min, 25% at 13.75 min, 100% at 15.00–20.00 min, and 0% at 20.01–25.00 min. Absorbance was read in a Shimadzu fluorescence detector, with excitation and emission being 360 nm and 455 nm, respectively. The concentration was expressed in μmol/L.22
Lactate, BDNF, and GDNF assays
The lactate level was measured using a commercial kit manufactured by Interteck‐Katal Biotechnology, Brasil. Calibration factors were determined using a standard of lithium lactate (4.44 mmol/L). Lactate concentration is expressed as mmol/L (24). The levels of BDNF and GDNF were assayed using commercially available enzyme‐linked immunosorbent assay kits (ELISA) (BDNF ref. DBNT00 and GDNF ref. DY212; R&D Systems Inc., Minneapolis, MN, USA.). The CSF concentrations of BDNF or GDNF were determined by measuring the absorbance of standard and samples at 450 nm with a SpectraMax M5 (Molecular Devices). The calibration curve was linear up to 1000 pg/mL for BDNF, and up to 2000 pg/mL to GDNF (25). The concentrations are expressed as pg/mL. All samples and standards were carried out in duplicate within the same experiment, and the variation within each duplicate was <5%.
Statistical analysis
We submitted data to Kolmogorov–Smirnov testing for normality evaluation. Glutamate, lactate, BDNF, and GDNF values were fitted in a standard distribution curve and were therefore subjected to nonparametric analyses. Comparisons between groups were performed by two‐tailed Mann–Whitney test. ROC curves were created to explore the ability of biomarkers to predict survival and nonsurvival patients after severe TBI. Estimates of the areas under the curves (AUCs) were obtained, with an AUC of 0.5 indicating no discrimination and an AUC of 1.0 indicating a perfect diagnostic test. Bivariate correlations among biomarkers were performed by Spearman correlation test. Spearman cross‐correlation analyses among biomarkers was also performed and corrected by Bonferroni Method. All numerical variables are presented as mean ± standard deviation (S.D.). P values at ≤ 0.05 were considered statistically significant. All analyses were performed using GraphPad Prism 5.0 or Matlab 2014b.
Results
CSF biomarkers glutamate, lactate, BDNF, and GDNF
The level of the excitatory neurotransmitter glutamate in TBI patients (nonsurvival: median [IQR]: 61.25 [41.40–88.20] μmol/L and survival: 25.35 [5.00–93.70] μmol/L) was significantly higher than control group (median [IQR]: 2.6 [2.1–3.3] μmol/L) (P < 0.0001). Also, glutamate levels were significantly increased in nonsurvival relative to survival patients (P = 0.0256) (Fig. 1A).
Figure 1.
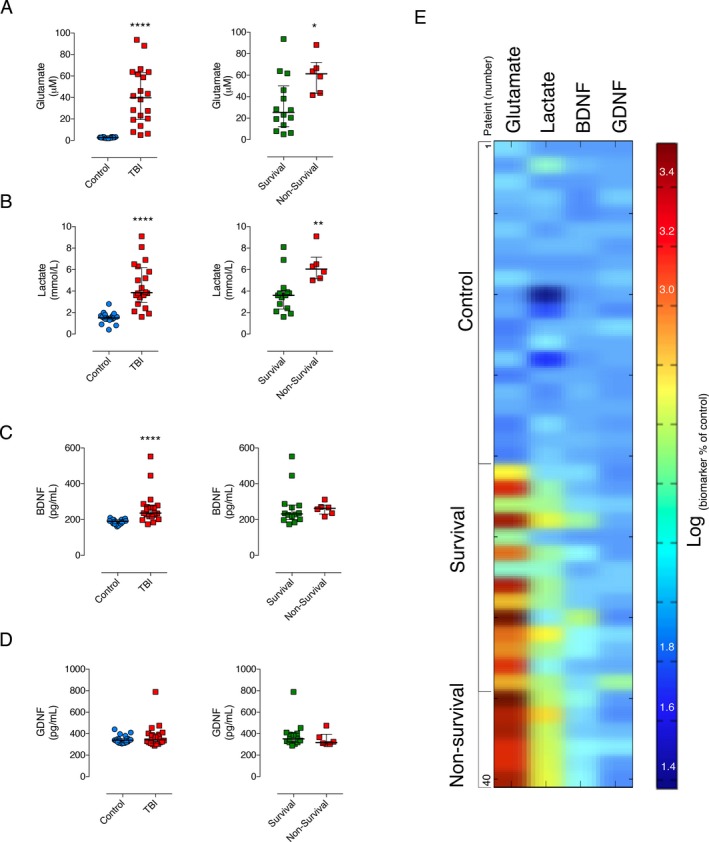
Cerebrospinal fluid biomarkers in controls and severe TBI patients (survival and nonsurvival). The CSF levels of glutamate (A), lactate (B), and BDNF (C) in TBI group at hospital admission were significantly different than control group (n = 20 per group, ****P > 0.0001). In addition, glutamate and lactate levels were significantly higher in nonsurvival (n = 6) relative to survival patients (n = 14) (*P = 0.0256 and **P = 0.0060, respectively). The GDNF level was not different between control and TBI groups, (D). Both BDNF (C, right panel) and GDNF (D, right panel) were not different between survival and nonsurvival TBI patients. The dynamic heatmap shows a neurochemical signature regarding the profile of the biomarkers in control subjects and TBI patients (E). Horizontal lines indicate median and interquartile range. The dynamic heatmap scale represents normalized (log) percent of change in relation to controls.
Similarly, we found that CSF lactate levels in TBI group were significantly increased compared with control group (median [IQR]: 1.5 [0.4–2.8] mmol/L) (P < 0.0001). Further, there was a significant difference in lactate level between nonsurvival (median [IQR]: 6.05 [5.00–9.10] mmol/L) relative to survival group (median [IQR]: 3.6 [1.6–8.1] mmol/L) (P = 0.0060) (Fig. 1B).
In contrast to glutamate and lactate, while CSF BDNF levels in TBI patients were significantly different from control group (median [IQR]: 190.8 [161.0–210.4] pg/mL (P = 0.0001), BDNF levels in nonsurvival (median [IQR]: 263.0 [216.5–311.9] pg/mL) were not significantly different from survival group (median [IQR] 231.7 [174.0–552.6] pg/mL) (P = 0.4072) (Fig. 1C).
Moreover, there was no significant difference in CSF GDNF levels between control (median [IQR]: 338.9 [306.8–440.9] pg/mL) and TBI, and between nonsurvival (median [IQR]: 317.7 [303.0–473.9] pg/mL) and survival patients (median [IQR] 351.7[287.6–788.7] pg/mL (P = 0.3015) (Fig. 1D).
The dynamic heat map (Fig. 1E) provides a visualization of the biomarker signatures. We can visualize that both lactate and glutamate displayed a distinct profile in the survival and nonsurvival patients, whereas BDNF and GDNF showed no evident discriminatory profile.
CSF biomarkers and bivariate correlation analysis
To provide insights regarding the quantitative relationship between biomarkers, we tested the correlation coefficient in the TBI and Control groups pooled together. There was significant statistical correlation between CSF levels of glutamate and lactate (r = 0.7051; P = 0.053; Fig. 2A), glutamate and BDNF (r = 0.6983; P = 0.024) (Fig. 2B), and BDNF and lactate (r = 0.7491; P = 0.039) (Fig. 2D). The correlations between GDNF and glutamate (r = −0.01794; P = 0.462) (Fig. 2C), GDNF and BDNF (r = −0.004; P = 0.574) (Fig. 2F), and GDNF and lactate (r = 0.1368; P = 0.513) (Fig. 2E) did not reach statistical significance.
Figure 2.
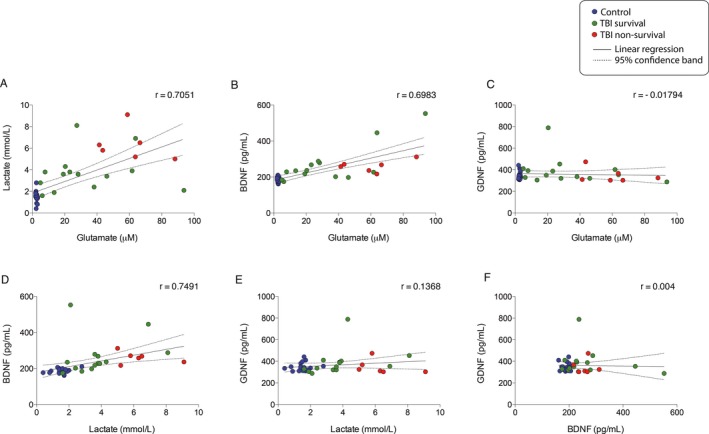
Spearman bivariate correlation analyses between cerebrospinal fluid biomarkers. There were statistically significant correlations between glutamate and lactate (A) (r = 0.7051; P = 0.053), glutamate and BDNF (B) (r = 0.6983; P = 0.024), Lactate and BDNF (D)(r = 0.7491; P = 0.039). GDNF did not show statistically significant correlations with glutamate (C), lactate (E), and BDNF (F).
The statistical correlations between the CSF biomarkers and clinical outcomes are showed in Table 2. There were no significant correlations between fluid biomarkers concentrations at admission and clinical outcomes measured up to 3 days after hospital admission. In contrast, glutamate and lactate were significantly correlated with modified Rankin Scale (mRS). Correlation coefficients for Glutamate vs. mRS, and Lactate vs. mRS were (r = 0.6427, P = 0.002) and (r = 0.4635, P = 0.03), respectively.
Table 2.
Correlation between CSF biomarkers and clinical outcomes in TBI survival and nonsurvival groups
| Variables | Spearman (r) | P ‐value |
|---|---|---|
| Glutamate vs. ICP | 0.34 | 0.15 |
| Lactate vs. ICP | 0.09 | 0.72 |
| BDNF vs. ICP | 0.16 | 0.50 |
| GDNF vs. ICP | 0.20 | 0.41 |
| Glutamate vs. MAP | −0.21 | 0.37 |
| Lactate vs. MAP | 0.01 | 0.98 |
| BDNF vs. MAP | −0.05 | 0.84 |
| GDNF vs. MAP | 0.21 | 0.37 |
| Glutamate vs. CPP | −0.36 | 0.12 |
| Lactate vs. CPP | −0.15 | 0.53 |
| BDNF vs. CPP | −0.18 | 0.46 |
| GDNF vs. CPP | 0.01 | 0.96 |
| Glutamate vs. GCS | −0.15 | 0.54 |
| Lactate vs. GCS | −0.39 | 0.09 |
| BDNF vs. GCS | 0.13 | 0.58 |
| GDNF vs. GCS | −0.16 | 0.49 |
ICP, Intracranial pressure; BDNF, brain‐derived neurotrophic factor; GDNF, glial cell line‐derived neurotrophic factor; MAP, Mean arterial pressure; CPP, Cerebral perfusion pressure; GCS, Glasgow coma scale.
ROC curves and biomarkers associations
Given the CSF concentrations of glutamate and lactate showed the highest level in nonsurvival patients, we evaluated the accuracy of those tests as early prognostic biomarkers of neurological deterioration. The area under the ROC curve (AUC) indicate how well glutamate and lactate can discriminate patients that will survive from patients that will evolve to death up 3 days in the intensive unit care. The ROC curve was achieved from glutamate and lactate up to 3 days from admission in relation to death (Fig. 3A and B). The specificity rate is showed in function of sensitivity rate at different cutoff points. Setting a cutoff of >57.75, glutamate provides 80% of sensitivity and 84.62% of specificity (AUC: 0.8214, 95% CI, CL: 54.55% to 98.08%) and with a cutoff of >4.650, lactate has 100% of sensitivity and 85.71% of specificity (AUC: 0.8810, 95% CI, CL: 54.55–98.08%). Glutamate and lactate provide high specificity and sensitivity, with almost identical power, for predicting brain death in TBI patients.
Figure 3.
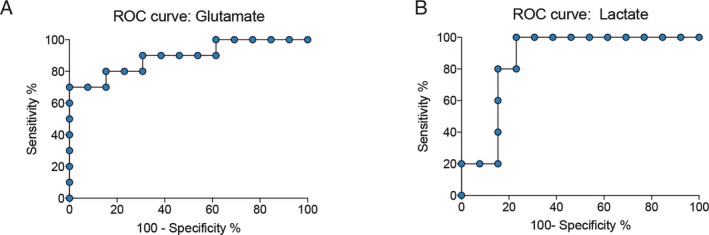
Diagnostic accuracy of biomarkers predicts brain death. Area under the receiver‐operating characteristic curve (AUC) for CSF glutamate (A) and lactate (B) concentrations. ROC curves based on glutamate and lactate were used as a predictor of death in TBI patients. The cutoff for glutamate was set at >57.75, providing 80% of sensitivity and 84.62% of specificity (AUC: 0.8214, 95% CI, CL: 54.55–98.08%). The cutoff for lactate was set at >4.650, providing 100% of sensitivity and 85.71% of specificity (AUC: 0.8810, 95% CI, CL: 54.55–98.08%). Both biomarkers presented high specificity and sensitivity for discriminating TBI survival and nonsurvival patients.
Additionally, aiming to corroborate the proposed functional interplay among biomarkers, we have developed a network using cross‐correlation analysis. We identified statistically significant associations among glutamate, lactate, and BDNF. One may propose that these associations suggest reactive neuronal and glial responses to injury, and ultimately may reflect some level of functional coupling (Fig. 4).
Figure 4.
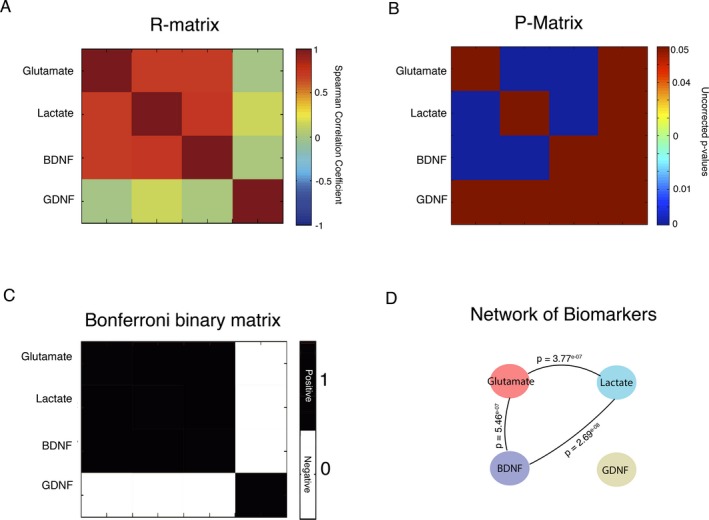
Association between biomarkers indexed by cross‐correlation analyses. (A) Symmetric matrix of correlation coefficient values between biomarkers; (B) Symmetric matrix of – values between biomarkers; (C) Binary matrix corrected by Bonferroni method at P < 0.05; (D) Network for the biomarkers indicating high levels of associations between glutamate, lactate and BDNF.
Discussion
This study used a multi‐biomarker approach in the CSF of severe TBI patients searching for neuroactive substances with potential clinical and prognostic relevance. Our results demonstrated that CSF levels of glutamate, lactate, and BDNF at hospital admission have potential value as biomarkers of severe head injury. Further, glutamate and lactate levels were able to predict with high sensitivity and specificity patients who will die within 3 days following severe TBI. Also, up to 3 days, the clinical outcomes including intracranial pressure (ICP), mean arterial pressure, cerebral perfusion pressure, and GCS were similar between TBI groups (nonsurvival vs. survival) and did not correlate with CSF biomarkers.
The role of glutamate as biomarker of brain injury and clinical outcomes has previously been explored in studies with severe TBI patients. In contrast to our findings, Brown et al., 199825 showed that despite the persistent increase in the CSF glutamate levels up to 5 days after severe TBI, these changes did not predict brain death or correlate with GCS and electrophysiological deterioration. However, the majority of clinical studies indicate that increased brain extracellular glutamate levels in association with comorbidities may predict poor functional outcomes and brain death.11, 12, 13, 14 For instance, secondary mechanisms associated with the brain tissue deformation including global ischemia, sustained increased intracranial pressure and focal contusions11, 13, and spontaneous brain hypothermia (<36°C)26 have been implicated in the increase in glutamate levels and acute neuronal death. Also, increased extracellular brain glutamate concentration after TBI is also influenced by decreased astrocytic glutamate uptake.27 This might indicate that different players associated to the tripartite glutamatergic synapse, likely contribute for the deterioration of brain function after severe TBI, such as disrupted energy support rapidly triggering neuronal death.
We found that CSF lactate levels at hospital admission predict severe TBI patients who will die within 3 days after hospitalization. As previously stated, astrocytes take up glutamate in the synaptic space thereby triggering glycolysis and lactate production, which is shuttled to neurons.15 The flux of these substrates in the body fluids has been explored in the clinical setting as biomarkers of neuroenergetic support, neuron‐to‐astrocyte interactions, and clinical outcomes.28 Recently, Thelin et al., 2014 showed that an increased brain extracellular levels of lactate and pyruvate after TBI were correlated with unfavorable neurological outcome based on Glasgow Outcome Scale29. Moreover, there is strong evidence showing that impaired cerebrovascular pressure reactivity, and decreased brain perfusion and oxygenation, are forces driving metabolic abnormalities related to lactate/pyruvate ratio. However, this oxygen deficiency is more pronounced in the perilesional tissue26, 30 and apparently does not explain the metabolic alterations in the whole brain after a TBI. Indeed, increased lactate production in the injured brain seems to be independent of oxygen availability, and therefore cannot be considered as a direct ischemic and hypoxic metabolic marker30. Instead, experimental and clinical studies highlight the existence of an acute hyperglycolysis after severe TBI followed by subacute decrease in glucose metabolism.31, 32 The early hyperglycolytic phase seems to reflect the energy demands for reversal of ionic imbalances caused by massive release of glutamate33, while in subacute phase, the decrease in glucose utilization may indicate impaired mitochondrial function.33, 34
Interestingly, we found a significant correlation between lactate and glutamate levels indicating they are strongly and positively associated. In comparison to our findings in CSF, Bouzat et al., 201416 demonstrated that hypertonic infusion of lactate in severe TBI patients improved metabolic profile and decreased glutamate levels in the brain extracellular microdyalisate. In addition, a previous study in severe TBI patients showed that brain glucose availability decreases locally and lactate increases in the absence of ischemia. In contrast, our study, suggests that lactate production may significantly increase even in the presence of a satisfactory glucose supply (Table 1) to attend brain metabolic needs. However, as proposed by Lama et al., 201428, the acute increase in the brain tissue lactate concentrations after severe TBI is associated to decreased neuronal uptake, probably leading to irreversible injury and pan‐necrosis. Therefore, although the high extracellular availability of lactate suggests that the metabolic machinery in astrocytes was responsive to the increased energy demands after TBI, neurons were apparently not capable of using lactate as substrate, ultimately contributing to the patient's death. This highlights a putative role for the monocarboxylated transporters expression in the metabolic uncoupling and neuronal death after severe TBI. From the clinical perspective, we propose that short‐term elevation of CSF glutamate and lactate levels after severe TBI has strong predictive value for unfavorable neurological outcome. Moreover, lactate and glutamate positively correlated with long‐term disability grade scale mRS).
In addition, we also assessed the ability of neurotrophins BDNF and GDNF in interrogate clinical outcomes in severe TBI patients. Data from preclinical studies indicate that BDNF modulates vesicular glutamate release and NMDA receptor sensitivity35, 36, and the availability of lactate for neurons through the monocarboxylate transporters‐2.21 As a CSF biomarker, BDNF displayed significant correlations with both glutamate and lactate; however, it was not predictive of imminent death within 3 days of hospitalization as were glutamate and lactate levels. Actually, measurements of BDNF levels in plasma and serum of severe TBI patients have provided controversial results. Simon et al., 2016 reported no predictive value for BDNF to discriminate survival and nonsurvival patients.37 Similar to our findings, day‐of‐injury serum BDNF was associated with TBI diagnosis; however, the authors also showed 6‐month prognostic value regarding recovery from TBI.38 Further, the CSF levels of BDNF predicted mortality within 7 days of hospitalization after severe TBI.39 Though preclinical studies proposes GDNF as therapeutic strategy to treat TBI, which highlights a neurobiological importance, its clinical value as fluid biomarker in patients has been little explored. In our study, CSF GDNF level was not a good diagnostic or predictive biomarker of poor clinical outcome. The main limitation of this study was the small sample size. Further, due to logistic difficulties, we did not have access to the biological samples of patients posthospitalization.
The CSF levels of glutamate and lactate have potential prognostic value to discriminate with high sensitivity and specificity severe TBI patients who will evolve to death within 3 days after hospitalization. Glutamate and lactate are promising CSF biomarkers of death after severe TBI.
Author Contribution
Dr. Stefani: study concept and design; revising the manuscript; patient's care, data and interpretation. Dr. Modkovski: study concept and design; revising the manuscript patient's care and data collection. Dr. Hansel: study concept and design; revising the manuscript; biological fluids processing and analyses, neurobiochemical assays and data interpretation. Dr. Zimmer: study concept and design; revising the manuscript; data analysis and interpretation. Ms. Kopczynski: study concept and design; revising the manuscript; neurobiochemical assays and data interpretation. Dr. Muller: study concept and design; revising the manuscript; data collection and analysis. Ms. Strogulski: study concept and design; revising the manuscript; analysis and interpretation. Ms. Rodolphi: study concept and design; revising the manuscript; analysis and interpretation. MSc. Carteri: study concept and design; revising the manuscript; statistical analysis and data interpretation. Dr. Schmidt: study concept and design; revising the manuscript; selection of control individuals; clinical and neurological evaluation and cerebrospinal fluid sampling. Dr. Oses: study concept and design; revising the manuscript; study supervision, data acquisition and interpretation. Dr. Smith: study concept and design; drafting the manuscript, study concept and design, analysis and interpretation of data. Dr. Portela: study concept and design; drafting the manuscript, interpretation of data and obtaining funding. All authors critically revised and approved the final version of the manuscript.
Conflict of Interest
Dr. Marco A. Stefani, Dr. Rafael Modkovski, Dr. Gisele Hansel, Dr. Eduardo R. Zimmer, Ms. Afonso Kopczynski, Dr. Alexandre P. Muller, Ms. Nathan R. Strogulski, Ms. Marcelo S. Rodolphi, Ms. Randhall K. Carteri, Dr. André P. Schmidt, Dr. Jean P. Oses, Dr. Douglas H. Smith and Dr. Luis V. Portela report no financial, personal or scientific conflict of interest.
Acknowledgements
The authors thank Dr. Ana Elisa Böhmer (Department of Pharmacology, Institute of Biomedical Sciences, University of São Paulo) for the technical support in HPLC analysis. This research was funded by resources from FAPERGS # 1010267, CAPES PNPD # 1663, Science without borders CNPQ # 4011645/2012‐6 and CNPQ INCTen Excitotoxicity and Neuroprotection # 573677/2008‐5. The authors have no financial, personal or scientific conflict of interest.
References
- 1. Roozenbeek B, Maas AI, Menon DK. Changing patterns in the epidemiology of traumatic brain injury. Nat Rev Neurol 2013;9:231–236. [DOI] [PubMed] [Google Scholar]
- 2. Bohmer AE, Oses JP, Schmidt AP, et al. Neuron‐specific enolase, S100B, and glial fibrillary acidic protein levels as outcome predictors in patients with severe traumatic brain injury. Neurosurgery 2011;68:1624–1630; discussion 30‐1. [DOI] [PubMed] [Google Scholar]
- 3. Johnson VE, Stewart W, Smith DH. Axonal pathology in traumatic brain injury. Exp Neurol 2013;246:35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hay JR, Johnson VE, Young AM, et al. Blood‐brain barrier disruption is an early event that may persist for many years after traumatic brain injury in humans. J Neuropathol Exp Neurol 2015;74:1147–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Faden AI, Demediuk P, Panter SS, Vink R. The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science 1989;244:798–800. [DOI] [PubMed] [Google Scholar]
- 6. Moro N, Ghavim SS, Harris NG, et al. Pyruvate treatment attenuates cerebral metabolic depression and neuronal loss after experimental traumatic brain injury. Brain Res 2016;1642:270–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rostami E, Krueger F, Plantman S, et al. Alteration in BDNF and its receptors, full‐length and truncated TrkB and p75(NTR) following penetrating traumatic brain injury. Brain Res 2014;1542:195–205. [DOI] [PubMed] [Google Scholar]
- 8. Zetterberg H, Smith DH, Blennow K. Biomarkers of mild traumatic brain injury in cerebrospinal fluid and blood. Nat Rev Neurol 2013;9:201–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shahim P, Tegner Y, Wilson DH, et al. Blood biomarkers for brain injury in concussed professional ice hockey players. JAMA Neurol 2014;71:684–692. [DOI] [PubMed] [Google Scholar]
- 10. Bonfoco E, Krainc D, Ankarcrona M, et al. Apoptosis and necrosis: two distinct events induced, respectively, by mild and intense insults with N‐methyl‐D‐aspartate or nitric oxide/superoxide in cortical cell cultures. Proc Natl Acad Sci USA 1995;92:7162–7166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zauner A, Bullock R, Kuta AJ, et al. Glutamate release and cerebral blood flow after severe human head injury. Acta Neurochir Suppl 1996;67:40–44. [DOI] [PubMed] [Google Scholar]
- 12. Bullock R, Zauner A, Woodward JJ, et al. Factors affecting excitatory amino acid release following severe human head injury. J Neurosurg 89:507–518. [DOI] [PubMed] [Google Scholar]
- 13. Koura SS, Doppenberg EM, Marmarou A, et al. Relationship between excitatory amino acid release and outcome after severe human head injury. Acta Neurochir Suppl 1998;71:244–246. [DOI] [PubMed] [Google Scholar]
- 14. Chamoun R, Suki D, Gopinath SP, et al. Role of extracellular glutamate measured by cerebral microdialysis in severe traumatic brain injury. J Neurosurg 2010;113:564–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pellerin L, Magistretti PJ. Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci USA 1994;91:10625–10629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bouzat P, Sala N, Suys T, et al. Cerebral metabolic effects of exogenous lactate supplementation on the injured human brain. Intensive Care Med 2014;40:412–421. [DOI] [PubMed] [Google Scholar]
- 17. Quintard H, Patet C, Zerlauth JB, et al. Improvement of neuroenergetics by hypertonic lactate therapy in patients with traumatic brain injury is dependent on baseline cerebral lactate/pyruvate ratio. J Neurotrauma 2016;33:681–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Glenn TC, Kelly DF, Boscardin WJ, et al. Energy dysfunction as a predictor of outcome after moderate or severe head injury: indices of oxygen, glucose, and lactate metabolism. J Cereb Blood Flow Metab 2003;23:1239–1250. [DOI] [PubMed] [Google Scholar]
- 19. Almeida RD, Manadas BJ, Melo CV, et al. Neuroprotection by BDNF against glutamate‐induced apoptotic cell death is mediated by ERK and PI3‐kinase pathways. Cell Death Differ 2005;12:1329–1343. [DOI] [PubMed] [Google Scholar]
- 20. Farrand AQ, Gregory RA, Scofield MD, et al. Effects of aging on glutamate neurotransmission in the substantia nigra of Gdnf heterozygous mice. Neurobiol Aging 2015;36:1569–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Robinet C, Pellerin L. Brain‐derived neurotrophic factor enhances the expression of the monocarboxylate transporter 2 through translational activation in mouse cultured cortical neurons. J Cereb Blood Flow Metab 2010;30:286–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schmidt AP, Tort AB, Silveira PP, et al. The NMDA antagonist MK‐801 induces hyperalgesia and increases CSF excitatory amino acids in rats: reversal by guanosine. Pharmacol Biochem Behav 2009;91:549–553. [DOI] [PubMed] [Google Scholar]
- 23. Zetterberg H, Blennow K. Fluid biomarkers for mild traumatic brain injury and related conditions. Nat Rev Neurol 2016;12:563–574. [DOI] [PubMed] [Google Scholar]
- 24. Huisman TA, Schwamm LH, Schaefer PW, et al. Diffusion tensor imaging as potential biomarker of white matter injury in diffuse axonal injury. AJNR Am J Neuroradiol 2004;25:370–376. [PMC free article] [PubMed] [Google Scholar]
- 25. Brown JI, Baker AJ, Konasiewicz SJ, Moulton RJ. Clinical significance of CSF glutamate concentrations following severe traumatic brain injury in humans. J Neurotrauma 1998;15:253–263. [DOI] [PubMed] [Google Scholar]
- 26. Timofeev I, Carpenter KL, Nortje J, et al. Cerebral extracellular chemistry and outcome following traumatic brain injury: a microdialysis study of 223 patients. Brain 2011;134(Pt 2):484–494. [DOI] [PubMed] [Google Scholar]
- 27. Goodrich GS, Kabakov AY, Hameed MQ, et al. Ceftriaxone treatment after traumatic brain injury restores expression of the glutamate transporter, GLT‐1, reduces regional gliosis, and reduces post‐traumatic seizures in the rat. J Neurotrauma 2013. Aug;30:1434–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lama S, Auer RN, Tyson R, et al. Lactate storm marks cerebral metabolism following brain trauma. J Biol Chem 2014;289:20200–20208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Thelin EP, Nelson DW, Ghatan PH, Bellander BM. Microdialysis monitoring of csf parameters in severe traumatic brain injury patients: a novel approach. Front Neurol 2014;5:159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lazaridis C, Andrews CM. Brain tissue oxygenation, lactate‐pyruvate ratio, and cerebrovascular pressure reactivity monitoring in severe traumatic brain injury: systematic review and viewpoint. Neurocrit Care 2014;21:345–355. [DOI] [PubMed] [Google Scholar]
- 31. Bergsneider M, Hovda DA, Shalmon E, et al. Cerebral hyperglycolysis following severe traumatic brain injury in humans: a positron emission tomography study. J Neurosurg 1997;86:241–251. [DOI] [PubMed] [Google Scholar]
- 32. Yoshino A, Hovda DA, Kawamata T, et al. Dynamic changes in local cerebral glucose utilization following cerebral conclusion in rats: evidence of a hyper‐ and subsequent hypometabolic state. Brain Res 1991;561:106–119. [DOI] [PubMed] [Google Scholar]
- 33. Katayama Y, Becker DP, Tamura T, Hovda DA. Massive increases in extracellular potassium and the indiscriminate release of glutamate following concussive brain injury. J Neurosurg 1990;73:889–900. [DOI] [PubMed] [Google Scholar]
- 34. Verweij BH, Muizelaar JP, Vinas FC, et al. Impaired cerebral mitochondrial function after traumatic brain injury in humans. J Neurosurg 2000;93:815–820. [DOI] [PubMed] [Google Scholar]
- 35. Black IB. Trophic regulation of synaptic plasticity. J Neurobiol 1999;41:108–118. [PubMed] [Google Scholar]
- 36. Kohara K, Kitamura A, Morishima M, Tsumoto T. Activity‐dependent transfer of brain‐derived neurotrophic factor to postsynaptic neurons. Science 2001;291:2419–2423. [DOI] [PubMed] [Google Scholar]
- 37. Simon D, Nascimento RI, Filho EM, et al. Plasma brain‐derived neurotrophic factor levels after severe traumatic brain injury. Brain Inj 2016;30:23–28. [DOI] [PubMed] [Google Scholar]
- 38. Korley FK, Diaz‐Arrastia R, Wu AH, et al. Circulating brain‐derived neurotrophic factor has diagnostic and prognostic value in traumatic brain injury. J Neurotrauma 2016;33:215–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Failla MD, Conley YP, Wagner AK. Brain‐derived neurotrophic factor (BDNF) in traumatic brain injury‐related mortality: interrelationships between genetics and acute systemic and central nervous system BDNF profiles. Neurorehabil Neural Repair 2016;30:83–93. [DOI] [PMC free article] [PubMed] [Google Scholar]