

Abstract
Objectives
Neuromyelitis optica spectrum disorder (NMOSD) is a severe inflammatory disorder of the central nervous system (CNS) targeted against aquaporin‐4 (AQP4). The origin and trafficking of AQP4‐specific B cells in NMOSD remains unknown.
Methods
Peripheral (n = 7) and splenic B cells (n = 1) recovered from seven NMOSD patients were sorted into plasmablasts, naïve, memory, and CD27‐IgD‐ double negative (DN) B cells, and variable heavy chain (VH) transcriptome sequences were generated by deep sequencing. Peripheral blood (PB) VH repertoires were compared to the same patient's single‐cell cerebrospinal fluid (CSF) plasmablast (PB) VH transcriptome, CSF immunoglobulin (Ig) proteome, and serum Ig proteome. Recombinant antibodies were generated from paired CSF heavy‐ and light chains and tested for AQP4 reactivity.
Results
Approximately 9% of the CSF VH sequences aligned with PB memory B cells, DN B cells, and plasmablast VH sequences. AQP4‐specific VH sequences were observed in each peripheral B‐cell compartment. Lineage analysis of clonally related VH sequences indicates that CSF AQP4‐specific B cells are closely related to an expanded population of DN B cells that may undergo antigen‐specific B‐cell maturation within the CNS. CSF and serum Ig proteomes overlapped with the VH sequences from each B‐cell compartment; the majority of matches occurring between the PB VH sequences and serum Ig proteome.
Interpretation
During an acute NMOSD relapse, a dynamic exchange of B cells occurs between the periphery and CNS with AQP4‐specific CSF B cells emerging from postgerminal center memory B cells and plasmablasts. Expansion of the PB DN B‐cell compartment may be a potential biomarker of NMOSD activity.
Introduction
B cells may play multiple roles in the pathogenesis of neuromyelitis optica spectrum disorders (NMOSD).1 In 75% of NMOSD patients, autoreactive B cells produce antibodies against the aquaporin‐4 (AQP4) water channel (AQP4‐IgG).2, 3 In the central nervous system (CNS), AQP4 is highly expressed on astrocyte end‐feet, and AQP4‐IgG has been shown to initiate an inflammatory cascade that ultimately leads to demyelination and neuronal injury.1, 4, 5 However, the location(s) of initial antigen presentation and affinity maturation, as well as the composition of migratory AQP4‐reactive B cells remains largely unknown.
Recently, we compared the CSF B‐cell variable heavy chain (VH) transcriptome (VH sequences) from NMOSD patients with their respective CSF and blood immunoglobulin (Ig) proteomes (Ig peptides).6 We found that a substantial proportion of CSF AQP4‐IgG is produced intrathecally by CSF B cells and cannot be accounted for by a passive influx of serum AQP4‐IgG. Clonal analysis of CSF B cells and the serum Ig proteome suggested that CSF AQP4‐reactive B cells arose in part from newly emerging germinal center clones.6 Here, we directly investigate the relationship between peripheral blood (PB) and CSF B‐cell populations in NMOSD patients using next‐generation sequencing, VH repertoire analysis, and Ig proteomics. Our results indicate that CD19 + CD27‐IgD‐ double negative (DN) B cells are closely linked to AQP4‐specific CSF plasmablasts and undergo further differentiation, and possibly affinity maturation, within the CNS compartment.
Methods
Standard protocol approvals, registrations, and patients
Patients were recruited in the Neurology Departments at the University of Colorado, Anschutz Medical Campus and the Technical University of Munich. All patients consented to the scientific use of their biologic samples. The study was approved by the University of Colorado School of Medicine Institutional Review Board. A total of seven NMOSD patients (ON07‐05, ON08‐08, ON09‐03, ON10‐01, ON 10‐03, ON11‐04, and ON09‐527) were recruited for Ig transcriptome and Ig proteome analyses. The clinical and CSF data have been presented previously.6 For additional FACS analyses of peripheral blood B‐cell populations, PBMCs from multiple sclerosis patients (n = 15), healthy controls (n = 15), and NMOSD patients (n = 4) were acquired from biobank samples stored in the Rocky Mountain MS Biorepository at the University of Colorado.
Specimen handling and routine CSF testing
CSF and blood were collected by lumbar puncture and venipuncture as previously described.6 Single CSF mononuclear cells (MNCs) were prepared as described previously.1 Peripheral blood was collected in CPT tubes and mononuclear cells isolated according to the manufacturer's instructions (BD Vacutainer, CPT cell preparation tubes with sodium acetate) and cryopreserved at −80°C for later sample analysis. The spleen of one NMOSD patient was obtained following informed consent prior to splenectomy for idiopathic neutropenia. The spleen was disrupted in RPMI media and passed through a 100‐micron cell strainer. Resuspended splenocytes were subsequently centrifuged through Ficol/Paque (Sigma) and the buffy coat collected. Residual red blood cells were lysed and the remaining mononuclear cells were washed in phosphate‐buffered saline (PBS), pH 7.4, resuspended in RPMI media, and cryopreserved.
CSF single‐cell analysis and recombinant antibody (rAb) production
CSF CD19 + CD138 + plasmablast heavy‐ (VH) and light‐chain (VL) variable region sequences were recovered by RT‐PCR and DNA sequencing as described previously.1, 7 Recombinant antibodies were produced in HEK293 cells (Invitrogen, Carlsbad, CA, R620‐07)7 and reactivity tested via immunofluorescence for binding to a permanent cell line expressing M23‐AQP4.8
Fluorescence activated cell sorting of peripheral blood B‐cell populations
Frozen peripheral blood MNCs were thawed at 37°C, washed in phosphate‐buffered saline (PBS) and incubated with the following antibodies: 20 μL CD27 PE (BD), 10 μL CD38 APCCy5.5 (Invitrogen), 15 μL CD19 PacBlue (AbDserotec), 5 μL IgD APC (Becton Dickinson), 20 μL CD14 FITC (AbDserotec), 5 μL CD56 FITC (Becton Dickinson), 10 μL CD20 FITC (Biolegend), 20 μL CD16 FITC (eBioscience), 20 μL CD3 FITC (Becton Dickinson). After another wash in PBS, PB B‐cell populations were sorted on a MoFlo flow cytometer (Cytomations, Fort Collins, CO) using the following surface markers: Naïve B cells CD19 + CD20 + CD27‐ CD38 + IgD+, Memory B cells CD19 + CD20 + CD27 + CD38 + , double negative (DN) B cells CD19 + CD20‐ CD27‐ IgD‐ CD38 + , and plasmablasts CD19 + , CD20‐ CD27 + + CD38high (Fig. 1). Sorted cells were collected by centrifugation and RNA was immediately extracted (Qiagen RNeasy Plus Micro Kit) according to the manufacturer's instructions. Plasmablasts and DN B cells were predominately found in the CD19+CD20low B cell pool and were further stratified according to IgD, CD27, and CD38 expression.
Figure 1.

Gating strategy for naïve, CD27 + memory, CD27‐ double negative B cells, and plasmablasts.
For FACS quantitation of PB B‐cell populations, frozen PBMC samples were used from NMOSD, multiple sclerosis, and healthy control patients. Plasmablasts showed significantly lower numbers following freeze‐thaw;9 paired analyses of fresh and frozen samples did not show any significant differences in the fraction of naïve B cells, DN B cells, and memory B cells.
Blood VH transcriptome library preparation and deep sequencing
For cDNA synthesis, we used the Clontech SMARTer Ultra Low RNA Kit for Illumina sequencing (high volume) for all seven samples. The SMARTer kit was applied according to the manufacturer's instructions; for the second strand synthesis, we added constant region primers representing each human Ig heavy‐chain class to pair with the Smarter RNA Kit universal 5′ primer. V‐region PCR was performed using heavy chain framework primers (VH families 1‐5) and a second set of internal constant region primers (IgA, IgG, IgM, and IgD), which incorporated specific sequences for Illumina Miseq (www.illumina.com) sequencing (Expand high fidelity PCR system, Roche). Individual reactions for each combination of framework and constant region primers were used to avoid bias toward a certain VH family or immunoglobulin isotype. Framework primers included unique indexed identifiers for each population. Primers also contained eight random nucleotides (unique molecular identifier = UMI) to minimize an amplification‐bias of certain transcripts during PCR reactions. After PCR, nucleotides were removed with a QIAquick PCR Purification Kit (Qiagen) and the PCR products eluted from agarose gels using the Min Elute Gel Extraction Kit (Qiagen) according to manufacturer's instructions. Prior to Illumina sequencing, PCR products were pooled according to the numbers of sorted B cells and the quality and quantity of the DNA was evaluated using a Bioanalyzer 2100 (Agilent Technologies), Qubit Fluorometer (Life technologies) and real‐time PCR. The finished libraries were diluted and loaded on the flow cell of the MiSeq system (Illumina) according to the manufacturer's instructions at a concentration of 10 pM with approximately 8% PhiX DNA (Illumina) to increase complexity. Samples were subjected to cluster generation and sequencing using a paired‐end 251‐bp cycle protocol; an average run generated 12 million reads with 85% passing a quality score of Q30.
Data analysis
The pRESTO workflow10 was used for data analysis and adopted to our specific primer design and usage of unique molecular identifiers (UMIs). Illumina sequencing reads in FASTQ format were presorted into the B‐cell populations according to their indexes. Raw, Illumina MiSeq high‐throughput sequencing reads were quality‐controlled, assembled, and filtered using pRESTO.10 VH variable‐diversity‐joining [V(D)J] germline segments were determined using IMGT/HighV‐QUEST.11 Functional V(D)J sequences were assigned into clones based on identical nucleic acid complementarity determining region‐3 (CDR3) sequence length, VH variable gene segment, VH joining gene segment, and ≥ 75% CDR3 sequence identity at the nucleotide level. The stringency for CDR3 identity was chosen based on prior analysis of NMOSD CSF plasmablast clones1 that showed significant intraclonal CDR3 sequence variability. A similar approach was taken in the analysis of antibody‐secreting cell populations in systemic lupus erythematosus (SLE).12 Lineage analysis (rooted B‐cell lineage trees) was performed using IgTree,13 kindly provided by Prof. Ramit Mehr.
Mass spectrometry of CSF and serum IgG serum and overlap analysis to peripheral Ig proteomes
CSF and serum IgG (0.5–2.0 mL) peptides were analyzed by mass spectrometry as described previously.6 Heavy‐chain proteins were digested with trypsin and the tryptic mixtures were extracted in 1% formic acid/50% acetonitrile (ACN). Samples were analyzed on a linear trap quadropole (LTQ) Orbitrap Velos mass spectrometer (Thermo Fisher Scientific) coupled to an Eksigent nanoLC‐2D system through a nanoelectrospray LC−MS interface using a 90 min gradient from 6% to 40% ACN. Peptide fragmentation was performed in a higher energy collisional dissociation cell with normalized collision energy of 40%, and tandem mass spectra were acquired in the Orbitrap mass analyzer. Data acquisition was performed using Xcalibur software (version 2.0.6). Tandem mass (MS/MS) spectra were converted into mgf files using an in‐house script. Mascot (version 2.2; Matrix Science Inc., London, UK) was used to perform database searches against our database containing translated blood B‐cell VH transcriptome CDR3 parts (+10 amino acids in front and after the CDR3 part). Peptide tolerance was set at ± 15 ppm with an MS/MS tolerance of ± 0.1 Da from spectra. Scaffold (version4, Portland, OR, USA) was used to validate each individual peptide and protein identifications. Peptide identifications were accepted at a ˃95.0% probability, protein identifications at a ˃90.0% probability.
Statistics
For comparison of the parameters between different B‐cell subsets the nonparametric Kruskal–Wallis test with multiple comparison correction (Dunn's procedure) was applied. For comparison of the percentage distribution between fresh and frozen B cells, a paired T‐test was used. The mutational distance between the different blood B‐cell populations and the CSF counterparts were analyzed by Mann–Whitney test, differences in the overlap between blood Ig sequences and CSF and serum Ig peptides were determined by Wilcoxon rank‐sum test.
Results
CSF and blood Ig repertoire analysis
In order to assess CSF VH transcriptome repertoires, single CD19 + CD138 + plasmablasts were recovered from the CSF of seven NMO patients following relapse. On average, we obtained CSF VH sequences from 55 cells per patient (range: 32–104 sequences), and most (mean: 47.3%; range: 27.3–81.0%) were contained within clonal populations. Paired heavy and light chains were used to generate recombinant monoclonal antibodies (rAbs) and tested for AQP4 reactivity using a cell‐binding assay. Sixty‐five percent of all recombinant antibodies and 74% of CSF plasmablast clones were AQP4‐specific.
To determine the distribution of VH sequences clonally related to AQP4‐specific CSF plasmablasts within the peripheral B‐cell pool, naïve (CD19 + CD20 + CD27‐IgD+), memory (CD19 + CD20 + CD27 + ), plasmablast (CD19 + CD20‐CD27 + IgD‐CD38 + +), and double negative (CD19 + CD20‐CD27‐IgD‐) B cells were sorted from peripheral blood of the same patient (Fig. 1), and immunoglobulin VH sequence repertoires generated using next‐generation deep sequencing. On average, 52789 sequences/ blood population (range: 107–113565 sequences) were processed through the pRESTO bioinformatics pipeline and the ensuing repertoires analyzed in consecutive steps to determine the total number of unique sequences (Table S1), VH family affiliation, Ig subclass distribution, and CDR3 diversity (Fig. S1). The VH family distribution of the naive B cells approximated the expected germline frequency with a slight overrepresentation of the VH5 family across the different populations (average distribution of VH1 19%, VH2 5%, VH3 34%, VH4: 23%, VH5: 12%). VH family distributions were similar to those reported by Glanville et al.14, 15 and demonstrated no significant differences between peripheral blood B‐cell populations (Fig. S1). The Ig isotypes were restricted to IgD (40%) and IgM (60%) in the naïve B‐cell population; whereas, in the CD27 + memory B‐cell population, the distribution was 40% IgM, 40% IgA, and 20% IgG. The distribution of Ig isotypes in the DN B‐cell and plasmablast populations showed no significant differences (Fig. S1); however, in one patient, a larger fraction of CD27 + memory, DN B cells, and plasmablasts expressed IgA. The absolute number of clonotypes (≥2 sequences) was lowest in the DN B‐cell population. The diversity index for the CDR3 regions (Shannon‐Wiener index) was also lower in the DN B‐cell population, but the differences did not reach statistical significance (Fig. S1).
Overlap between CSF and peripheral B‐cell Ig repertoires on Ig transcriptome level
We compared the CSF and peripheral blood Ig VH sequence repertoires in each of the seven NMOSD patients. For patient ON09‐03, additional Ig VH sequences from splenic B cells were available for comparison. B‐cell VH sequences were deemed related when they had identical VJ gene segments, identical CDR3 length, and at least a 75% match in the CDR3 nucleotide sequence. Prior analysis of CSF plasmablast clones in our NMOSD patients had demonstrated a variation of up to 75% at the nucleotide level.
We identified an overlap of clonally related B cells across the blood‐brain barrier (Table S2) in five out of seven patients. On average, 9% of the CSF Ig sequences could be linked to peripheral Ig sequences. In total, across all patients, 101 peripheral Ig VH sequences were identified that overlapped with 36 CSF Ig VH transcripts (Table 1). These sequences were contained within 14 clonal populations. Overlapping B‐cell sequences were class‐switched (isotypes: IgG 52%, IgA 31%, IgM 17%) and detected within each of the peripheral blood B‐cell compartments: memory, DN memory, and plasmablast. Within the splenic VH repertoire of patient ON09‐03, only memory B cells and plasmablasts matched CSF VH sequences. Peripheral blood VH sequences clonally related to CSF AQP4‐specific VH sequences were also divided among the peripheral blood memory, DN memory, and plasmablast compartments, the majority being recovered from the CD27 + memory cell compartment (Table 1). While the largest fraction of clonally related sequences was identified in the plasmablast compartment, only 7% were AQP4‐specific. In contrast, 39% and 100% of the CD27 + memory and DN memory B cells, respectively, were AQP4‐specific. No AQP4‐specific transcript was identified within the splenic VH repertoire.
Table 1.
Peripheral blood B cell clones matching CSF Ig sequences
| Memory B cells | Double negative B cells | Plasmablasts | |
|---|---|---|---|
| Number of sequencesa (% Total) | |||
| All clonally related sequences | 38 (38) | 5 (5) | 58 (57) |
| AQP4 + clonally related sequences | 15 (62) | 5 (21) | 4 (17) |
| Average mutational distanceb | |||
| All clonally related sequences | 71 | 10 | 64 |
| AQP4 + clonally related sequences | 16 | 10 | 13 |
Total number of sequences for the five NMOSD patients with overlapping peripheral and CSF VH sequences.
Mutational distance is calculated as the number of nucleotide differences in VH sequences between clonally related peripheral and CSF B cells.
Lineage analysis of overlapping CSF and blood Ig sequences
To further define B‐cell maturation and migration pathways between the periphery and the CNS, we evaluated the patterns of somatic hypermutations among clonally related CSF and peripheral blood VH sequences. Based on the number of shared mutations compared to germline, hierarchical maturation trees were developed using IgTree. Most of the clonally related blood and CSF VH sequences were linked by an unrecovered precursor. In a few cases, however, blood and CSF VH sequences were directly linked. In most instances, blood VH sequences were closer to germline (Fig. 2). VH sequences derived from DN B cells often preceded related blood memory and plasmablast VH sequences in hierarchical trees indicating that DN B cells might develop into these cell types. In one Ig tree, we observed a blood plasmablast VH sequence directly followed by another mutated CSF plasmablast sequence. Since plasmablasts do not undergo further somatic hypermutation, it is likely that a common precursor for both sequences failed to be sampled.
Figure 2.

Hierarchical maturation diagrams (trees) of related CSF and blood VH sequences. Trees are rooted to germline sequences (GL), and the number of mutations between VH sequences are noted (a single mutation is left blank). Unknown intermediates are symbolized as white circles. (A) The most common representative VH tree showing an unknown intermediate linking a CSF plasmablast to peripheral B cells (12/14 VH Trees). (B) A single peripheral blood B‐cell VH sequence directly links to a clonally related CSF plasmablast (2/14 Ig Trees). (C) CSF plasmablast VH sequence hierarchically linked to a peripheral blood VH sequence (1/14 Ig Trees). GL, Germline; MEM, memory B cell; DN, double negative B cells; PBL, plasmablast, SP PBL, spleen plasmablast.
We calculated the shortest mutational distance between the different peripheral blood B‐cell VH sequences and their matching CSF counterparts (Table 1). The mutational distance between peripheral DN B cell and CSF plasmablast VH sequences was lower than related, overlapping plasmablast VH sequences (P = 0.001) and memory B‐cell sequences (P = 0.06). Overlapping peripheral DN B cells were restricted to AQP4‐specific sequences.
Overlap between peripheral blood Ig transcriptome and CSF/blood Ig proteome
We next examined the contribution of different peripheral blood B‐cell populations to their paired blood and CSF Ig peptide libraries (Ig proteome). We matched serum Ig peptides to the translated CDR3 region of peripheral blood B‐cell VH sequences (Ig transcriptome) from the same patient (Table 2). On average, only 1% of the PB Ig sequences showed an exact match to the corresponding CSF and serum Ig peptides. Most, approximately two‐thirds, of the matching Ig peptides were contained within the serum Ig fraction. The remainder was confined to the CSF Ig peptides, and only a small fraction was observed within both compartments (3.5%) (Table 2). The peripheral blood plasmablast Ig transcriptome repertoire showed the greatest number of VH sequences matching both serum and CSF CDR3 Ig peptides (30%) (Table 2); however, differences to the other B‐cell populations were not statistically significant. Interestingly, while most peripheral blood VH sequences predominantly overlapped with the serum Ig peptides, an equal number of CSF and blood peptides aligned to the DN B cells VH sequences. None of the CSF/blood proteome Ig peptides could specifically be aligned to sequences of AQP4‐specific Igs.
Table 2.
Percentage CSF/Blood Ig peptides matching peripheral blood VH repertoire sequences
| Naïve Ig sequences | Memory Ig sequences | Double negative Ig sequences | Plasmablasts Ig sequences | Total Ig sequences | |
|---|---|---|---|---|---|
| CSF Ig proteome (# peptides) | 4.9% (10) | 7.9% (16) | 10.9% (22) | 11.4% (23) | 35.1% (71) |
| CSF and blood Ig proteome (# peptides) | 0.5% (1) | 1.0% (2) | 0.5% (1) | 1.5% (3) | 3.5% (7) |
| Blood Ig proteome (# peptides) | 18.3% (37) | 13.4% (27) | 10.9% (22) | 18.8% (38) | 61.4% (124) |
Two hundred and two of 6965 Ig peptides (2.9%) could specifically be aligned to the CDR3 domain sequence of 17482 Ig transcripts (8458 ‐ naive; 4384 ‐ memory; 2127 ‐ double negative; 2513 ‐ plasmablasts).
Abbreviations: Ig ‐immunoglobulin; CSF ‐ cerebrospinal fluid.
Double negative B cells are up‐regulated in the peripheral blood of NMOSD patients
Previous studies have documented elevated circulating plasmablasts in NMOSD patients.16, 17 Due to the close linear relationship between clonal populations of circul ating peripheral blood DN B cells and AQP4‐reactive CSF plasmablasts, we examined the fraction of DN B cells in the peripheral blood of 11 NMOSD patients (seven in this study), 15 multiple sclerosis (MS) patients, and 15 healthy controls by FACS (Fig. 3, Table S3). The fraction of DN B cells in NMOSD patients was significantly elevated when compared to MS patients and healthy controls. While we observed elevated circulating plasmablasts in NMOSD patients relative to healthy controls, the plasmablast fraction was not different from that observed in the MS patient cohort. The fraction of memory B cells in NMOSD patients was significantly lower than the other groups.
Figure 3.
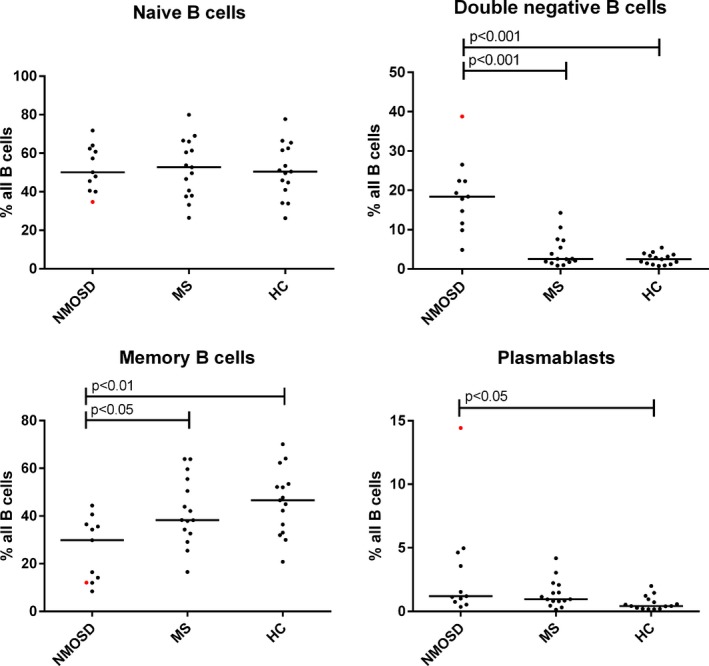
Percentage distribution of peripheral blood B‐cell populations (naïve, memory, and double negative B cells, plasmablasts) in neuromyelitis spectrum disease (NMOSD), healthy controls (HC), multiple sclerosis (MS) subjects. Red dots indicate NMOSD patient treated with rituximab.
Discussion
B cells may play multiple roles in NMOSD pathogenesis.1 We used Ig transcriptome and Ig proteome analyses to evaluate the clonal relationship between different peripheral B‐cell populations and AQP4‐specific B cells in the CSF of active seropositive NMO patients. In contrast to prior studies,16, 17 we identified clonal relationships between CSF B cells and circulating class‐switched memory, DN memory, and plasmablast populations. We also observed an overlap between CSF B cells and memory B cells and plasmablasts from the spleen of one patient. Lineage analyses of related peripheral blood and CSF B cells provide indirect evidence of possible B‐cell maturation into antibody‐secreting cells (ASCs) within the CSF of NMOSD patients.
Our results show some similarities to a recent analysis of CSF and peripheral blood B cells in MS patients where clonal relationships were observed between CSF B cells and peripheral (class‐switched) memory, double negative B cells and plasmablasts.18 In both disorders, the patterns of mutations in VH sequences in bicompartmental clones are consistent with a migration of multiple antigen‐experienced peripheral B‐cell populations across the blood‐brain barrier. In MS, lineage analysis suggests a bidirectional exchange of B‐cell clones.18, 19, 20 While B cell maturation into ASCs occurs in both compartments, antigen‐specific maturation predominates in the periphery.20 These analyses and this study, however, are hampered by relatively limited sampling of peripheral blood and CSF VH sequences. The result is incomplete lineage trees that might skew the apparent relationship of clonal sequences. In some lineages, we observed peripheral blood B cells that were immediately linked to their CSF counterparts, supporting direct migration of B cells from the periphery to the CSF. However, in most cases, the bicompartmental NMOSD B‐cell clones are connected by an unknown precursor. While this is most likely the result of limited sampling of a unidirectional migratory stream from the periphery to the CNS, we cannot exclude other mechanisms, such as the release of multiple independent clones from recent germinal center activity, activation of independent precursors in both compartments, recirculation of B cells between the CNS and periphery, or contemporaneous compartmental‐specific maturation of a common progenitor.
In previous work, we demonstrated that a substantial proportion of CSF Ig transcripts and peptides in acute relapsing NMO patients were exclusive to the CNS6. The current analysis of the overlap of peripheral blood VH sequences with the CSF and blood Ig peptides further supports our conclusion that the humoral immune response in active NMO patients involves a migration and differentiation of AQP4‐specific antibody‐producing cells in the CNS. In addition to a direct migration of AQP4 autoantibody‐secreting plasmablasts into the CNS compartment, we observed connections between CSF Ig peptides and peripheral blood memory, DN memory, and naïve VH transcripts indicating that additional maturation and differentiation of peripheral blood B cells into ASCs likely occurs within the CNS. Interestingly, a high degree of connectivity was observed between ASCs and activated naïve B cells in active systemic lupus erythematosus patients.12 Our data suggest that naïve B cells may also be actively recruited into the enhanced ASC response associated with NMOSD clinical relapse.16 As circulating IgG is the product of long‐lived bone marrow‐resident plasma cells, these newly emerging AQP4‐specific B‐cell clones likely contribute little to the serum AQP4‐IgG that passively crosses the blood–brain barrier during relapse. Indeed, only a small proportion of CSF and blood IgG peptides matched peripheral blood VH sequences within our NMOSD patients.
Analysis of the origin and trafficking of AQP4‐specific CSF B cells reveal a potential role for class‐switched CD27‐IgD‐, double negative B cells in the proinflammatory B‐cell response during active disease. Although we identified AQP4‐specific CSF B cells related to peripheral memory B cells, DN B cells, and plasmablasts, bicompartmental DN (CD27‐IgD‐) clones were restricted to AQP4‐specific CSF B cells and peripheral DN B cells showed the closest mutational distance to their CSF counterparts. Indeed, in one cluster, a DN B cell was the direct precursor of a CSF plasmablast. DN B cell VH sequences that match CDR3 Ig peptides are equally distributed between CSF and blood, whereas VH sequences from other B‐cell populations preferentially correspond to serum‐derived peptides. The strong connection between AQP4‐specific peripheral blood DN B cells and CSF plasmablasts suggests that DN B cells may play a role in initiating NMOSD disease activity.
DN B cells are thought to represent a transient memory subset that may originate from incomplete germinal centers or extrafollicular reactions21, providing an expanded source of IgG+ plasmablasts.22 CD27 + and CD27‐ DN class‐switched memory cells share overlapping clones23, and the transition of B cells between these memory pools may be due to downregulation of CD27 secondary to chronic antigen stimulation.24 Recent work in rheumatoid arthritis indicates that the expansion of relatively short‐lived DN B cells reflects a state of chronic B‐cell hyperactivity that is responsive to IL‐6R inhibition.25 IL6R inhibition has shown early promise for the treatment of NMOSD26 and is currently under investigation in Phase 3 clinical trials (NCT02073279, NCT02028884).
As observed in SLE, the percentage of DN B cells is significantly elevated in the peripheral blood of NMOSD patients, and the DN B‐cell VH repertoire shows a lower CDR3 diversity.27 This suggests that SLE and NMOSD may share similar underlying immune pathophysiology. An enhanced ASC response is presumed to drive pathologic autoantibody production in SLE, and DN B‐cell expansion is reported to resolve after B‐cell depletion therapy.28 We speculate that a similar vigorous ASC response is resulting in the release and migration of AQP4‐specific DN B cells from disease‐relevant germinal center reactions to the CNS and may provide some explanation for the reported efficacy of B‐cell depletion in NMOSD.29 Interestingly, CD20 is low to absent on the surface of NMOSD DN B cells raising the possibility that asynchronous depletion of this subpopulation of B cells following anti‐CD20 therapy may contribute to paradoxical NMO relapses after the infusion of rituximab.30 Interestingly, in study patient ON09‐527, who was relapsing on rituximab therapy, CD20‐ DN B cells were the most abundant circulating peripheral B‐cell population (Fig. S1).
The overlap of CSF plasmablasts with B cells in both the peripheral blood memory and plasmablast compartments contrasts with results previously reported by Chihara and colleagues (2013). Their analysis may have been skewed due to the limited number of Ig transcripts obtained from single peripheral blood and CSF plasmablasts, the small number of NMOSD patients surveyed, and the focused attention on the expanded peripheral blood plasmablast compartment. In addition, the identification of identical VDJ clones in multiple NMOSD patients raises concerns for cross‐contamination.17 In contrast, our extensive sampling of the peripheral B‐cell compartment indicates that during disease activity, AQP4‐specific CSF B cells transit to the CSF from multiple postgerminal center populations. The relationship between these compartments may be fluid. Indeed, memory B cells appear to transit between the CD27 + and CD27‐ compartments.31 DN memory B cells may acquire CD27 following polyclonal stimulation with CpG ligand32 and may play an important role in maintaining the CD27 + memory population. It remains unclear when DN peripheral B cells begin to express CD27 after transiting to the CNS. Evaluating the surface markers of circulating DN B cells in NMOSD patients may provide important clues on their role in populating the CNS compartment during NMOSD flares.
Our analysis of the overlap between the CSF and peripheral blood B cells in active NMOSD patients was limited by several issues. First, our CSF VH repertoires were restricted by the single‐cell approach for identifying CSF B cells with AQP4 specificity. Second, we used frozen PBMCs for generation of Ig libraries which limited the recovery of B cells and VH transcripts from individual peripheral B‐cell pools. Third, while lineage analysis provides a unique tool to study trafficking patterns of B cells in NMOSD and MS, mass sequencing of B cell Ig transcripts only allows a snapshot of the B‐cell repertoire at a certain time and does not allow a fully representative picture of the dynamic relationship among transiting B cells. Lastly, despite safeguards such as high fidelity PCR and UMIs,33 sequencing errors and overamplification of transcripts may result in artificial or overrepresented B‐cell clusters or clones. Further advances in sequencing technology, longitudinal analyses of blood /CSF Ig repertoires, and sampling of relevant lymph nodes could help to overcome these limitations and provide a clearer picture of B‐cell maturation in NMOSD.
Despite these limitations, our deep sequence analyses, in combination with our recent comparison of the CSF and peripheral Ig transcriptomes and proteomes,6 provide multiple lines of evidence that the primary autoimmune response against AQP4 initiates with the release of AQP4‐specific memory B cells, DN B cells, and plasmablasts into the peripheral blood (Fig. 4). Lineage analysis suggests that DN B cells may represent the most direct migratory population; however, we cannot exclude that intermediates in the CD27 + memory and plasmablast populations were missed in our small sample of peripheral blood. Polyclonal stimulation in the periphery or CNS may be a primary mechanism for CD27 acquisition in the DN population and may explain the exacerbation of NMO disease activity associated with inter‐current illness. Clonally related CD27 + memory B cells and plasmablasts B cells may migrate to peripheral plasma cell niches in the bone marrow where they may produce serum AQP4‐IgG. Since a large fraction of the CSF Ig proteome is composed of sequences unique to the CSF Ig transcriptome, migrating DN memory B cells may represent an important proinflammatory B‐cell population involved in the initiation of new CNS disease activity in NMOSD.
Figure 4.

B‐cell trafficking and antibody production in NMOSD. Antigen‐experienced (AQP4‐reactive) B cells migrate between both compartments. While some B cells undergo clonal expansion and affinity maturation in the CNS, most of the AQP4‐specific germinal center reactions take place in the periphery. AQP4‐specific antibodies are produced locally by antibody secreting cells in the CNS; however, a substantial component transits from the serum through an open blood–brain barrier.
Conflict of Interest
None declared.
Supporting information
Figure S1. VH family, Ig subclass, Shannon‐Wiener index, and clonotype number in peripheral blood B cell populations.
Table S1. Number of sorted cells and processed Ig sequences for each patient and B cell population.
Table S2. VH sequence CDR3 overlap between CSF, blood and spleen B cell populations.
Table S3. Demographic and clinical data on patients for additional FACS analysis.
Acknowledgments
M. C. Kowarik was supported by the Deutsche Forschungsgesellschaft (DFG, Ko 4367/1‐1) and SyNergy. J.L.B. and G.P.O. were supported by the National Institutes of Health (EY022936, UM1AI110498, NS072141) and the Guthy‐Jackson Charitable Foundation.
References
- 1. Bennett JL, Lam C, Kalluri SR, et al. Intrathecal pathogenic anti‐aquaporin‐4 antibodies in early neuromyelitis optica. Ann Neurol 2009;66:617–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lennon VA, Wingerchuk DM, Kryzer TJ, et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet 2004;364:2106–2112. [DOI] [PubMed] [Google Scholar]
- 3. Lennon VA, Kryzer TJ, Pittock SJ, et al. IgG marker of optic‐spinal multiple sclerosis binds to the aquaporin‐4 water channel. J Exp Med 2005;202:473–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ratelade J, Zhang H, Saadoun S, et al. Neuromyelitis optica IgG and natural killer cells produce NMO lesions in mice without myelin loss. Acta Neuropathol 2012;123:861–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Papadopoulos MC, Bennett JL, Verkman AS. Treatment of neuromyelitis optica: state‐of‐the‐art and emerging therapies. Nat Rev Neurol 2014;10:493–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kowarik MC, Dzieciatkowska M, Wemlinger S, et al. The cerebrospinal fluid immunoglobulin transcriptome and proteome in neuromyelitis optica reveals central nervous system‐specific B cell populations. J Neuroinflammation 2015;12:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Owens GP, Ritchie AM, Burgoon MP, et al. Single‐cell repertoire analysis demonstrates that clonal expansion is a prominent feature of the B cell response in multiple sclerosis cerebrospinal fluid. J Immunol 2003;171:2725–2733. [DOI] [PubMed] [Google Scholar]
- 8. Crane JM, Lam C, Rossi A, et al. Binding affinity and specificity of neuromyelitis optica autoantibodies to aquaporin‐4 M1/M23 isoforms and orthogonal arrays. J Biol Chem 2011;286:16516–16524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kyu SY, Kobie JH, Yang H, et al. Frequencies of Human Influenza‐specific Antibody Secreting Cells or Plasmablasts post Vaccination from Fresh and Frozen Peripheral Blood Mononuclear Cells. J Immunol Methods 2009;340:42–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Van der Heiden JA, Yaari G, Uduman M, et al. pRESTO: a toolkit for processing high‐throughput sequencing raw reads of lymphocyte receptor repertoires. Bioinformatics 2014;30:1930–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Alamyar E, Duroux P, Lefranc MP, Giudicelli V. IMGT((R)) tools for the nucleotide analysis of immunoglobulin (IG) and T cell receptor (TR) V‐(D)‐J repertoires, polymorphisms, and IG mutations: IMGT/V‐QUEST and IMGT/HighV‐QUEST for NGS. Methods Mol Biol 2012;882:569–604. [DOI] [PubMed] [Google Scholar]
- 12. Tipton CM, Fucile CF, Darce J, et al. Diversity, cellular origin and autoreactivity of antibody‐secreting cell population expansions in acute systemic lupus erythematosus. Nat Immunol 2015;16:755–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Barak M, Zuckerman NS, Edelman H, et al. IgTree: creating Immunoglobulin variable region gene lineage trees. J Immunol Methods 2008;338(1–2):67–74. [DOI] [PubMed] [Google Scholar]
- 14. Glanville J, Zhai W, Berka J, et al. Precise determination of the diversity of a combinatorial antibody library gives insight into the human immunoglobulin repertoire. Proc Natl Acad Sci USA 2009;106:20216–20221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Glanville J, Kuo TC, von Büdingen HC, et al. Naive antibody gene‐segment frequencies are heritable and unaltered by chronic lymphocyte ablation. Proc Natl Acad Sci USA 2011;108:20066–20071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chihara N, Aranami T, Sato W, et al. Interleukin 6 signaling promotes anti‐aquaporin 4 autoantibody production from plasmablasts in neuromyelitis optica. Proc Natl Acad Sci USA 2011;108:3701–3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chihara N, Aranami T, Oki S, et al. Plasmablasts as migratory IgG‐producing cells in the pathogenesis of neuromyelitis optica. PLoS ONE 2013;8:e83036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Palanichamy A, Apeltsin L, Kuo TC, et al. Immunoglobulin class‐switched B cells form an active immune axis between CNS and periphery in multiple sclerosis. Sci Transl Med 2014;6:248ra106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. von Büdingen HC, Kuo TC, Sirota M, et al. B cell exchange across the blood‐brain barrier in multiple sclerosis. J Clin Invest 2012;122:4533–4543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stern JN, Yaari G, Vander Heiden JA, et al. B cells populating the multiple sclerosis brain mature in the draining cervical lymph nodes. Sci Transl Med 2014;6:248ra107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sanz I, Wei C, Lee FE, Anolik J. Phenotypic and functional heterogeneity of human memory B cells. Semin Immunol 2008;20:67–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fleischer SJ, Giesecke C, Mei HE, et al. Increased frequency of a unique spleen tyrosine kinase bright memory B cell population in systemic lupus erythematosus. Arthritis Rheumatol 2014;66:3424–3435. [DOI] [PubMed] [Google Scholar]
- 23. Wu YC, Kipling D, Dunn‐Walters DK. The relationship between CD27 negative and positive B cell populations in human peripheral blood. Front Immunol 2011;26:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. De Jong R, Brouwer M, Hooibrink B, et al. The CD27‐ subset of peripheral blood memory CD4 + lymphocytes contains functionally differentiated T lymphocytes that develop by persistent antigenic stimulation in vivo. Eur J Immunol 1992;22:993–999. [DOI] [PubMed] [Google Scholar]
- 25. Mahmood Z, Muhammad K, Schmalzing M, et al. CD27‐IgD‐ memory B cells are modulated by in vivo interleukin‐6 receptor (IL‐6R) blockade in rheumatoid arthritis. Arthritis Res Ther 2015;14:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ayzenberg I, Kleiter I, Schröder A, et al. Interleukin 6 receptor blockade in patients with neuromyelitis optica nonresponsive to anti‐CD20 therapy. JAMA Neurol 2013;70:394–397. [DOI] [PubMed] [Google Scholar]
- 27. Wei C, Anolik J, Cappione A, et al. A new population of cells lacking expression of CD27 represents a notable component of the B cell memory compartment in systemic lupus erythematosus. J Immunol 2007;178:6624–6633. [DOI] [PubMed] [Google Scholar]
- 28. Anolik JH, Barnard J, Cappione A, et al. Rituximab improves peripheral B cell abnormalities in human systemic lupus erythematosus. Arthritis Rheum 2004;50:3580–3590. [DOI] [PubMed] [Google Scholar]
- 29. Kim SM, Park J, Kim SH, et al. Factors associated with the time to next attack in neuromyelitis optica: accelerated failure time models with random effects. PLoS ONE 2013;16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nakashima I, Takahashi T, Cree BA, et al. Transient increases in anti‐aquaporin‐4 antibody titers following rituximab treatment in neuromyelitis optica, in association with elevated serum BAFF levels. J Clin Neurosci 2011;18:997–998. [DOI] [PubMed] [Google Scholar]
- 31. Pauli NT, Henry Dunand CJ, Wilson PC. Exploiting human memory B cell heterogeneity for improved vaccine efficacy. Front Immunol 2011;2:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bernasconi NL, Traggiai E, Lanzavecchia A. Maintenance of serological memory by polyclonal activation of human memory B cells. Science 2002;298:2199–2202. [DOI] [PubMed] [Google Scholar]
- 33. Tan AY, Michaeel A, Liu G, et al. Molecular diagnosis of autosomal dominant polycystic kidney disease using next‐generation sequencing. J Mol Diagn 2014;16:216–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. VH family, Ig subclass, Shannon‐Wiener index, and clonotype number in peripheral blood B cell populations.
Table S1. Number of sorted cells and processed Ig sequences for each patient and B cell population.
Table S2. VH sequence CDR3 overlap between CSF, blood and spleen B cell populations.
Table S3. Demographic and clinical data on patients for additional FACS analysis.