

Abstract
Inclusion Body Myositis (IBM) is a relatively common acquired inflammatory myopathy in patients above 50 years of age. Pathological hallmarks of IBM are intramyofiber protein inclusions and endomysial inflammation, indicating that both myodegenerative and inflammatory mechanisms contribute to its pathogenesis. Impaired protein degradation by the autophagic machinery, which regulates innate and adaptive immune responses, in skeletal muscle fibers has recently been identified as a potential key pathomechanism in IBM. Immunotherapies, which are successfully used for treating other inflammatory myopathies lack efficacy in IBM and so far no effective treatment is available. Thus, a better understanding of the mechanistic pathways underlying progressive muscle weakness and atrophy in IBM is crucial in identifying novel promising targets for therapeutic intervention. Here, we discuss recent insights into the pathomechanistic network of mutually dependent inflammatory and degenerative events during IBM.
Introduction
Inclusion body myositis (IBM) is a progressive slow‐onset inflammatory myopathy that is characterized by the concomitant presence of multi‐focal myofiber‐surrounding lymphocytic infiltrates as well as vacuolar myodegeneration.1, 2, 3, 4 Together with dermatomyositis (DM), polymyositis (PM), necrotizing autoimmune myositis (NAM) and overlap myositis (OM), IBM belongs to the heterogenous group of inflammatory myopathies and, amongst individuals 50 years of age and older, it is considered as a relatively frequent disorder.3, 5 The underlying interrelationship between the inflammatory component of the disease and the observed multi‐protein aggregation remains elusive and subject to vigorous debate.3, 6, 7, 8 Unlike other inflammatory myopathies, IBM presents mainly refractory toward immunosuppressive therapy and at present, there is no effective treatment available.5, 9, 10 In this review, we will focus on the current knowledge about the interrelationship of inflammatory and myodegenerative pathomechanisms in IBM.
Clinical Presentation
The disease commonly commences slowly‐progressive, sometimes over decades. The clinical presentation is heterogenous and at times difficult to distinguish from other inflammatory myopathies (muscle weakness and atrophy), motor‐neuron disease (asymmetry), and muscular dystrophies (slowly progressive disease).3 In two large observational studies, the mean age of onset has been reported to be 59 ± 9 and 61 years, respectively.9, 11 The cardinal symptom of this highly debilitating disease is the late‐onset steady acquisition of muscular weakness and atrophy over a long period of time whilst sensory function is completely preserved. The decline of muscle strength ranges between 3.5 and 5.5% per annum.9, 11 Unlike other myopathies, during which proximal muscles are initially affected, IBM shows early involvement of distal muscles. Classical manifestation patterns frequently include the quadriceps, deep finger flexors, foot extensors, and often presents asymmetrically at the beginning. Frequent falls may be an early clinical sign of IBM. Paraspinal and axial muscles may be affected, resulting in head drop and camptocormia.12 Depending on the study, oropharyngeal dysphagia is reported in up to 40–86% of IBM cases, mostly due to upper esophageal sphincter dysfunction.13, 14, 15, 16 It develops insidiously, leading to frequent choking episodes and is, alongside pneumonia as a result of immobility, considered to be a potentially fatal complication of IBM. Importantly, dysphagia may be an isolated, initial manifestation and IBM should be considered by the examining physician as differential diagnosis for new onset of dysphagia in the elderly.13, 14, 17, 18, 19, 20, 21 Early in the disease course, tendon reflexes remain unaffected, however, hyporeflexia may occur at later stages of the disease due to significant muscle atrophy.19 The heart muscle remains usually unaffected and the incidence of cardial muscle abnormalities does not exceed the expected incidence for the respective age group.22 There is no evidence for increased cancer risk in IBM patients.23
Epidemiology
IBM affects males more frequently (3:1), shows an overall prevalence of approximately 4‐15/1000000 (35‐71/1000000 > age 50, respectively), is noticably frequent in Western Australia, Japan, Norway, Olmsted County (Minnesota, USA), and is especially rare in Turkey and India.3, 24, 25, 26, 27, 28, 29, 30, 31 In Japan, the number of diagnosed IBM cases has steadily increased since 1991, whereas the number of PM cases has remained constant.32 Prolonged life span and concomitant increase of the fraction of elderly people as well as westernization of dietary habits in Japan might be contributing factors for this observation.33 Although it was suggested that mortality is increased in IBM patients, solid evidence is still insufficient and the matter remains subject to larger studies.17, 34
Current data prompt that IBM meets the criteria to be catagorized as an orphan disease. However, it is likely that the prevalence of IBM is still underestimated. Although heavily debated, it is conceivable that a significant number of patients diagnosed with PM might in fact suffer from IBM.35, 36, 37, 38 Aside from erroneous diagnoses, the slowly progressive nature of the disease course and the heterogenicity in its clinical presentation make the condition prone to delayed diagnosis. Increasing awareness and continous efforts to optimize diagnostic criteria for IBM are of utmost importance in ensuring ample care to patients.
Diagnosis
The chronic disease progression of IBM makes it challenging to detect the condition at an early time point and on average there is a 5‐year delay in diagnosis.9, 39 Creatine kinase levels in serum can be normal to only mildly elevated and will not exceed 10‐fold increase above the upper limit of normal. Muscle biopsies of affected areas typically show CD8+ T cells surrounding nonnecrotic, healthy appearing muscle fibers that express major histocompatibility complex (MHC) class I. Additionally, ragged‐red‐, ragged‐blue‐ and cytochrome oxidase‐negative fibers, as well as autophagic vacuoles and congophilic amyloid deposits are regularly observed.3
The original diagnostic criteria according to Griggs et al. strongly relied on histopathological features of the disease.40 However, it is now apparent that a given muscle biopsy will rarely show all pathological changes that go along with IBM. Basing a definitive diagnosis on the prerequisite of detecting all formerly described histopathological alterations will likely lead to underdiagnosis of the disease. It has been previously described that some patients that fit clinical categorization of IBM, lack canonical biopsy features of IBM.41 It has become clear that histopathological abnormalities in IBM are likely to appear scattered and patchy in a spatio‐temporal manner. The increasing research efforts over the past 45 years, together with accumulated clinical experience, allows physicians today to reliably diagnose the disease not exclusively due to histopathological changes in muscle biopsies but rather through an integrated approach, using clinical and histological observations alike. Therefore, more recently defined diagnostic criteria do not call for the presence of all typical pathological hallmarks but employ the presence of defined patterns of clinical, laboratory, and histological features to categorize the diagnosis into either clinicopathologically defined IBM, clinically defined IBM or probable IBM.12, 42 One study applying machine learning algorithms to construct data‐derived IBM diagnostic criteria claims that the combinational approach of finger flexor or quadriceps weakness, endomysial inflammation, and either invasion of nonnecrotic muscle fibers or rimmed vacuoles, performed with a 90% sensitivity and 96% specificity among 371 patients.43
Pathomechanisms in IBM
Histopathological hallmarks of IBM muscle feature both myodegenerative multi‐protein aggregates as well as endomysial lymphocytic infiltrates.2, 3, 4 Several lines of evidence suggest that inflammatory mechanisms precede myodegeneration,8 but so far a precise answer and sound evidence is lacking as discussed below in detail. Currently, it remains unresolved and controversially discussed if the inflammatory changes observed in IBM muscle are a direct result of primary myodegeneration or if protein aggregation is secondary to initial inflammatory events. The solution of this conundrum is key to identify an appropriate remedy for this debilitating disease.
Immunopathomechanisms in IBM
Endomysial lymphocytic infiltrations in IBM muscle are usually found at perivascular sites and appear scattered. Similar to PM, the mononuclear infiltrates in IBM predominantly consist of CD8+ cytotoxic T cells (CTLs) surrounding nonnecrotic muscle fibers. More than 30% of all invading cells and around 50% of invading CD8+ T cells depict activation marker positivity.44 Unlike in healthy individuals, scattered clusters of nonnecrotic muscle fibers ectopically express MHC class I molecules in a moderate to strong degree on their surface3, 45 and infiltrating CD8+ T cells form close contacts with these MHC class I expressing fibers (Fig. 1). While a considerable amount of muscle fibers with cytoplasmic abnormalities (such as lined vacuoles) do not express MHC class I, regenerating muscle fibers in IBM muscle do show sarcolemmal expression of this molecule.45 Although macrophages constitute only a minor fraction of the mononuclear infiltrates invading nonnecrotic muscle fibers, they account for up to 80% of the infiltrates surrounding necrotic fibers.44 Distinct from DM but consistent with lymphocytic infiltrates observed in PM, muscle‐invading CD8+ T cells stain positive for pore‐forming and cytolytic molecules such as perforin, granzyme A, and granulysin.46, 47, 48 It has been demonstrated that perforin‐polarization within endomysial CD8+ T cells occurs toward target myofibers indicative of immunosynapse formation and arguing strongly for a possible recognition of specific antigens presented via MHC class I expressing myofibers.49 In line with this, muscle fibers in IBM patients express co‐stimulatory molecules such as ICOS‐L, CD276, and BB1 on their surface.48, 50, 51
Figure 1.
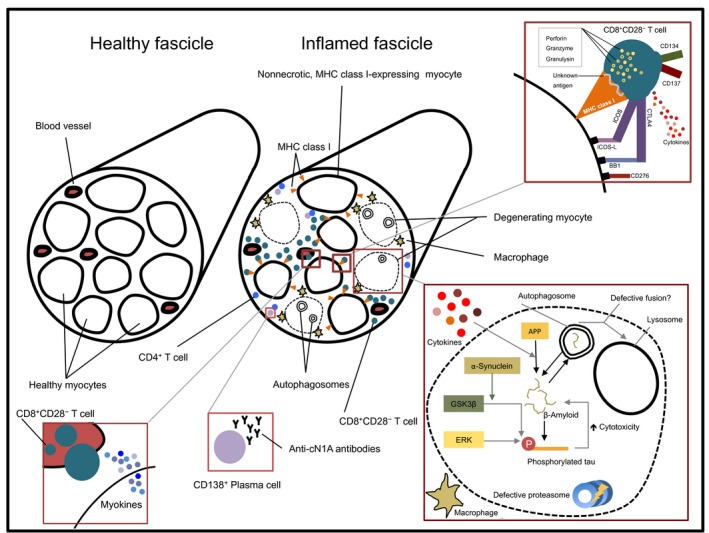
Scheme of the pathological changes in inclusion body myositis compared to healthy muscle. Mainly nonnecrotic, MHC class I‐expressing myofibers are surrounded by invading CD8+ CD28− T cells, which is the predominant immune subset in the endomysial infiltrates. These CD8+ CD28− T cells form immunological synapses with MHC class I bearing myofibers, contain cytolytic proteins, release proinflammatory cytokines, and express costimulatory molecules corresponding to complementary molecules on the surface of myofibers. Additionally, myofibers themselves are immunologically active via releasing myokines. Albeit they are found less frequently, also CD4+ T cells and CD138+ plasma cells are present in the endomysium and may contribute to the myoinflammatory environment. Degenerating myofibers are mainly surrounded by macrophages and contain APP‐derived β‐amyloid and phosphorylated tau. If the disruption of proteostasis by virtue of impaired macroautophagy and defective proteasomal degradation is an upstream event in the pathomechanism of IBM or if it follows the increasing aggregation of aberrant proteins, remains a matter of debate.
Immunohistochemical and RT‐PCR analyses revealed preferential usage of certain CD8+ T cell receptor (TCR) variable segments in endomysial infiltrates compared to peripheral CD8+ T cell‐TCR profiles in IBM patients.52, 53 Although conclusive proof is still lacking, this myo‐peripheral discrepancy of TCR restriction suggests that CD8+ T cells patrol the muscle in a stochastical manner and only upon recognition of their cognate antigen clonally expand in situ. In line with this, endomysial T cells depict expression of proliferation marker Ki‐67 suggestive of a pervasive antigen‐driven response within the muscle compartment.54 However, specific recruitment to the muscle compartment remains a possibility. Using the combination of RT‐PCR, immunohistochemistry and TCR Vβ chain CDR3 spectratyping in three sequential muscle biopsies of three IBM patients, Amemiya et al. found clonal persistence of CD8+ T cells in subsequent muscle biopsies. This is supportive of earlier studies and suggests that IBM might be maintained by a continuous antigen‐driven T cell response.55, 56 Additionally, a more recent CDR3 spectratyping study of CD8+ T cell‐TCR Vβ chains in 12 IBM patients identified Vβ 9, 10, 11, 16, 18, 23, and 24 as subfamilies with the strongest degree in myo‐restriction. Indicative of determinant spreading, follow‐up muscle biopsies (after 12 months) confirmed persisting CD8+ T cell clonality, while the pattern of expanded Vβ subfamilies had changed.57
Viruses
By analogy to numerous autoimmune diseases, a viral contribution to the etiology of IBM has been discussed for as long as the condition has been identified.58, 59 Presently, it is not ultimately clear by which exact mechanism(s) viruses may trigger autoimmunity. Host‐inherent anti‐viral responses comprise a meticulously regulated mounting of the immune system. Erroneous and faulty progression of such antiviral responses may lead to subsequent break‐down of self‐tolerance with concomitant epitope spreading and recognition of auto‐antigens60 (Fig. 2). Other potential virus‐mediated mechanisms include bystander activation and immortalization of low‐affinity autoaggressive effector cells due to unphysiological exposure and subsequent presentation of self‐antigens in the context of a strong antiviral response.60 Despite considerable effort, so far no virus could be isolated and amplified from affected muscle tissue of IBM patients and no conclusive evidence for a viral trigger of this myopathy exists.3, 61 However, an association with human immunodeficiency virus (HIV)62 and human T lymphotropic virus (HTLV)63, 64 seropositivity has been clearly demonstrated and in the case of HIV as many as 10% of infiltrating CD8+ T cells showed specificity for human leukocyte antigen‐A* 0201‐HIV‐gag. Worthy of note, in both cases HIV‐ and HTLV‐derived viral antigens could not be detected in muscle fibers but exclusively on endomysial macrophages.62, 63 Furthermore, an association between hepatitis C virus (HCV) infection and IBM was recently reported in a Japanese case–control study that included 114 IBM and 44 PM patients.65 While the frequency of PM patients that also carried anti‐HCV antibodies was comparable to the general population that of IBM patients was significantly increased.65 However, these data need to be interpreted carefully and it appears to be unlikely that HCV is a key determinant in the development if IBM. The increasing incidence of IBM in Japan is in strong contrast to the decreasing incidence in HCV infections.66, 67, 68 Moreover, countries with relatively high incidence rates of HCV infections belong to the regions that are stricken the least by IBM.26, 30, 61, 69, 70, 71 It is conceivable that nonpersistent contact with a pathogen suffices to trigger autoimmunity.72 At this point, a viral contribution to the etiopathogenesis of IBM cannot be ruled out, yet more conclusive evidence is clearly needed.
Figure 2.
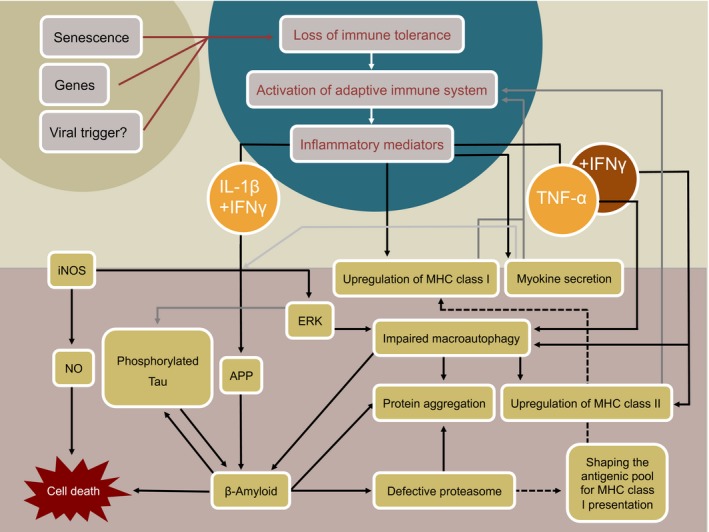
Schematic overview of a possible crosstalk between key pathological mechanisms during IBM. Genetic predisposition, aging, and exposure to a yet unidentified viral trigger may each individually or in combination lead to breakdown of immune tolerance with subsequent activation of the adaptive immune system. Invasion of myoantigen‐specific T cells into the endomysium could establish and maintain a pro‐inflammatory environment in the muscle. Upregulation of MHC class I and II molecules on myofibers and release of myokines in response to inflammation may serve as a feedback loop that helps to perpetuate disease. Disturbed proteostasis may result in response to specific pro‐inflammatory mediators. Conversely, it is argued, that a primary event within myofibers leads to degenerative changes that entail inflammation as a secondary event, yet evidence for this latter scenario is lacking.
CD8+ T cells
With regards to differentiation, surface marker expression and functionality, the CD8+ T cell compartment displays considerable heterogenicity. Short‐telomer‐bearing CD8+CD28− T cells are thought to comprise a highly differentiated oligoclonal subset arising from chronic antigen exposure as hypothesized for IBM19, 49, 73, 74, 75, 76, 77 and several autoimmune conditions are accompanied by increased frequencies of CD8+CD28− T cells.78, 79, 80 Furthermore, muscle‐infiltrating CD8+ T cells in patients suffering from PM and DM have been reported to be mainly CD28−.81 Two recent studies showed that frequencies of highly cytotoxic CD8+CD28− T cells in inflamed muscle and in peripheral blood of IBM patients are significantly increased and their capability to secrete IFNγ was superior compared to healthy controls.82, 83 CD8+CD28− T cells are devoid of costimulatory interaction between CD80:CD28, however, it is reported that CD8+CD28− T cells after CD3 ligation in turn upregulate alternative costimulatory molecules such as inducible costimulator (ICOS), CD134 and CD13784 which could facilitate T cell: muscle fiber interaction and is in keeping with the observed upregulation of ICOS‐L on muscle fibers during IBM.48
Aside from lacking CD28‐expression, expression of the terminally sulfated glycan carbohydrate CD57 is generally regarded as a marker for terminal differentiation and clonal exhaustion on CD8+ T cells and this T cell subset is commonly oligoclonally expanded during conditions of chronic immune activation.85, 86, 87 CD8+CD57+ T cell frequencies are especially increased in the elderly, they have strong cytotoxic potential, high expression of adhesion molecules, strong migratory potential toward nonlymphoid organs and – indicative of a cytotoxic effector memory phenotype – they depict expression of CX3CR1. It is believed that CD8+CD57+ T cells are highly differentiated antigen‐driven effector cells in a state of replicative senescence with limited capacities to proliferate.88, 89, 90, 91, 92 Recent reports, however, suggest that these CD8+CD57+CD28− T cells might comprise a rather heterogeneous group of highly antigen‐experienced cells that, depending on the immunobiological context and stimuli, differ in their susceptibility to apoptosis and their capability to proliferate and expand.84, 93, 94, 95
During T cell‐large granular lymphocytic leukemia (T‐LGL leukemia) clonally expanded large granular CD8+CD57+ CTLs can be found in peripheral blood, spleen and bone marrow.96 Interestingly, a recent study describes a previously undiscovered association between IBM and T‐LGL leukemia.97 T‐LGL leukemia is a rare condition within the spectrum of lymphoproliferative disorders on the interface between neoplasia and extensive antigen‐driven CTL response and is frequently associated with autoimmune disorders.98, 99, 100 According to this new study, the clinical criteria for an expanded LGL population in association to an autoimmune disorder101 were met in more than half of the 38 investigated IBM patients. Greenberg and colleagues controversially argue that, at least in some cases of IBM, an initial autoimmune process might transform into a neoplasia‐like condition with extensive clonal expansion of large granular CTLs resembling those of T‐LGL leukemia.97 This is especially intriguing in light of the fact that IBM is refractory to common immunotherapies. Frequently, this lack of efficacy in targeting the immune system has given rise to the assumption that IBM might be primarily a myodegenerative disorder and the inflammatory component little more than an etiopathogenetic epiphenomenon.7, 102, 103 Although further investigation into the matter is needed, this recent study offers a different narrative, which would have substantial implications toward both the diagnosis and therapy of IBM.104
Other infiltrating immune cells
Aside from the previously described cytotoxic CD8+ T cells, inflammatory infiltrates in IBM additionally harbor myeloid cells,105 plasma cells,106 and CD4+ T cells.44, 48, 82, 83, 107, 108 Early studies have previously revealed that the antigen‐presenting properties of human myocytes exceed the mere bearing of MHC class I molecules. In fact, muscle fibers can be categorized as facultative antigen‐presenting cells, that in a proinflammatory milieu can upregulate MHC class II molecules, express intercellular adhesion molecule (ICAM)‐1 and ICOS ligand (ICOS‐L).108, 109, 110 In line with this, MHC class II expressing muscle fibers are found in IBM.108 In fact, up to 66.7% of muscle fibers in IBM show high positivity for MHC class II as opposed to lower counts in other IIMs (PM: 23.7%; DM: 20%).111 Interestingly, microdissection studies revealed that HLA‐DR, HLA‐DB, and CIITA are predominantly upregulated in infiltrated but not in healthy appearing muscle fibers.112 Comparable to their CD8+ counterpart, CD4+ T cells in IBM are mostly devoid of CD28‐expression, display a striking TCR Vβ restriction, and are expanded in the peripheral blood as well as in inflamed muscle tissue.83 Similar to CTLs, they depict a strong proinflammatory phenotype and cytotoxic properties which might be executed toward MHC class II‐bearing muscle fibers.83 Additionally, local presentation of antigen via professional antigen presenting cells (APCs) or MHC class II bearing muscle fibers has been suggested.105, 108 The pathological role of CD4+CD28− T cells during IBM, therefore, might have been underappreciated so far.
Tregs
FOXP3+CD4+ T regulatory cells (Tregs) constitute a unique lymphocyte subset that holds the capacity to control and limit immune responses mounted against self‐ and foreign antigens in order to retain immune homeostasis and self‐tolerance.113, 114 It has become evident that distinct tissue‐specific Treg populations with unique phenotypical and functional properties exist. In skeletal muscle, they arise from a small pool of resident Tregs and strongly accumulate following muscle damage.115, 116 Under the control of interleukin (IL)‐33, these myophil Tregs execute essential functions in promoting and orchestrating local regeneration upon muscle injury.117, 118 Importantly, they are significantly diminished in aged mice, leading to insufficient muscle repair upon injury.118 A critical role for Tregs during myositis had already been postulated in an experimental autoimmune myositis model during which antibody‐mediated depletion of Tregs leads to significant increase of the histopathological disease score and a more diffuse muscle inflammation pattern.119 On the contrary, in vitro expanded adoptively transferred polyclonal Tregs are able to decrease the severity in this model. These findings are extended by a study that, employing a new model, adoptively transferred FOXP3/synaptotagmin VII double mutant‐derived lymphocytes into RAG‐1−/− mice together with muscle antigens. This entails strong myositis reflected by myofiber infiltrating CD4+ and CD8+ T cells and macrophages. Coadministration of functional Tregs fully protects animals from developing myositis.120 Furthermore, the capacity of Tregs to dampen CD8+ T cell cytotoxicity directed against human myoblasts has been confirmed in vitro121 and immunohistochemical studies in IBM muscle revealed presence of Tregs in close spatial association to other infiltrating mononuclear cells. The amount of Tregs positively correlated with the amount of total CD3+ cells. However, these results are not specific for IBM but could be obtained in PM and DM muscle as well.121 A more recent report shows a significant decrease in circulating Tregs in IBM patients compared to nonmyositic controls.82 Functionality of the remainder of peripheral Tregs with regard to proliferation‐suppression of autologous T cells, however, was unaffected.82 This study also confirmed the previous finding that Tregs are indeed present in inflamed muscle of IBM patients.82 To which degree this report is specific to IBM or if similar results were to be obtained in other inflammatory myopathies remains to be investigated. The physiological role of muscle‐resident Tregs and their contribution during myositis has only begun to unravel. It becomes apparent that few studies so far addressed the presence and subcategorization of CD4+ T cells, including Tregs (which constitute up to 60% of CD4+ T cells in muscle upon injury116), in muscle infiltrates of IBM. Such work could help to better understand the role of these cells.
B cells, plasma cells, and autoantibodies
Despite the often‐proclaimed predominant role of CD8+ T cells in IBM, several reports suggest an underrated humoral component in the immunopathology of inflammatory myopathies. Sera from IBM patients contain increased amounts of muscle antigen‐reactive monoclonal antibodies122 and although CD20+ B cells are scarce, substantial numbers of transcriptionally active CD138+ plasma cells can be detected in inflamed muscle of IBM patients.106 Immunoglobulin heavy chain gene transcript analyses in IBM, PM, and DM revealed that these cells undergo isotype switching, oligoclonal expansion and somatic hypermutation which suggests local affinity maturation of antibodies,123 a process that usually occurs in germinal centers under the aid of follicular dendritic cells (fDCs) and follicular B helper T cells.124, 125, 126 In fact, an early study characterized nodular lymphocytic accumulations in inflamed muscle and found microanatomical organization patterns as well as adhesion molecule expression reminiscent of those in secondary lymphoid organs.127 Others, however, have reported that these nodular accumulations lack B cell follicles and presence of DRC+ fDCs characteristic for lymphoid germinal centers.128
B cell maturation is highly dependent on bidirectional interactions with cognate CD4+ T cells.124, 125, 126 Amongst others, ICOS:B7RP‐1 ligation is essential for the successful execution of this crosstalk.129 In line with this, ICOS+CD4+ T cells have been reported to be present in IBM infiltrates.48 B cell activating factor of the TNF superfamily (Baff) is a cytokine crucial for B cell survival and has been implicated in autoantibody formation in patients suffering from autoimmune diseases.130 Serum levels of Baff are elevated in some patients suffering from IIMs including IBM patients.131 Furthermore, Baff transcripts are markedly increased in muscle tissue from IIM patients compared to nonmyositic controls (IBM>PM>DM).128 However, serum Baff levels were highest in IIM patients that also had detectable levels of anti‐histidyl‐tRNA‐synthetase antibody Jo‐1, an autoantibody that is extremely rare in IBM.132, 133, 134 Additionally, Baff serum levels seem to positively correlate with serum CK levels, which can be normal to only moderately elevated in IBM.131
The presence of specific autoantibodies is not only relevant with regard to possible therapeutic options but has immediate implications for diagnosis. Autoantibodies associated with myositis have been identified in more than half of the patients suffering from myositis.3, 135, 136 In 2011, a previously undetected circulating antibody against a muscle‐derived protein was found in 52% of IBM patients (13/25) but was absent in control individuals (PM, DM, healthy volunteers).137 Shortly thereafter, the group and others identified the target of these antibodies to be cytosolic 5′‐nucleotidase 1A (the antibody is now commonly referred to as anti‐cN1A antibody).138, 139 Moderate reactivity of anti‐cN1A antibodies was reported to be 70% sensitive and 92% specific for the diagnosis of IBM.136 Another study reported similar numbers, detecting anti‐cN1A antibodies in 61% of IBM patients but on the other hand also in 5% of PM, 23% of Sjögren's syndrome patients (SS), and 14% of systemic lupus erythematosus (SLE) patients, even in absence of any muscular symptoms.140 In a subsequent report however, the frequency of seropositive IBM patients was only 34.8% (24/69).141 Circulating anti‐cN1A antibodies may aid in distinguishing IBM (37%) from PM and DM (<5%), however, the picture becomes less clear when acknowledging that these antibodies are also detected in autoimmune conditions such as SS (36%) and SLE (20%).142 The presence of anti‐cN1A antibodies is neither associated with gender nor malignancy and appears to be independent of specific HLA‐DR alleles.141 Two recent reports also found anti‐cN1A antibodies in 33% (102/311)143 and 35.8% (24/67)144 of IBM patients, respectively. The anti‐cN1A positive IBM patients showed a higher adjusted mortality risk and depicted more cytochrome oxidase deficient muscle fibers as compared to sero‐negative patients.143 Moreover, passive immunization with purified IgG fractions derived from either anti‐cN1A‐positive or anti‐cN1A‐negative IBM patients in in vitro and in vivo models, led to myodegenerative changes (such as p62 protein aggregation), resembling those observed in IBM muscle.144 Whether pathogenicity is directly transferred via anti‐cN1A antibodies or if presence of these autoantibodies is simply indicative of other, yet unidentified mechanisms is so far unclear.145
Taken together, the above‐mentioned findings argue for B cell activation with subsequent production of autoantibodies against muscle epitopes in IBM. However, the pathogenetic role of B cells, their specificity and relevance needs further investigation.6
Inflammatory mediators – cytokines, chemokines, and myokines
Signal peptides secreted by invading leukocytes and resident myofibers alike are an integral part of the inflammatory milieu in muscle and are believed to directly contribute to the pathology of IBM via induction of surface molecules (on myofibers and invading leukocytes), chemotaxis of myoaggressive immune cells and subsequent muscle injury. The interplay of soluble factors and expression patterns of their respective surface receptors is complex and dynamic in its nature and a plethora of key suspects have been suggested.146
An early immunocytochemistry study that evaluated expression of inflammatory mediators in myositis, found predominant presence of IL‐1α (in endothelial cells), IL‐1β and TGF‐β (both in inflammatory cells), albeit no apparent difference in the expression pattern was observed between DM, PM and IBM.147 Similarly, De Bleeker and colleagues detected TNF‐α in macrophages, endothelial cells, and central myonuclei in IBM, DM, and PM muscle but not in that of nonmyositic controls.148 Chronic administration of TNF‐α via osmotic minipumps has shown to be already sufficient to attract neutrophils and macrophages to the muscle compartment.149 A more recent study found mRNA levels of GM‐CSF, IL‐4, IL‐10, IL‐12, IL‐13, IL‐23, IL‐1β and TNF‐α to be significantly increased in IBM muscle compared to healthy controls. Although this was also true for PM muscle, DM muscle did not present with increased levels of these cytokines and TNF‐α showed the highest values in IBM patients.150
Thrombospondin‐1 (TSP‐1) has been reported to function as a chemoattractant for leukocytes to sites of inflammation and interaction with its ligands activates and perpetuates autoaggressive T cell expansion.151, 152 Furthermore, expression of TSP‐1 and its binding partners CD36 and CD47 is upregulated on mRNA and protein level in IBM153 and TNF‐α can induce TSP‐1 and CD47 expression on human myoblasts in vitro.153
A crucial role for IFNγ in the pathoetiology of IBM has been proposed as well.136 During IBM, muscle fibers ubiquitously express MHC class I on their surface and to a higher degree than DM or PM muscle.154 However, not all MHC class I bearing myofibers depict presence of immune infiltrates and although MHC class I expression seems to sustain CD8+ T cell myoinfiltration in the case of IBM, MHC class I expression by itself seems not to be sufficient to entail infiltration of cytotoxic T cells (as demonstrated during DM, where muscle fibers express MHC class I but no CD8+ T cell infiltrates can be detected).45, 155 Ivanidze et al. reported segmental upregulation of IFNGR2, exclusively on attacked MHC class I‐expressing myofibers vs. MHC class I bearing myofibers that did not have infiltrates (nonattacked myofibers). The expression of IFNGR2 positively correlated with the amount of infiltrating CD8+ T cells.112
This strongly argues for MHC class I upregulation upstream and independent of IFNγ signaling during IBM. It is possible that, following this ubiquitous expression of MHC class I on myofibers upon a so far unknown trigger, CD8+ T cells might recognize cognate antigen in a stochastical manner, become activated, expand and secrete proinflammatory cytokines which, in turn, may induce IFNGR expression and perpetuate susceptibility toward further myocytotoxicity.112 In line with this, CD8+CD28− T cells found in the peripheral blood of IBM patients are more prone to produce IFNγ and IFNγ‐inducable chemoattractant mediators such as CXCL‐9, CXCL‐10 and IL‐12 are increased in serum of IBM patients compared to nonmyositic controls.82 Aside from infiltrating leukocytes, myofibers themselves might also actively participate in secreting proinflammatory mediators. Upregulated mRNA expression levels of CXCL‐9 and CXCL‐10 in muscle biopsies of IBM patients had been reported before and the same study demonstrated synthesis of CXCL‐9 and CXCL‐10 by human muscle fibers after IFNγ incubation in vitro.156 However, as with other cytokines discussed previously, CXCL‐9 and CXCL‐10 regulation is confirmed for other IIMs as well and these changes seem to reflect a general inflammatory milieu and maintenance of such within the muscle compartment.157
One conceivable alternative mechanism responsible for initial upregulation of MHC class I (upstream of IFNγ signalling) includes viral genesis (as discussed above)62, 63, 64, 158 or other proinflammatory cytokines like TNF‐α and IL‐1β.112, 159 Interestingly, in addition to IFNGR2 expression, transcripts of RANTES and Stat3 are reported to be increased in attacked myofibers vs. nonattacked myofibers as well.112 RANTES is produced in response to TNF‐α and synergistic effects between TNF‐α and IFNγ with regards to RANTES synthesis are reported.160, 161 Additionally, pro‐inflammatory cytokines like IL‐1β and, in particular, TNF‐α might hamper myoregeneration in IBM (and other IIMs) by suppressing myogenic microRNAs such as miR‐1, miR‐133a and miR‐133b.150 TNF‐like weak inducer of apoptosis (TWEAK) is a recently described member of the TNF superfamily. The proinflammatory cytokine signals through binding to its receptor Fn14 and activates NFκb in a TGFβ‐activated kinase 1‐(TAK1‐) dependent manner.162 TWEAK is expressed in a wide variety of cell types including monocytes and macrophages, dendritic cells and T cells163, 164, 165, 166, 167 and its implications in controlling muscle tissue repair and regeneration have reaffirmed its role as a key regulator of myogenesis.164, 168 As opposed to DM, PM and healthy mesoangioblasts, IBM mesoangioblasts fail to fully differentiate into skeletal myotubes.169 A recent study found increased TWEAK‐Fn14‐expression in IBM muscle compared to DM and PM muscle. Moreover, culture media from IBM‐derived differentiating mesoangioblasts show significantly higher levels of TWEAK as compared to nonmyositic or DM controls and IBM‐derived mesoangioblasts depict higher Fn14‐expression than those derived from other IIMs.170 During chronic inflammatory conditions, TWEAK has been shown to mediate proliferation of precursor cells while prohibiting their terminal differentiation.171 Furthermore, a critical role for TWEAK/Fn14 in fostering muscle atrophy has been proposed.172, 173 Therefore, disbalance of the TWEAK/Fn14 axis may, similarly to what has been reported for TNF‐α and IL‐1β, block myogenic differentiation through NFκb‐signalling174, 175 and, additionally, promote progressive muscle wasting and atrophy during IBM. Interestingly, in a colitis model, TWEAK, IL‐13, and TNF‐α act in concert and synergistically promote intestinal epithelial cell injury and induction of fibroblast proliferation.176, 177 Although a possible role for IL‐13 in the pathomechanism of IBM has not been addressed thus far, mRNA levels in muscle derived from nonmyositic controls and different IIMs depicted the highest and most consistent levels of IL‐13 in IBM samples.150
Finally, potential regulatory roles in the pathomechanism of IIM have been ascribed to IL‐17A and IL‐15.178, 179 However, most studies have so far focused on PM and DM and little data are currently available in IBM. Given the similarities in the inflammatory muscle milieu, especially that of PM, IL‐15 and IL‐17A should be further investigated for their involvement in IBM.
The differential interaction of selected cytokines with degenerative pathomechanisms during IBM will be discussed further below.
Degenerative pathomechanisms in IBM
Aside from the previously discussed inflammatory component of IBM, its pathoetiology includes distinct myodegenerative changes including, but not limited to, vacuolization, abnormal posttranslational modifications of proteins with subsequent congophilic misfolded multiprotein aggregates and dysfunctional mitochondrial activity.7, 103 The observation that IBM behaves largely refractory to anti‐inflammatory treatment gave rise to the proposition that inflammatory myofiber infiltrates are largely an epiphenomenon to age‐related primary myodegenerative events similar to neurodegenerative disorders such as Alzheimer's disease (AD) and Parkinson's disease (PD).7, 103 In the following section, we will discuss the degenerative changes observed in IBM muscle and evaluate the possible pathomechanistic interrelationship with inflammatory processes.
Amyloid‐β, α‐Synuclein, Presenilin, Tau
Detection of intracellular accumulation of amyloid precursor protein (APP)‐derived amyloid‐β (Aβ) peptides as congophilic inclusions was amongst the first evidence for defective myoproteostasis in IBM muscle.180, 181, 182 Aβ is usually generated as an either 40 or 42 amino acid‐long peptide. The more hydrophobic 42 amino acid long isoform exhibits a stronger tendency to self‐associate into insoluble fibrils, oligomerize and cluster into aggregates than Aβ 1‐40. 183, 184, 185 It is therefore considered to be more cytotoxic and the predominant isoform to be accumulated as oligomers in IBM muscle.186, 187 Congophilic Aβ is detected in up to 70% of IBM muscle fibers and mostly found in nonvacuolated areas.188 Aβ peptides are generated via the sequential cleavage of the transmembrane glycoprotein APP by the protease β‐site of the APP cleaving enzyme 1 (BACE1) and the γ‐secretase complex.189, 190, 191 Components of the sequential cleavage machinery of APP, such as BACE1, are upregulated in IBM muscle.192, 193 Recently, a γ‐secretase activating protein (GSAP) has been characterized, which selectively mediates Aβ generation via facilitating the interaction between γ‐secretase complex members and APP‐CTF.194 IBM muscle fibers depict increased protein and mRNA expression of GSAP in comparison to nonmyositic controls.193 Consequently, the members of the γ‐secretase complex that catalyzes the final step of the Aβ generation, such as nicastrin, presenilin‐1 (PS‐1) and presenilin enhancer 2 are increased on protein and mRNA level in IBM muscle.193 Phosphorylation of APP by glycogen synthase kinase 3β (GSK3β) facilitates increased generation of cytotoxic Aβ.195, 196 In line with these findings, GSK3β is activated and APP is found to be highly phosphorylated in IBM muscle.197
One study evaluated plasma levels of Aβ in IBM patients compared to myositic and nonmyositic controls and although Aβ plasma levels were increased in IBM compared to PM, levels were also elevated in DM disqualifying the assay as an appropriate diagnostic tool.198 A more recent report, however, found that plasma levels of BACE1, PS‐1 and soluble APP are increased in IBM patients compared to healthy controls and patients diagnosed with PM and DM.199
Aside from extracellular amyloid plaques, also intraneuronal neurofibrillary tangles mainly comprised of the microtubule‐associated protein tau, are a morphological feature of AD brains.200, 201, 202 There is evidence that Aβ partly executes its cytotoxicity upstream of tau hyperphosphorylation and subsequent self‐assembly.203, 204 Aspects of Aβ cytotoxicity are tau‐dependent, indicating a reciprocal, self‐enhancing component during the interaction of the two.205 Cytoplasmic hyperphosphorylated tau tangles in AD brains consist predominantly of 15–21 nm long paired helical filaments (PHF).201 Similarly, in IBM muscle, accumulations of hyperphosphorylated tau‐containing PHF are observed and kinases such as extracellular signal‐regulated kinase (ERK) or GSK3β, which have been reported to phosphorylate tau, are increased and colocalized with tau in IBM muscle fibers.206, 207, 208, 209, 210
α‐Synuclein, another aberrant protein that is present as insoluble cytoplasmic aggregates in neurodegenerative brain disorders also abnormally accumulates in IBM muscle fibers.211, 212, 213, 214, 215 Expression and toxicity of the small protein is increased under conditions of oxidative stress but is negatively regulated by the activity of heat shock proteins.216, 217, 218 Interestingly, α‐synuclein has been reported to facilitate phosphorylation of tau by the above mentioned kinase GSK3β.219
In addition to Aβ, hyperphosphorylated tau and α‐synuclein also ApoE,220, 221 p62/SQSTM1222 and prion protein223, 224, 225 are found to be aggregated in IBM muscle fibers, all indicative of protein dyshomeostasis being a distinctive feature of IBM. In support of this, a recent study found that treatment with arimoclomol, a coinducer of heat shock responses, significantly ameliorated IBM‐like phenotype in vitro and in vivo and appeared to be safe in a proof‐of‐concept study with IBM patients.226 The clinical efficacy of arimoclomol is currently tested in a clinical trial (NCT02753530).
Faulty protein disposal: the proteasome & autophagy
Although there is now a plethora of convincing evidence for severe defects in myoproteostasis, the individual specificity of the aforementioned aggregated proteins in IBM pathology remains to be further evaluated. The pathoetiology of IBM appears to follow a dynamic pattern and a given muscle biopsy at a given stage of the disease may greatly differ from those taken at different time points or even locations. In fact, the paradox is not limited to IBM. In AD, which regularly serves skeptics as a paramount example for a bona fide amyloid‐disorder, more than one‐third of ApoE noncarriers that clinically present with mild to moderate cognitive deficit, do not show significant cerebral amyloidosis in positron emission tomography.227, 228, 229
The underlying causative event that promotes and propagates self‐aggregation of aberrant proteins in IBM myofibers has yet to be elucidated. A delicately regulated surveillance of protein turnover is especially crucial in postmitotic cells such as neurons and myocytes.230, 231, 232, 233, 234 Eukaryotic cells employ two predominant molecular systems to keep a tight balance between translation and degradation of cellular proteins, namely the proteasomal system and autophagy.235, 236, 237 Dysfunction in either of these two proteolytic systems, and subsequent imbalance of protein homeostasis, is one of nine defining characteristics of cellular aging.238 However, severity, time of onset, and acceleration of these pathomechanisms determine to which degree these changes will meet the pathological spectrum.235, 237, 238
The proteasome
In the ubiquitin‐proteasomal system (UPS), sequentially polyubiquitylated proteins are targeted toward the barrel‐shaped multipartite 26S proteasome which executes caspase‐like, trypsin‐like, and chymotrypsin‐like proteolytic activities located on its β‐subunits (β1, β2, and β5, respectively).239 The proteasomal system is tightly controlled by regulatory molecules and executes ubiquitin‐dependent and ‐independent proteolytic degradation.240 Polyubiquitination‐independent recognition and subsequent degradation of oxidized substrates by the core particle 20S proteasome is especially relevant in the context of aging cells.235, 241
Fratta et al. have reported that the 26S proteasome co‐stains with Aβ, phosphorylated tau, ubiquitin, and heat shock protein 70 (Hsp70) in muscle biopsies from IBM patients.242 While protein expression of proteasomal subunits 19S, 20Sα, and 20Sβ is greatly increased in IBM muscle compared to age‐matched controls, proteolytic activity of the proteasomal machinery is significantly impaired in IBM muscle. In accordance with this, inhibition of the proteasome in human myofibers in vitro leads to formation of aberrant multiprotein aggregates.242
Particularly cells of the hematopoietic system harbor a unique form of the proteasome, termed the immunoproteasome, in which the catalytic β subunits β1, β2, β5 have been replaced by β1i, β2i, and β5i resulting in increased enzymatic cleavage following hydrophobic residues and decreased cleavage following acidic residues, respectively.243, 244, 245 Exchange of constitutive proteasome sub‐units with immunoproteasome sub‐units distinctively shapes the pool of MHC class I ligands and can be facilitated via exposure to proinflammatory cytokines like IFNγ and TNF‐α.243, 244 A recent study found that the immunoproteasome sub‐units β1i and β5i are upregulated and colocalized with MHC class I molecules in IBM muscle.246 In vitro experiments show that exposure to TNF‐α and IFNγ increases replacement toward immunoproteasomal sub‐units in primary human myoblasts and the selective inhibition of proteasomal subunit β5i in myoblasts results in increased expression of TNF‐α and IFNγ‐dependent myokines like IL‐1β, IL‐6, CXCL‐9, and CXCL‐10.246 However, these results were also obtained in other IIM such as DM and immune‐mediated necrotizing myopathy, indicating a downstream effect of preceding myoinflammatory events. It remains unclear if proteasomal dysfunction is a primary event in IBM pathology, or if soluble intermediates of aggregation‐prone proteins facilitate proteasomal inhibition.
Autophagy
Autophagy comprises a set of intracellular catabolic pathways that degrade cytoplasmic content by means of the lysosomal system.247, 248 While occupying pivotal roles during host defense against microbes, induction of tolerance, antigen‐presentation, and tissue differentiation, a key function of autophagy pathways is to maintain a well‐balanced proteostasis and provision of metabolic building blocks and energy sources in response to nutrient deprivation and other cellular stressors248, 249, 250, 251, 252 As opposed to the proteasomal system, autophagy, in addition to removing aberrant proteins, aids in the removal of defective or excess mitochondria, lysosomes and peroxisomes and keeps, thereby, homeostasis on the level of macromolecules and whole organelles alike.253, 254, 255, 256 Consequently, defective autophagy pathways have been ascribed a pathological role in degenerative diseases of the brain236, 237, 257 and emerging evidence implicates autophagy in the pathoetiology of IBM. Initial hints about malregulated autophagy in IBM were introduced in an early study in 1980.258 However, it took another 24 years for evidence that members of the autophagy machinery (Atg5 and Atg12) are upregulated on mRNA level in IBM muscle as compared to healthy and amyotrophic lateral sclerosis muscle.259 We demonstrated that accumulated APP and its proteolytic fragment Aβ in skeletal muscel fibers are targeted for lysosomal degradation via macroautophagy.260 We observed APP/Aβ‐containing autophagosomes at increased frequency in muscle fibers of IBM muscle biopsies, but not in nonmyopathic muscle or nonvacuolated myopathic controls. Moreover, Aβ‐containing autophagosomes were almost exclusively observed in degenerating muscle fibers of the type II (fast‐twitching) and in part associated with overexpression of MHC class I and II on myofibers and invasion by CD4+ and CD8+ cells.260 A more recent immunohistochemistry study reports overexpression of the autophagy proteins ATG5, microtubule‐associated protein light chain 3 (LC3) and Beclin‐1 in IBM muscle biopsies. Interestingly, lymphocytic infiltrates were predominantly found surrounding Beclin‐1+ myofibers.261 Recently, components of chaperone‐mediated autophagy were identified to be increased in IBM as well.262 Güttsches et al. identified an overrepresentation of rare missense coding variants of an autophagic adaptor protein facilitating autophagosome trafficking, FYCO1, in IBM patients and suggested that a failure in autophagosome/endosome trafficking may underlie IBM pathogenesis.263 In addition to FYCO1, missense pathogenic variants responsible for autophagosome maturation and degradation (VCP and p62/SQSTM1) have been found in patients with IBM.264, 265 We could previously show that autophagy is constitutively active in human myocytes and can be upregulated via the proinflammatory cytokines TNF‐α 108 or IFNγ together with IL‐1β.266 Interestingly, composite exposure to TNF‐α and IFNγ leads to significant autophagy‐dependent translocation of intracellular MHC class II to the cell surface in myocytes and more than 40% of muscle fibers in IBM that costain for autophagosomes and MHC class II have contact to CD4+ and CD8+ infiltrating T cells.108 Dengjel and colleagues reported that upregulation of autophagic activity, by means of altered lysosomal processing, significantly increases the fraction of intracellular source protein‐derived peptides presented on MHC class II.267 These findings suggest that the proinflammatory environment in IBM muscle promotes induction of autophagy in myofibers and subsequently enhances surface MHC class II, thereby maintaining CD4+ T cell infiltrates via the presentation of yet unknown self‐peptides. Intracellular antigen presentation via MHC class I molecules is also regulated by the autophagy machinery, because autophagy‐related proteins enhance MHC class I internalization for degradation and thereby diminish antigen display on the cell surface.268 Indeed, in vivo studies have primarily found enhanced CD8+ T cell responses in mice with defective autophagy in antigen‐presenting cells268, 269, 270 A potential mechanism leading to such increased CD8+ T cell expansions is that, in the absence of autophagy, more substrate becomes available for canonical MHC class I loading271 or by decreased endocytosis and degradation of cell surface MHC class I molecules.268 Thus, defective autophagy in skeletal muscle fibers could drive increased MHC class I expression and CD8+ T cell accumulation.
Brain biopsies of early (preclinical) and moderate stage AD patients exhibit impaired neuronal autophagic activity represented by increased numbers of LC3+ autophagosomes and diminished fusion of these vesicles with lysosomes into autolysosomes.272 Moreover, autophagosomes purified from an APP695‐transfected murine fibroblast‐like cell line, contain copious amounts of APP, PS1, nicastrin, and γ‐secretase complex with functional amyloidogenesis at the site of the autophagosome, resulting in Aβ 1‐40 and Aβ 1‐42 peptides.272 Importantly, these generated peptides were not instable intermediates as they did not seem to undergo additional cleavage after further 24 h incubation in autophagosomes as opposed to their Aβ‐specific cleavage in lysosomal fractions.272 In line with this, in vitro exposure of human muscle cells to autophagy inducers TNF‐α or rapamycin leads to marked increase of intracellular APP and Aβ oligomers. Specific siRNA‐mediated knockdown of the essential autophagy gene Atg12 prevents the assembly of autophagosomes and abbrogates TNF‐α‐mediated accumulation of Aβ in muscle cells.273 These findings appear to be in contrast to a subsequent study in which Nogalska et al. report increased accumulation of Aβ oligomers upon inhibition of autophagy in cultured human myofibers.187 However, these results should be carefully interpreted since inhibition of autophagy was carried out by using chloroquine and bafilomycin A1 (a specific inhibitor of the V‐ATPase), both of which lack specificity and are believed to inhibit lysosomal acidification and thereby subsequent fusion of the autophagosome with lysosomes rather than the assembly of the autophagosome,274 which results in impaired autophagosome maturation and accumulation of autophagosomes. In the case of bafilomycin A1, the actual inhibitory potential with regard to blocking autophagosome‐lysosome fusion has been doubted previously and bafilomycin A1 increases LC3 lipidation to a similar degree as autophagy‐inducer rapamycin.275, 276 Conversely, others have confirmed increased Aβ generation associated with preceding autophagosome accumulation in different mammalian cell types.277, 278, 279
Autophagy and the proteasomal system are unequivocally colligated in their endeavor to keep proteostasis. Selective transport of target molecules toward degrading vesicles or macromolecular structures like the proteasome requires multifunctional adaptor molecules. Polyubiquitylated proteins can be targeted via p62/SQSTM1 or neighbor of BRCA1 gene 1 (NBR1) for degradation via the proteasomal or the autophagy/lysosomal system.280, 281, 282, 283 In IBM muscle, both p62/SQSTM1 and NBR1 are upregulated on protein and mRNA level and colocalize with phosphorylated tau in protein aggregates.222, 284 The cargo protein p62/SQSTM1 can bind Lys63‐linked ubiquitin and phosphorylation at Ser403 of p62 enhances the binding affinity of p62 to ubiquitin.285 A recent study demonstrates that aggregated p62/SQSTM1 is largely phosphorylated at Ser403 in muscles of IBM patients and Lys63‐linked ubiquitin colocalized with p62/SQSTM1 aggregates, suggesting impaired initiation of selective autophagy targeting ubiquitinated proteins.286
Generally, as a result of active cellular synthesis processes, an unpreventable fraction of misfolded proteins, the so‐called defective ribosomal products (DRiPs) arise and need to be subsequently cleared from the cytosol in order to avoid cell stress and cytotoxicity. In one proposed model, DRiPs are polyubiquitylated and subsequently subjected to proteasomal degradation. The resulting peptides are fed into the MHC class I presenting pathway and will be surveilled by CD8+ T cells.287 Upon impairment of autophagy in HeLa cells, its substrates accumulate in p62/SQSTM1‐positive aggresome‐like induced structures and are fed into the proteasomal pathway with subsequent presentation via MHC class I.271 Differential activity of autophagy might, therefore, shape the peptide pool presented on MHC class I and it is tempting to speculate that impairment of this control mechanism in IBM muscle abets invasion of myoaggressive immune cells.
Differential diagnosis can be challenging facing the PM‐IBM spectrum of T cell‐rich inflammatory myopathies. The recent advances in identifying autophagy as a relevant malregulated process in IBM has already yielded practical application in that using a combination of LC3 (sensitive) and transactive response DNA‐binding protein 43 kDa (TDP‐43) (specific) stainings was found to be effective in discriminating IBM from PM.288
Interrelationship between inflammation, cell stress and myodegeneration
It has been proposed that muscle invasion of peripheral immune cells and progressive myodegeneration are closely linked in the development of IBM.289 However, the precise sequence of events remains incompletely understood. In IBM muscle, but not in PM or DM, IL‐1β is spatially associated with Aβ and degenerative changes positively correlate with the degree of inflammation in IBM patients.290 Furthermore, exposure of human myotubes to IL‐1β leads to upregulation of APP and accumulation of Aβ in vitro and this effect can be synergistically promoted by composite exposure with IFNγ.290 Pro‐inflammatory stimuli, such as IL‐1β, TNF‐α, and IFNγ augment expression of inducible nitric oxide synthase (iNOS), and composite exposure of murine muscle cells with IFNγ and Aβ peptides provoke robust nitric oxide (NO) production.291, 292, 293 Expression of iNOS in IBM muscle has been reported several years ago294 and more recently confirmed and extended: iNOS expression and concomitant NO production was enhanced in IBM muscle compared to DM and PM muscle.295 More importantly, nitrotyrosine, the product of tyrosine nitration in the presence of metabolically active NO, colocalized with Aβ in IBM muscle fibers. In vitro assays revealed that exposure of primary human muscle cells to IL‐1β together with IFNγ elicits strong NO production, followed by necrotic cell death.295 Conversely, the pharmacological inhibition of iNOS prevented cytokine‐mediated accumulation of Aβ and necrotic cell death indicative of iNOS being at the interface of proinflammatory stress and degenerative changes in IBM muscle. Taken together, these data revealed a crucial role for IBM‐relevant pro‐inflammatory mediators in the promotion of amyloidogenesis in the muscle. In double‐transgenic MCK‐APP/PS1 mice, an animal model for IBM, chronic exposure to inflammatory stimuli significantly increases deposition of the insoluble and cytotoxic Aβ 1‐42 in myofibers whereas Aβ 1‐40 levels remain unchanged. In addition, chronic inflammation by virtue of TNF‐α, IL‐6, and IL‐1β, facilitated and enhanced GSK3β‐mediated phosphorylation of tau in myofibers resulting in pronounced motor impairment.210
The small heat shock protein αB crystallin is constitutively expressed in human skeletal muscle cells, binds misfolded proteins in order to avert their aggregation and its expression is sensitive to TNF‐α‐mediated induction.296, 297, 298 During AD pathology expression of αB crystallin is increased in CNS resident glial cells that are found in close spatial proximity to extracellular amyloid and neurofibrillary tangles.299 Human myotubes exposed to the combination of IL‐1β and IFNγ show marked induction of αB crystallin and APP.300 More importantly, expression of αB crystallin is increased in IBM muscle (and to a lower degree in PM and DM) not only in muscle fibers with structural abnormalities but also in normal appearing myofibers, suggesting an early event in IBM pathogenesis that links pro‐inflammatory cell stress to accumulation of aberrant proteins.300, 301
The neuronal receptor for advanced glycation endproduct (RAGE) has been implicated in a pro‐inflammatory pathway of AD pathology in that binding of its ligand Aβ facilitates NFκB‐dependent M‐GSF production and subsequent chemotaxis of myeloid cells.302 High mobility group box 1 (HMGB1), another ligand for RAGE, is a nuclear DNA‐binding protein that can be actively secreted or passively released upon necrotic cell death (but not upon apoptosis) and exert proinflammatory effects by triggering myeloid cells to secrete substantial amounts of TNF‐α, IL‐1β, IL‐6, IL‐8, macrophage inflammatory protein (MIP)‐1α, MIP‐1β.303, 304, 305 HMGB1 is expressed and released by human skeletal muscle cells upon muscle injury and via binding to RAGE expressed on the surface of myoblasts faciliates myogenesis and muscle regeneration.306, 307
RAGE, in association with reactive oxygen species‐ and NFκB‐dependent pathways and HMGB1 are overexpressed in myositis.308, 309, 310 In IBM muscle, RAGE and HMGB1 colocalize with Aβ and neurofilament/tau and composite exposure of human muscle cells with IFNγ and IL‐1β leads to cytoplasmic translocation and subsequent release of HMGB1 into the extracellular space.310 Furthermore, exposure of human muscle cells to exogenous HMGB1 is equally pontent in triggering Aβ accumulation as IFNγ/IL‐1β.310 These findings strongly suggest a facilitator role for the HMGB1‐RAGE‐Aβ‐axis in interconnecting inflammatory and degenerative events during IBM.310
In a possible pathological setting, necrosis‐undergoing myofibers might release vast amounts of HMGB1. Excessive presence of this mediator might overwrite its pro‐myoregenerative function and rather promote protein aggregation in RAGE‐expressing muscle cells as well as release of proinflammatory cytokines by infiltrating immune cells. This cascade perpetuates and amplifies the myoaggressive microenvironment in IBM.
Collectively, mounting evidence from in vitro studies, animal models, and human muscle samples suggests that inflammation in IBM can trigger and sustain cell stress in skeletal muscle with subsequent accumulation of unwanted proteins and irreversible muscle fiber damage.
Author Contributions
All three authors have made substantial, intellectual, and equally valuable contribution to the work and approved it for publication.
Conflict of Interest
The authors declare that there is no financial or other relationships that might lead to a perceived conflict of interest.
Acknowledgments
C.W.K. was supported by a scholarship provided by the German Research Foundation (DFG grant KE 1831/1‐1) and a scholarship by the University of Zürich (Forschungskredit FK‐14‐021). J.D.L. was supported by the Swiss National Science Foundation (31003A‐169664), the Novartis Foundation for medical‐biological research, the Sassella Foundation, the Hartmann Müller Foundation, and the Swiss Multiple Sclerosis Society.
References
- 1. Carstens P‐O, Schmidt J. Diagnosis, pathogenesis and treatment of myositis: recent advances. Clin Exp Immunol 2014;175:349–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dalakas MC. Polymyositis, dermatomyositis and inclusion‐body myositis. N Engl J Med 1991;325:1487–1498. [DOI] [PubMed] [Google Scholar]
- 3. Dalakas MC. Inflammatory muscle diseases. N Engl J Med 2015;372:1734–1747. [DOI] [PubMed] [Google Scholar]
- 4. Yunis EJ, Samaha FJ. Inclusion body myositis. Lab Invest 1971;25:240–248. [PubMed] [Google Scholar]
- 5. Schmidt J, Dalakas MC. Inclusion‐body myositis in the elderly: an update. Aging Health 2010;6:687–694. https://doi.org/10.2217/ahe.10.64;6(6):687-694. [Google Scholar]
- 6. Schmidt J, Dalakas MC. Inclusion body myositis: from immunopathology and degenerative mechanisms to treatment perspectives. Expert Rev Clin Immunol 2013;9:1125–1133. [DOI] [PubMed] [Google Scholar]
- 7. Askanas V, Engel WK, Nogalska A. Sporadic inclusion‐body myositis: A degenerative muscle disease associated with aging, impaired muscle protein homeostasis and abnormal mitophagy. BBA ‐ Molecular Basis of Disease 2014;4:1–11. [DOI] [PubMed] [Google Scholar]
- 8. Benveniste O, Stenzel W, Hilton‐Jones D, et al. Amyloid deposits and inflammatory infiltrates in sporadic inclusion body myositis: the inflammatory egg comes before the degenerative chicken. Acta Neuropathol 2015;129:611–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Benveniste O, Guiguet M, Freebody J, et al. Long‐term observational study of sporadic inclusion body myositis. Brain 2011;134(Pt 11):3176–3184. [DOI] [PubMed] [Google Scholar]
- 10. Breithaupt M, Schmidt J. Update on treatment of inclusion body myositis. Curr Rheumatol Rep 2013;15:329. [DOI] [PubMed] [Google Scholar]
- 11. Cox FM, Titulaer MJ, Sont JK, et al. A 12‐year follow‐up in sporadic inclusion body myositis: an end stage with major disabilities. Brain 2011;134(Pt 11):3167–3175. [DOI] [PubMed] [Google Scholar]
- 12. Needham M, Mastaglia FL. Sporadic inclusion body myositis: a review of recent clinical advances and current approaches to diagnosis and treatment. Clin Neurophysiol 2016;127:1764–1773. [DOI] [PubMed] [Google Scholar]
- 13. Mulcahy KP, Langdon PC, Mastaglia F. Dysphagia in inflammatory myopathy: self‐report, incidence, and prevalence. Dysphagia 2012;27:64–69. [DOI] [PubMed] [Google Scholar]
- 14. Cox FM, Verschuuren JJ, Verbist BM, et al. Detecting dysphagia in inclusion body myositis. J Neurol 2009;256:2009–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Olthoff A, Carstens P‐O, Zhang S, et al. Evaluation of dysphagia by novel real‐time MRI. Neurology 2016;87:2132–2138. [DOI] [PubMed] [Google Scholar]
- 16. Murata K‐Y, Kouda K, Tajima F, Kondo T. A dysphagia study in patients with sporadic inclusion body myositis (s‐IBM). Neurol Sci 2012;33:765–770. [DOI] [PubMed] [Google Scholar]
- 17. Molberg Ø, Dobloug C. Epidemiology of sporadic inclusion body myositis. Curr Opin Rheumatol 2016;28:657–660. [DOI] [PubMed] [Google Scholar]
- 18. Catalán M. Selva‐O'Callaghan A, Grau JM. Diagnosis and classification of sporadic inclusion body myositis (sIBM). Autoimmun Rev 2014;13:1–4. [DOI] [PubMed] [Google Scholar]
- 19. Dalakas MC. Sporadic inclusion body myositis–diagnosis, pathogenesis and therapeutic strategies. Nat Clin Pract Neurol 2006;2:437–447. [DOI] [PubMed] [Google Scholar]
- 20. Ko EH, Rubin AD. Dysphagia due to inclusion body myositis: case presentation and review of the literature. Ann Otol Rhinol Laryngol 2014;123:605–608. [DOI] [PubMed] [Google Scholar]
- 21. Wintzen AR, Bots GT, de Bakker HM, et al. Dysphagia in inclusion body myositis. J Neurol Neurosurg Psychiatr 1988;51:1542–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cox FM, Delgado V, Verschuuren JJ, et al. The heart in sporadic inclusion body myositis: a study in 51 patients. J Neurol 2010;257:447–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dobloug GC, Garen T, Brunborg C, et al. Survival and cancer risk in an unselected and complete Norwegian idiopathic inflammatory myopathy cohort. Semin Arthritis Rheum 2015;45:301–308. [DOI] [PubMed] [Google Scholar]
- 24. Badrising UA, Maat‐Schieman M, van Duinen SG, et al. Epidemiology of inclusion body myositis in the Netherlands: a nationwide study. Neurology 2000;55:1385–1387. [DOI] [PubMed] [Google Scholar]
- 25. Dobloug GC, Antal EA, Sveberg L, et al. High prevalence of inclusion body myositis in Norway; a population‐based clinical epidemiology study. Eur J Neurol 2015;22:672–e41. [DOI] [PubMed] [Google Scholar]
- 26. Khadilkar SV, Patil SG, Amin SN. Study of idiopathic inflammatory myopathies with special reference to borderland between idiopathic inflammatory myopathies and muscular dystrophies. Neurol India 2008;56:356–362. [DOI] [PubMed] [Google Scholar]
- 27. Meyer A, Meyer N, Schaeffer M, et al. Incidence and prevalence of inflammatory myopathies: a systematic review. Rheumatology (Oxford) 2015;54:50–63. [DOI] [PubMed] [Google Scholar]
- 28. Needham M, Corbett A, Day T, et al. Prevalence of sporadic inclusion body myositis and factors contributing to delayed diagnosis. J Clin Neurosci 2008;15:1350–1353. [DOI] [PubMed] [Google Scholar]
- 29. Needham M, James I, Corbett A, et al. Sporadic inclusion body myositis: phenotypic variability and influence of HLA‐DR3 in a cohort of 57 Australian cases. J Neurol Neurosurg Psychiatr 2008;79:1056–1060. [DOI] [PubMed] [Google Scholar]
- 30. Oflazer PS, Deymeer F, Parman Y. Sporadic‐inclusion body myositis (s‐IBM) is not so prevalent in Istanbul/Turkey: a muscle biopsy based survey. Acta Myol 2011;30:34–36. [PMC free article] [PubMed] [Google Scholar]
- 31. Prieto S, Grau JM. The geoepidemiology of autoimmune muscle disease. Autoimmun Rev 2010;9:A330–A334. [DOI] [PubMed] [Google Scholar]
- 32. Suzuki N, Aoki M, Mori‐Yoshimura M, et al. Increase in number of sporadic inclusion body myositis (sIBM) in Japan. J Neurol 2012;259:554–556. [DOI] [PubMed] [Google Scholar]
- 33. Nakanishi H, Koike H, Matsuo K, et al. Demographic features of Japanese patients with sporadic inclusion body myositis: a single‐center referral experience. Intern Med 2013;52:333–337. [DOI] [PubMed] [Google Scholar]
- 34. Price MA, Barghout V, Benveniste O, et al. Mortality and Causes of Death in Patients with Sporadic Inclusion Body Myositis: survey Study Based on the Clinical Experience of Specialists in Australia, Europe and the USA. J Neuromuscul Dis 2016;3:67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Amato AA, Griggs RC. Unicorns, dragons, polymyositis, and other mythological beasts. Neurology 2003;61:288–289. [DOI] [PubMed] [Google Scholar]
- 36. Bronner IM, Linssen WHJP, van der Meulen MFG, et al. Polymyositis: an ongoing discussion about a disease entity. Arch Neurol 2004;61:132–135. [DOI] [PubMed] [Google Scholar]
- 37. Hengstman GJD, vanEngelen BGM . Polymyositis: an overdiagnosed entity. Neurology 2004;63:402–3‐ author reply 403. [PubMed] [Google Scholar]
- 38. van der Meulen MFG, Bronner IM, Hoogendijk JE, et al. Polymyositis: an overdiagnosed entity. Neurology 2003;61:316–321. [DOI] [PubMed] [Google Scholar]
- 39. Machado PM, Ahmed M, Brady S, et al. Ongoing developments in sporadic inclusion body myositis. Curr Rheumatol Rep 2014;16:477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Griggs RC, Askanas V, DiMauro S, et al. Inclusion body myositis and myopathies. Ann Neurol 1995;38:705–713. [DOI] [PubMed] [Google Scholar]
- 41. Chahin N, Engel AG. Correlation of muscle biopsy, clinical course, and outcome in PM and sporadic IBM. Neurology 2008;70:418–424. [DOI] [PubMed] [Google Scholar]
- 42. Rose MR. ENMC IBM Working Group. 188th ENMC International Workshop: inclusion Body Myositis, 2‐4 December 2011, Naarden, The Netherlands. Neuromuscul Disord 2013;23:1044–1055. [DOI] [PubMed] [Google Scholar]
- 43. Lloyd TE, Mammen AL, Amato AA, et al. Evaluation and construction of diagnostic criteria for inclusion body myositis. Neurology 2014;83:426–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Engel AG, Arahata K. Monoclonal antibody analysis of mononuclear cells in myopathies. II: phenotypes of autoinvasive cells in polymyositis and inclusion body myositis. Ann Neurol 1984;16:209–215. [DOI] [PubMed] [Google Scholar]
- 45. Karpati G, Pouliot Y, Carpenter S. Expression of immunoreactive major histocompatibility complex products in human skeletal muscles. Ann Neurol 1988;23:64–72. [DOI] [PubMed] [Google Scholar]
- 46. Ikezoe K, Ohshima S, Osoegawa M, et al. Expression of granulysin in polymyositis and inclusion‐body myositis. J Neurol Neurosurg Psychiatr 2006;77:1187–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Orimo S, Koga R, Goto K, et al. Immunohistochemical analysis of perforin and granzyme A in inflammatory myopathies. Neuromuscul Disord 1994;4:219–226. [DOI] [PubMed] [Google Scholar]
- 48. Schmidt J, Rakocevic G, Raju R, Dalakas MC. Upregulated inducible co‐stimulator (ICOS) and ICOS‐ligand in inclusion body myositis muscle: significance for CD8+ T cell cytotoxicity. Brain 2004;127(Pt 5):1182–1190. [DOI] [PubMed] [Google Scholar]
- 49. Goebels N, Michaelis D, Engelhardt M, et al. Differential expression of perforin in muscle‐infiltrating T cells in polymyositis and dermatomyositis. J Clin Invest 1996;97:2905–2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Murata K, Dalakas MC. Expression of the costimulatory molecule BB‐1, the ligands CTLA‐4 and CD28, and their mRNA in inflammatory myopathies. Am J Pathol 1999;155:453–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Waschbisch A, Wintterle S, Lochmuller H, et al. Human muscle cells express the costimulatory molecule B7‐H3, which modulates muscle‐immune interactions. Arthritis Rheum 2008;58:3600–3608. [DOI] [PubMed] [Google Scholar]
- 52. Fyhr IM, Moslemi AR, Tarkowski A, et al. Limited T‐cell receptor V gene usage in inclusion body myositis. Scand J Immunol 1996;43:109–114. [DOI] [PubMed] [Google Scholar]
- 53. Lindberg C, Oldfors A, Tarkowski A. Restricted use of T cell receptor V genes in endomysial infiltrates of patients with inflammatory myopathies. Eur J Immunol 1994;24:2659–2663. [DOI] [PubMed] [Google Scholar]
- 54. Lindberg C, Oldfors A, Tarkowski A. Local T‐cell proliferation and differentiation in inflammatory myopathies. Scand J Immunol 1995;41:421–426. [DOI] [PubMed] [Google Scholar]
- 55. Amemiya K, Granger RP, Dalakas MC. Clonal restriction of T‐cell receptor expression by infiltrating lymphocytes in inclusion body myositis persists over time. Studies in repeated muscle biopsies. Brain 2000;123(Pt 10):2030–2039. [DOI] [PubMed] [Google Scholar]
- 56. O'Hanlon TP, Dalakas MC, Plotz PH, Miller FW. The alpha beta T‐cell receptor repertoire in inclusion body myositis: diverse patterns of gene expression by muscle‐infiltrating lymphocytes. J Autoimmun 1994;7:321–333. [DOI] [PubMed] [Google Scholar]
- 57. Salajegheh M, Rakocevic G, Raju R, et al. T cell receptor profiling in muscle and blood lymphocytes in sporadic inclusion body myositis. Neurology 2007;69:1672–1679. [DOI] [PubMed] [Google Scholar]
- 58. Chou SM. Myxovirus‐like structures in a case of human chronic polymyositis. Science 1967;158:1453–1455. [DOI] [PubMed] [Google Scholar]
- 59. Dalakas MC. Inflammatory, immune, and viral aspects of inclusion‐body myositis. Neurology 2006;66(2 Suppl 1):S33–S38. [DOI] [PubMed] [Google Scholar]
- 60. Getts DR, Chastain EML, Terry RL, Miller SD. Virus infection, antiviral immunity, and autoimmunity. Immunol Rev 2013;255:197–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Dalakas MC, Schmidt J. Viruses in IBM: hit‐and‐run, hide and persist, or irrelevant? Neurology 2016;86:204–205. [DOI] [PubMed] [Google Scholar]
- 62. Dalakas MC, Rakocevic G, Shatunov A, et al. Inclusion body myositis with human immunodeficiency virus infection: four cases with clonal expansion of viral‐specific T cells. Ann Neurol 2007;61:466–475. [DOI] [PubMed] [Google Scholar]
- 63. Cupler EJ, Leon‐Monzon M, Miller J, et al. Inclusion body myositis in HIV‐1 and HTLV‐1 infected patients. Brain 1996;119(Pt 6):1887–1893. [DOI] [PubMed] [Google Scholar]
- 64. Ozden S, Gessain A, Gout O, Mikol J. Sporadic inclusion body myositis in a patient with human T cell leukemia virus type 1‐associated myelopathy. Clin Infect Dis 2001;32:510–514. [DOI] [PubMed] [Google Scholar]
- 65. Uruha A, Noguchi S, Hayashi YK, et al. Hepatitis C virus infection in inclusion body myositis: a case‐control study. Neurology 2016;86:211–217. [DOI] [PubMed] [Google Scholar]
- 66. Sy T, Jamal MM. Epidemiology of hepatitis C virus (HCV) infection. Int J Med Sci 2006;3:41–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tanaka H, Imai Y, Hiramatsu N, et al. Declining incidence of hepatocellular carcinoma in Osaka, Japan, from 1990 to 2003. Ann Intern Med 2008;148:820–826. [DOI] [PubMed] [Google Scholar]
- 68. Yamaguchi K, Kiyokawa H, Machida J, et al. Seroepidemiology of hepatitis C virus infection in Japan and HCV infection in haemodialysis patients. FEMS Microbiol Rev 1994;14:253–258. [DOI] [PubMed] [Google Scholar]
- 69. Gower E, Estes C, Blach S, et al. Global epidemiology and genotype distribution of the hepatitis C virus infection. J Hepatol 2014;61(1 Suppl):S45–S57. [DOI] [PubMed] [Google Scholar]
- 70. Messina JP, Humphreys I, Flaxman A, et al. Global distribution and prevalence of hepatitis C virus genotypes. Hepatology 2015;61:77–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Mastaglia FL. Sporadic inclusion body myositis: variability in prevalence and phenotype and influence of the MHC. Acta Myol 2009;28:66–71. [PMC free article] [PubMed] [Google Scholar]
- 72. Hohlfeld R. Biotechnological agents for the immunotherapy of multiple sclerosis. Principles, problems and perspectives. Brain 1997;120(Pt 5):865–916. [DOI] [PubMed] [Google Scholar]
- 73. Arosa FA. CD8+CD28‐ T cells: certainties and uncertainties of a prevalent human T‐cell subset. Immunol Cell Biol 2002;80:1–13. [DOI] [PubMed] [Google Scholar]
- 74. Vallejo AN. CD28 extinction in human T cells: altered functions and the program of T‐cell senescence. Immunol Rev 2005;205:158–169. [DOI] [PubMed] [Google Scholar]
- 75. Dalakas MC. Understanding the immunopathogenesis of inclusion‐body myositis: present and future prospects. Rev Neurol (Paris) 2002;158(10 Pt 1):948–958. [PubMed] [Google Scholar]
- 76. Greenberg SA. Proposed immunologic models of the inflammatory myopathies and potential therapeutic implications. Neurology 2007;69:2008–2019. [DOI] [PubMed] [Google Scholar]
- 77. Greenberg SA. Theories of the pathogenesis of inclusion body myositis. Curr Rheumatol Rep 2010;12:221–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Pedroza‐Seres M, Linares M, Voorduin S, et al. Pars planitis is associated with an increased frequency of effector‐memory CD57+ T cells. Br J Ophthalmol 2007;91:1393–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Schirmer M, Goldberger C, Würzner R, et al. Circulating cytotoxic CD8(+) CD28(‐) T cells in ankylosing spondylitis. Arthritis Res 2002;4:71–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Sun Z, Zhong W, Lu X, et al. Association of Graves’ disease and prevalence of circulating IFN‐gamma‐producing CD28(‐) T cells. J Clin Immunol 2008;28:464–472. [DOI] [PubMed] [Google Scholar]
- 81. Fasth AER, Dastmalchi M, Rahbar A, et al. T cell infiltrates in the muscles of patients with dermatomyositis and polymyositis are dominated by CD28null T cells. J Immunol 2009;183:4792–4799. [DOI] [PubMed] [Google Scholar]
- 82. Allenbach Y, Chaara W, Rosenzwajg M, et al. Th1 response and systemic treg deficiency in inclusion body myositis. PLoS ONE 2014;9:e88788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Pandya JM, Fasth AER, Zong M, et al. Expanded T cell receptor Vβ‐restricted T cells from patients with sporadic inclusion body myositis are proinflammatory and cytotoxic CD28null T cells. Arthritis Rheum 2010;62:3457–3466. [DOI] [PubMed] [Google Scholar]
- 84. Plunkett FJ, Franzese O, Finney HM, et al. The loss of telomerase activity in highly differentiated CD8+CD28‐CD27‐ T cells is associated with decreased Akt (Ser473) phosphorylation. J Immunol 2007;178:7710–7719. [DOI] [PubMed] [Google Scholar]
- 85. Wang EC, Lawson TM, Vedhara K, et al. CD8high+ (CD57+) T cells in patients with rheumatoid arthritis. Arthritis Rheum 1997;40:237–248. [DOI] [PubMed] [Google Scholar]
- 86. Sze DM, Giesajtis G, Brown RD, et al. Clonal cytotoxic T cells are expanded in myeloma and reside in the CD8(+)CD57(+)CD28(‐) compartment. Blood 2001;98:2817–2827. [DOI] [PubMed] [Google Scholar]
- 87. Wang EC, Borysiewicz LK. The role of CD8+, CD57+ cells in human cytomegalovirus and other viral infections. Scand J Infect Dis Suppl 1995;99:69–77. [PubMed] [Google Scholar]
- 88. Tarazona R, DelaRosa O, Alonso C, et al. Increased expression of NK cell markers on T lymphocytes in aging and chronic activation of the immune system reflects the accumulation of effector/senescent T cells. Mech Ageing Dev 2000;121:77–88. [DOI] [PubMed] [Google Scholar]
- 89. Focosi D, Bestagno M, Burrone O, Petrini M. CD57+ T lymphocytes and functional immune deficiency. J Leukoc Biol 2010;87:107–116. [DOI] [PubMed] [Google Scholar]
- 90. Brenchley JM, Karandikar NJ, Betts MR, et al. Expression of CD57 defines replicative senescence and antigen‐induced apoptotic death of CD8+ T cells. Blood 2003;101:2711–2720. [DOI] [PubMed] [Google Scholar]
- 91. Le Priol Y, Puthier D, Lécureuil C, et al. High cytotoxic and specific migratory potencies of senescent CD8+ CD57+ cells in HIV‐infected and uninfected individuals. J Immunol 2006;177:5145–5154. [DOI] [PubMed] [Google Scholar]
- 92. Böttcher JP, Beyer M, Meissner F, et al. Functional classification of memory CD8(+) T cells by CX3CR1 expression. Nat Commun 2015;6:8306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Chong LK, Aicheler RJ, Llewellyn‐Lacey S, et al. Proliferation and interleukin 5 production by CD8hi CD57+ T cells. Eur J Immunol 2008;38:995–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Strioga M, Pasukoniene V, Characiejus D. CD8+ CD28‐ and CD8+ CD57+ T cells and their role in health and disease. Immunology 2011;134:17–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Traitanon O, Gorbachev A, Bechtel JJ, et al. IL‐15 induces alloreactive CD28(‐) memory CD8 T cell proliferation and CTLA4‐Ig resistant memory CD8 T cell activation. Am J Transplant 2014;14:1277–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Koskela HLM, Eldfors S, Ellonen P, et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. N Engl J Med 2012;366:1905–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Greenberg SA, Pinkus JL, Amato AA, et al. Association of inclusion body myositis with T cell large granular lymphocytic leukaemia. Brain 2016;139(Pt 5):1348–1360. [DOI] [PubMed] [Google Scholar]
- 98. Burks EJ, Loughran TP. Pathogenesis of neutropenia in large granular lymphocyte leukemia and Felty syndrome. Blood Rev 2006;20:245–266. [DOI] [PubMed] [Google Scholar]
- 99. Loughran TP, Kadin ME, Starkebaum G, et al. Leukemia of large granular lymphocytes: association with clonal chromosomal abnormalities and autoimmune neutropenia, thrombocytopenia, and hemolytic anemia. Ann Intern Med 1985;102:169–175. [DOI] [PubMed] [Google Scholar]
- 100. Steinway SN, LeBlanc F, Loughran TP. The pathogenesis and treatment of large granular lymphocyte leukemia. Blood Rev 2014;28:87–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Bareau B, Rey J, Hamidou M, et al. Analysis of a French cohort of patients with large granular lymphocyte leukemia: a report on 229 cases. Haematologica 2010;95:1534–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Askanas V, Engel WK. Inclusion‐body myositis: a myodegenerative conformational disorder associated with Abeta, protein misfolding, and proteasome inhibition. Neurology 2006;66(2 Suppl 1):S39–S48. [DOI] [PubMed] [Google Scholar]
- 103. Askanas V, Engel WK, Nogalska A. Inclusion body myositis: a degenerative muscle disease associated with intra‐muscle fiber multi‐protein aggregates, proteasome inhibition, endoplasmic reticulum stress and decreased lysosomal degradation. Brain Pathol 2009;19:493–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Hohlfeld R, Schulze‐Koops H. Cytotoxic T cells go awry in inclusion body myositis. Brain 2016;139(Pt 5):1312–1314. [DOI] [PubMed] [Google Scholar]
- 105. Greenberg SA, Pinkus GS, Amato AA, Pinkus JL. Myeloid dendritic cells in inclusion‐body myositis and polymyositis. Muscle Nerve 2007;35:17–23. [DOI] [PubMed] [Google Scholar]
- 106. Greenberg SA, Bradshaw EM, Pinkus JL, et al. Plasma cells in muscle in inclusion body myositis and polymyositis. Neurology 2005;65:1782–1787. [DOI] [PubMed] [Google Scholar]
- 107. Englund P, Wahlström J, Fathi M, et al. Restricted T cell receptor BV gene usage in the lungs and muscles of patients with idiopathic inflammatory myopathies. Arthritis Rheum 2007;56:372–383. [DOI] [PubMed] [Google Scholar]
- 108. Keller CW, Fokken C, Turville SG, et al. TNF‐alpha induces macroautophagy and regulates MHC class II expression in human skeletal muscle cells. J Biol Chem 2011;286:3970–3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Goebels N, Michaelis D, Wekerle H, Hohlfeld R. Human myoblasts as antigen‐presenting cells. J Immunol 1992;149:661–667. [PubMed] [Google Scholar]
- 110. Wiendl H, Mitsdoerffer M, Schneider D, et al. Muscle fibres and cultured muscle cells express the B7.1/2‐related inducible co‐stimulatory molecule, ICOSL: implications for the pathogenesis of inflammatory myopathies. Brain 2003;126(Pt 5):1026–1035. [DOI] [PubMed] [Google Scholar]
- 111. Jain A, Sharma MC, Sarkar C, et al. Major histocompatibility complex class I and II detection as a diagnostic tool in idiopathic inflammatory myopathies. Arch Pathol Lab Med 2007;131:1070–1076. [DOI] [PubMed] [Google Scholar]
- 112. Ivanidze J, Hoffmann R, Lochmuller H, et al. Inclusion body myositis: laser microdissection reveals differential up‐regulation of IFN‐γ signaling cascade in attacked versus nonattacked myofibers. Am J Pathol 2011;179:1347–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Josefowicz SZ, Lu L‐F, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol 2012;30:531–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Pesenacker AM, Cook L, Levings MK. The role of FOXP3 in autoimmunity. Curr Opin Immunol 2016;43:16–23. [DOI] [PubMed] [Google Scholar]
- 115. Schiaffino S, Pereira MG, Ciciliot S, Rovere‐Querini P. Regulatory T cells and skeletal muscle regeneration. FEBS J 2016;4:517–524. [DOI] [PubMed] [Google Scholar]
- 116. Panduro M, Benoist C, Mathis D. Tissue Tregs. Annu Rev Immunol 2016;34:609–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Burzyn D, Kuswanto W, Kolodin D, et al. A special population of regulatory T cells potentiates muscle repair. Cell 2013;155:1282–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Kuswanto W, Burzyn D, Panduro M, et al. Poor Repair of Skeletal Muscle in Aging Mice Reflects a Defect in Local, Interleukin‐33‐Dependent Accumulation of Regulatory T Cells. Immunity 2016;44:355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Allenbach Y, Solly S, Grégoire S, et al. Role of regulatory T cells in a new mouse model of experimental autoimmune myositis. Am J Pathol 2009;174:989–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Young NA, Sharma R, Friedman AK, et al. Aberrant muscle antigen exposure in mice is sufficient to cause myositis in a Treg cell‐deficient milieu. Arthritis Rheum 2013;65:3259–3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Waschbisch A, Schwab N, Ruck T, et al. FOXP3+ T regulatory cells in idiopathic inflammatory myopathies. J Neuroimmunol 2010;225:137–142. [DOI] [PubMed] [Google Scholar]
- 122. Dalakas MC, Illa I, Gallardo E, Juarez C. Inclusion body myositis and paraproteinemia: incidence and immunopathologic correlations. Ann Neurol 1997;41:100–104. [DOI] [PubMed] [Google Scholar]
- 123. Bradshaw EM, Orihuela A, McArdel SL, et al. A local antigen‐driven humoral response is present in the inflammatory myopathies. J Immunol 2007;178:547–556. [DOI] [PubMed] [Google Scholar]
- 124. Corcoran LM, Tarlinton DM. Regulation of germinal center responses, memory B cells and plasma cell formation‐an update. Curr Opin Immunol 2016;39:59–67. [DOI] [PubMed] [Google Scholar]
- 125. DeFranco AL. The germinal center antibody response in health and disease. F1000Res 2016;5:999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Victora GD, Nussenzweig MC. Germinal centers. Annu Rev Immunol 2012;30:429–457. [DOI] [PubMed] [Google Scholar]
- 127. De Bleecker JL, Engel AG, Butcher EC. Peripheral lymphoid tissue‐like adhesion molecule expression in nodular infiltrates in inflammatory myopathies. Neuromuscul Disord 1996;6:255–260. [DOI] [PubMed] [Google Scholar]
- 128. Salajegheh M, Pinkus JL, Amato AA, et al. Permissive environment for B‐cell maturation in myositis muscle in the absence of B‐cell follicles. Muscle Nerve 2010;42:576–583. [DOI] [PubMed] [Google Scholar]
- 129. Akiba H, Takeda K, Kojima Y, et al. The role of ICOS in the CXCR5+ follicular B helper T cell maintenance in vivo. J Immunol 2005;175:2340–2348. [DOI] [PubMed] [Google Scholar]
- 130. Mackay F, Schneider P, Rennert P, Browning J. BAFF AND APRIL: a tutorial on B cell survival. Annu Rev Immunol 2003;21:231–264. [DOI] [PubMed] [Google Scholar]
- 131. Krystufková O, Vallerskog T, Helmers SB, et al. Increased serum levels of B cell activating factor (BAFF) in subsets of patients with idiopathic inflammatory myopathies. Ann Rheum Dis 2009;68:836–843. [DOI] [PubMed] [Google Scholar]
- 132. Hengstman GJ, van Engelen BG, Badrising UA, et al. Presence of the anti‐Jo‐1 autoantibody excludes inclusion body myositis. Ann Neurol 1998;44:423–423. [DOI] [PubMed] [Google Scholar]
- 133. Hengstman GJ, Ter Laak HJ, van Engelen BG, van Venrooij BG. Anti‐Jo‐1 positive inclusion body myositis with a marked and sustained clinical improvement after oral prednisone. J Neurol Neurosurg Psychiatr 2001;70:706–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Koffman BM, Rugiero M, Dalakas MC. Immune‐mediated conditions and antibodies associated with sporadic inclusion body myositis. Muscle Nerve 1998;21:115–117. [DOI] [PubMed] [Google Scholar]
- 135. Casciola‐Rosen L, Mammen AL. Myositis autoantibodies. Curr Opin Rheumatol 2012;24:602–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Zong M, Lundberg IE. Pathogenesis, classification and treatment of inflammatory myopathies. Nat Rev Rheumatol 2011;7:297–306. [DOI] [PubMed] [Google Scholar]
- 137. Salajegheh M, Lam T, Greenberg SA. Autoantibodies against a 43 KDa muscle protein in inclusion body myositis. PLoS ONE 2011;6:e20266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Benjamin Larman H, Salajegheh M, Nazareno R, et al. Cytosolic 5′‐nucleotidase 1A autoimmunity in sporadic inclusion body myositis. Ann Neurol 2013;73:408–418. [DOI] [PubMed] [Google Scholar]
- 139. Pluk H, van Hoeve BJA, van Dooren SHJ, et al. Autoantibodies to cytosolic 5′‐nucleotidase 1A in inclusion body myositis. Ann Neurol 2013;73:397–407. [DOI] [PubMed] [Google Scholar]
- 140. Lloyd TE, Christopher‐Stine L, Pinal‐Fernandez I, et al. Cytosolic 5′‐Nucleotidase 1A As a Target of Circulating Autoantibodies in Autoimmune Diseases. Arthritis Care Res (Hoboken) 2016;68:66–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Limaye VS, Lester S, Blumbergs P, Greenberg SA. Anti‐ C N1A antibodies in South Australian patients with inclusion body myositis. Muscle Nerve 2016;53:654–655. [DOI] [PubMed] [Google Scholar]
- 142. Herbert MK, Stammen‐Vogelzangs J, Verbeek MM, et al. Disease specificity of autoantibodies to cytosolic 5′‐nucleotidase 1A in sporadic inclusion body myositis versus known autoimmune diseases. Ann Rheum Dis 2016;75:696–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Lilleker JB, Rietveld A, Pye SR, et al. Cytosolic 5′‐nucleotidase 1A autoantibody profile and clinical characteristics in inclusion body myositis. Ann Rheum Dis 2017;76:862–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Tawara N, Yamashita S, Zhang X, et al. Pathomechanisms of anti‐cytosolic 5′‐nucleotidase 1A autoantibodies in sporadic inclusion body myositis. Ann Neurol 2017;259(suppl 4):554. [DOI] [PubMed] [Google Scholar]
- 145. Greenberg SA. Inclusion body myositis pathogenesis: Steady progress. Ann Neurol 2017;158(pt 11):1453. [DOI] [PubMed] [Google Scholar]
- 146. De Paepe B, Zschüntzsch J. Scanning for Therapeutic Targets within the Cytokine Network of Idiopathic Inflammatory Myopathies. Int J Mol Sci 2015;16:18683–18713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Lundberg I, Ulfgren AK, Nyberg P, et al. Cytokine production in muscle tissue of patients with idiopathic inflammatory myopathies. Arthritis Rheum 1997;40:865–874. [DOI] [PubMed] [Google Scholar]
- 148. De Bleecker JL, Meire VI, Declercq W, Van Aken EH. Immunolocalization of tumor necrosis factor‐alpha and its receptors in inflammatory myopathies. Neuromuscul Disord 1999;9:239–246. [DOI] [PubMed] [Google Scholar]
- 149. Peterson JM, Feeback KD, Baas JH, Pizza FX. Tumor necrosis factor‐alpha promotes the accumulation of neutrophils and macrophages in skeletal muscle. J Appl Physiol 2006;101:1394–1399. [DOI] [PubMed] [Google Scholar]
- 150. Georgantas RW, Streicher K, Greenberg SA, et al. Inhibition of myogenic MicroRNAs 1, 133, and 206 by inflammatory cytokines links inflammation and muscle degeneration in adult inflammatory myopathies. Arthritis Rheumatol 2014;66:1022–1033. [DOI] [PubMed] [Google Scholar]
- 151. Vallejo AN, Mügge LO, Klimiuk PA, et al. Central role of thrombospondin‐1 in the activation and clonal expansion of inflammatory T cells. J Immunol 2000;164:2947–2954. [DOI] [PubMed] [Google Scholar]
- 152. Vallejo AN, Yang H, Klimiuk PA, et al. Synoviocyte‐mediated expansion of inflammatory T cells in rheumatoid synovitis is dependent on CD47‐thrombospondin 1 interaction. J Immunol 2003;171:1732–1740. [DOI] [PubMed] [Google Scholar]
- 153. Salajegheh M, Raju R, Schmidt J, Dalakas MC. Upregulation of thrombospondin‐1(TSP‐1) and its binding partners, CD36 and CD47, in sporadic inclusion body myositis. J Neuroimmunol 2007;187:166–174. [DOI] [PubMed] [Google Scholar]
- 154. van der Pas J, Hengstman GJD, Ter Laak HJ, et al. Diagnostic value of MHC class I staining in idiopathic inflammatory myopathies. J Neurol Neurosurg Psychiatr 2004;75:136–139. [PMC free article] [PubMed] [Google Scholar]
- 155. Emslie‐Smith AM, Arahata K, Engel AG. Major histocompatibility complex class I antigen expression, immunolocalization of interferon subtypes, and T cell‐mediated cytotoxicity in myopathies. Hum Pathol 1989;20:224–231. [DOI] [PubMed] [Google Scholar]
- 156. Raju R, Vasconcelos O, Granger R, Dalakas MC. Expression of IFN‐gamma‐inducible chemokines in inclusion body myositis. J Neuroimmunol 2003;141:125–131. [DOI] [PubMed] [Google Scholar]
- 157. De Paepe B, De Keyzer K, Martin J‐J, De Bleecker JL. Alpha‐chemokine receptors CXCR1‐3 and their ligands in idiopathic inflammatory myopathies. Acta Neuropathol 2005;109:576–582. [DOI] [PubMed] [Google Scholar]
- 158. Bao S, King NJ, Dos Remedios CG. Flavivirus induces MHC antigen on human myoblasts: a model of autoimmune myositis?. Muscle Nerve 1992;15:1271–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Chevrel G, Granet C, Miossec P. Contribution of tumour necrosis factor alpha and interleukin (IL) 1beta to IL6 production, NF‐kappaB nuclear translocation, and class I MHC expression in muscle cells: in vitro regulation with specific cytokine inhibitors. Ann Rheum Dis 2005;64:1257–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Casola A, Henderson A, Liu T, et al. Regulation of RANTES promoter activation in alveolar epithelial cells after cytokine stimulation. Am J Physiol Lung Cell Mol Physiol 2002;283:L1280–L1290. [DOI] [PubMed] [Google Scholar]
- 161. Kovacic JC, Gupta R, Lee AC, et al. Stat3‐dependent acute Rantes production in vascular smooth muscle cells modulates inflammation following arterial injury in mice. J Clin Invest 2010;120:303–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Kumar M, Makonchuk DY, Li H, et al. TNF‐like weak inducer of apoptosis (TWEAK) activates proinflammatory signaling pathways and gene expression through the activation of TGF‐beta‐activated kinase 1. J Immunol 2009;182:2439–2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Chicheportiche Y, Bourdon PR, Xu H, et al. TWEAK, a new secreted ligand in the tumor necrosis factor family that weakly induces apoptosis. J Biol Chem 1997;272:32401–32410. [DOI] [PubMed] [Google Scholar]
- 164. Girgenrath M, Weng S, Kostek CA, et al. TWEAK, via its receptor Fn14, is a novel regulator of mesenchymal progenitor cells and skeletal muscle regeneration. EMBO J 2006;25:5826–5839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Kaplan MJ, Lewis EE, Shelden EA, et al. The apoptotic ligands TRAIL, TWEAK, and Fas ligand mediate monocyte death induced by autologous lupus T cells. J Immunol 2002;169:6020–6029. [DOI] [PubMed] [Google Scholar]
- 166. Maecker H, Varfolomeev E, Kischkel F, et al. TWEAK attenuates the transition from innate to adaptive immunity. Cell 2005;123:931–944. [DOI] [PubMed] [Google Scholar]
- 167. Nakayama M, Ishidoh K, Kojima Y, et al. Fibroblast growth factor‐inducible 14 mediates multiple pathways of TWEAK‐induced cell death. J Immunol 2003;170:341–348. [DOI] [PubMed] [Google Scholar]
- 168. Dogra C, Changotra H, Mohan S, Kumar A. Tumor necrosis factor‐like weak inducer of apoptosis inhibits skeletal myogenesis through sustained activation of nuclear factor‐kappaB and degradation of MyoD protein. J Biol Chem 2006;281:10327–10336. [DOI] [PubMed] [Google Scholar]
- 169. Morosetti R, Mirabella M, Gliubizzi C, et al. MyoD expression restores defective myogenic differentiation of human mesoangioblasts from inclusion‐body myositis muscle. Proc Natl Acad Sci 2006;103:16995–17000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Morosetti R, Gliubizzi C, Sancricca C, et al. TWEAK in inclusion‐body myositis muscle: possible pathogenic role of a cytokine inhibiting myogenesis. Am J Pathol 2012;180:1603–1613. [DOI] [PubMed] [Google Scholar]
- 171. Burkly LC, Michaelson JS, Hahm K, et al. TWEAKing tissue remodeling by a multifunctional cytokine: role of TWEAK/Fn14 pathway in health and disease. Cytokine 2007;40:1–16. [DOI] [PubMed] [Google Scholar]
- 172. Mittal A, Bhatnagar S, Kumar A, et al. The TWEAK‐Fn14 system is a critical regulator of denervation‐induced skeletal muscle atrophy in mice. J Cell Biol 2010;188:833–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Tajrishi MM, Shin J, Hetman M, Kumar A. DNA methyltransferase 3a and mitogen‐activated protein kinase signaling regulate the expression of fibroblast growth factor‐inducible 14 (Fn14) during denervation‐induced skeletal muscle atrophy. J Biol Chem 2014;289:19985–19999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Langen RC, Schols AM, Kelders MC, et al. Inflammatory cytokines inhibit myogenic differentiation through activation of nuclear factor‐kappaB. FASEB J 2001;15:1169–1180. [DOI] [PubMed] [Google Scholar]
- 175. Langen RCJ, Van Der Velden JLJ, Schols AMWJ, et al. Tumor necrosis factor‐alpha inhibits myogenic differentiation through MyoD protein destabilization. FASEB J 2004;18:227–237. [DOI] [PubMed] [Google Scholar]
- 176. Kawashima R, Kawamura YI, Oshio T, et al. Interleukin‐13 damages intestinal mucosa via TWEAK and Fn14 in mice‐a pathway associated with ulcerative colitis. Gastroenterology 2011;141:2119–2129.e8. [DOI] [PubMed] [Google Scholar]
- 177. Son A, Oshio T, Kawamura YI, et al. TWEAK/Fn14 pathway promotes a T helper 2‐type chronic colitis with fibrosis in mice. Mucosal Immunol 2013;6:1131–1142. [DOI] [PubMed] [Google Scholar]
- 178. Huang P‐L, Hou M‐S, Wang S‐W, et al. Skeletal muscle interleukin 15 promotes CD8(+) T‐cell function and autoimmune myositis. Skelet Muscle 2015;5:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Moran EM, Mastaglia FL. The role of interleukin‐17 in immune‐mediated inflammatory myopathies and possible therapeutic implications. Neuromuscul Disord 2014;1–10. [DOI] [PubMed] [Google Scholar]
- 180. Askanas V, Alvarez RB, Engel WK. beta‐Amyloid precursor epitopes in muscle fibers of inclusion body myositis. Ann Neurol 1993;34:551–560. [DOI] [PubMed] [Google Scholar]
- 181. Askanas V, Engel WK, Alvarez RB, Glenner GG. beta‐Amyloid protein immunoreactivity in muscle of patients with inclusion‐body myositis. The Lancet 1992;339:560–561. [DOI] [PubMed] [Google Scholar]
- 182. Askanas V, Engel WK, Alvarez RB. Light and electron microscopic localization of beta‐amyloid protein in muscle biopsies of patients with inclusion‐body myositis. Am J Pathol 1992;141:31–36. [PMC free article] [PubMed] [Google Scholar]
- 183. Bitan G, Kirkitadze MD, Lomakin A, et al. Amyloid beta ‐protein (Abeta) assembly: abeta 40 and Abeta 42 oligomerize through distinct pathways. Proc Natl Acad Sci 2003;100:330–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Chen Y‐R, Glabe CG. Distinct early folding and aggregation properties of Alzheimer amyloid‐beta peptides Abeta40 and Abeta42: stable trimer or tetramer formation by Abeta42. J Biol Chem 2006;281:24414–24422. [DOI] [PubMed] [Google Scholar]
- 185. Serpell LC. Alzheimer's amyloid fibrils: structure and assembly. Biochim Biophys Acta 2000;1502:16–30. [DOI] [PubMed] [Google Scholar]
- 186. Vattemi G, Nogalska A, King Engel W, et al. Amyloid‐beta42 is preferentially accumulated in muscle fibers of patients with sporadic inclusion‐body myositis. Acta Neuropathol 2009;117:569–574. [DOI] [PubMed] [Google Scholar]
- 187. Nogalska A, D'Agostino C, Engel WK, et al. Novel demonstration of amyloid‐β oligomers in sporadic inclusion‐body myositis muscle fibers. Acta Neuropathol 2010;120:661–666. [DOI] [PubMed] [Google Scholar]
- 188. Askanas V, Engel WK. Sporadic inclusion‐body myositis: conformational multifactorial ageing‐related degenerative muscle disease associated with proteasomal and lysosomal inhibition, endoplasmic reticulum stress, and accumulation of amyloid‐β42 oligomers and phosphorylated tau. Presse Med 2011;40(4 Pt 2):e219–e235. [DOI] [PubMed] [Google Scholar]
- 189. Scheuner D, Eckman C, Jensen M, et al. Secreted amyloid beta‐protein similar to that in the senile plaques of Alzheimer”s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer”s disease. Nat Med 1996;2:864–870. [DOI] [PubMed] [Google Scholar]
- 190. Siman R, Mistretta S, Durkin JT, et al. Processing of the beta‐amyloid precursor. Multiple proteases generate and degrade potentially amyloidogenic fragments. J Biol Chem 1993;268:16602–16609. [PubMed] [Google Scholar]
- 191. Vetrivel KS, Thinakaran G. Amyloidogenic processing of beta‐amyloid precursor protein in intracellular compartments. Neurology 2006;66(2 Suppl 1):S69–S73. [DOI] [PubMed] [Google Scholar]
- 192. Vattemi G, Engel WK, McFerrin J, et al. Presence of BACE1 and BACE2 in muscle fibres of patients with sporadic inclusion‐body myositis. The Lancet 2001;358:1962–1964. [DOI] [PubMed] [Google Scholar]
- 193. Nogalska A, D'Agostino C, Engel WK, Askanas V. Activation of the γ‐secretase complex and presence of γ‐secretase‐activating protein may contribute to Aβ42 production in sporadic inclusion‐body myositis muscle fibers. Neurobiol Dis 2012;48:141–149. [DOI] [PubMed] [Google Scholar]
- 194. He G, Luo W, Li P, et al. Gamma‐secretase activating protein is a therapeutic target for Alzheimer's disease. Nature 2010;467:95–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Aplin AE, Gibb GM, Jacobsen JS, et al. In vitro phosphorylation of the cytoplasmic domain of the amyloid precursor protein by glycogen synthase kinase‐3beta. J Neurochem 1996;67:699–707. [DOI] [PubMed] [Google Scholar]
- 196. Shin R‐W, Ogino K, Shimabuku A, et al. Amyloid precursor protein cytoplasmic domain with phospho‐Thr668 accumulates in Alzheimer's disease and its transgenic models: a role to mediate interaction of Abeta and tau. Acta Neuropathol 2007;113:627–636. [DOI] [PubMed] [Google Scholar]
- 197. Terracciano C, Nogalska A, Engel WK, Askanas V. In AbetaPP‐overexpressing cultured human muscle fibers proteasome inhibition enhances phosphorylation of AbetaPP751 and GSK3beta activation: effects mitigated by lithium and apparently relevant to sporadic inclusion‐body myositis. J Neurochem 2010;112:389–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Abdo WF, van Mierlo T, Hengstman GJ, et al. Increased plasma amyloid‐beta42 protein in sporadic inclusion body myositis. Acta Neuropathol 2009;118:429–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Catalán‐García M, Garrabou G, Morén C, et al. BACE‐1, PS‐1 and sAPPβ levels are increased in plasma from sporadic inclusion body myositis patients: surrogate biomarkers among inflammatory myopathies. Mol Med 2015;21:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Bloom GS. Amyloid‐β and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol 2014;71:505–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Honson NS, Kuret J. Tau aggregation and toxicity in tauopathic neurodegenerative diseases. J Alzheimers Dis 2008;14:417–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Karran E, Mercken M, De Strooper B. The amyloid cascade hypothesis for Alzheimer's disease: an appraisal for the development of therapeutics. Nat Rev Drug Discov 2011;10:698–712. [DOI] [PubMed] [Google Scholar]
- 203. Götz J, Chen F, vanDorpe J , Nitsch RM. Formation of neurofibrillary tangles in P301 l tau transgenic mice induced by Abeta 42 fibrils. Science 2001;293:1491–1495. [DOI] [PubMed] [Google Scholar]
- 204. Lewis J, Dickson DW, Lin WL, et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 2001;293:1487–1491. [DOI] [PubMed] [Google Scholar]
- 205. Rapoport M, Dawson HN, Binder LI, et al. Tau is essential to beta ‐amyloid‐induced neurotoxicity. Proc Natl Acad Sci 2002;99:6364–6369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Mirabella M, Alvarez RB, Bilak M, et al. Difference in expression of phosphorylated tau epitopes between sporadic inclusion‐body myositis and hereditary inclusion‐body myopathies. J Neuropathol Exp Neurol 1996;55:774–786. [DOI] [PubMed] [Google Scholar]
- 207. Askanas V, Engel WK, Bilak M, et al. Twisted tubulofilaments of inclusion body myositis muscle resemble paired helical filaments of Alzheimer brain and contain hyperphosphorylated tau. Am J Pathol 1994;144:177–187. [PMC free article] [PubMed] [Google Scholar]
- 208. Wilczynski GM, Engel WK, Askanas V. Association of active extracellular signal‐regulated protein kinase with paired helical filaments of inclusion‐body myositis muscle suggests its role in inclusion‐body myositis tau phosphorylation. Am J Pathol 2000;156:1835–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Nakano S, Shinde A, Kawashima S, et al. Inclusion body myositis: expression of extracellular signal‐regulated kinase and its substrate. Neurology 2001;56:87–93. [DOI] [PubMed] [Google Scholar]
- 210. Kitazawa M, Trinh DN, LaFerla FM. Inflammation induces tau pathology in inclusion body myositis model via glycogen synthase kinase‐3beta. Ann Neurol 2008;64:15–24. [DOI] [PubMed] [Google Scholar]
- 211. Angelova PR, Abramov AY. Alpha‐synuclein and beta‐amyloid ‐ different targets, same players: calcium, free radicals and mitochondria in the mechanism of neurodegeneration. Biochem Biophys Res Commun 2016;4:1110–1115. [DOI] [PubMed] [Google Scholar]
- 212. Spillantini MG, Schmidt ML, Lee VM, et al. Alpha‐synuclein in Lewy bodies. Nature 1997;388:839–840. [DOI] [PubMed] [Google Scholar]
- 213. Goedert M, Masuda‐Suzukake M, Falcon B. Like prions: the propagation of aggregated tau and α‐synuclein in neurodegeneration. Brain 2016;Pt 2, 266–278:aww230. [DOI] [PubMed] [Google Scholar]
- 214. Kim WS, Kågedal K, Halliday GM. Alpha‐synuclein biology in Lewy body diseases. Alzheimers Res Ther 2014;6:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Askanas V, Engel WK, Alvarez RB, et al. Novel immunolocalization of alpha‐synuclein in human muscle of inclusion‐body myositis, regenerating and necrotic muscle fibers, and at neuromuscular junctions. J Neuropathol Exp Neurol 2000;59:592–598. [DOI] [PubMed] [Google Scholar]
- 216. Xiang W, Schlachetzki JCM, Helling S, et al. Oxidative stress‐induced posttranslational modifications of alpha‐synuclein: specific modification of alpha‐synuclein by 4‐hydroxy‐2‐nonenal increases dopaminergic toxicity. Mol Cell Neurosci 2013;54:71–83. [DOI] [PubMed] [Google Scholar]
- 217. Xiong R, Zhou W, Siegel D, et al. A Novel Hsp90 Inhibitor Activates Compensatory Heat Shock Protein Responses and Autophagy and Alleviates Mutant A53T α‐Synuclein Toxicity. Mol Pharmacol 2015;88:1045–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Jones DR, Moussaud S, McLean P. Targeting heat shock proteins to modulate α‐synuclein toxicity. Ther Adv Neurol Disord 2014;7:33–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Kawakami F, Suzuki M, Shimada N, et al. Stimulatory effect of α‐synuclein on the tau‐phosphorylation by GSK‐3β . FEBS J 2011;278:4895–4904. [DOI] [PubMed] [Google Scholar]
- 220. Askanas V, Mirabella M, Engel WK, et al. Apolipoprotein E immunoreactive deposits in inclusion‐body muscle diseases. The Lancet 1994;343:364–365. [DOI] [PubMed] [Google Scholar]
- 221. Mirabella M, Alvarez RB, Engel WK, et al. Apolipoprotein E and apolipoprotein E messenger RNA in muscle of inclusion body myositis and myopathies. Ann Neurol 1996;40:864–872. [DOI] [PubMed] [Google Scholar]
- 222. Nogalska A, Terracciano C, D'Agostino C, et al. p62/SQSTM1 is overexpressed and prominently accumulated in inclusions of sporadic inclusion‐body myositis muscle fibers, and can help differentiating it from polymyositis and dermatomyositis. Acta Neuropathol 2009;118:407–413. [DOI] [PubMed] [Google Scholar]
- 223. Sarkozi E, Askanas V, Engel WK. Abnormal accumulation of prion protein mRNA in muscle fibers of patients with sporadic inclusion‐body myositis and hereditary inclusion‐body myopathy. Am J Pathol 1994;145:1280–1284. [PMC free article] [PubMed] [Google Scholar]
- 224. Askanas V, Bilak M, Engel WK, et al. Prion protein is abnormally accumulated in inclusion‐body myositis. NeuroReport 1993;5:25–28. [DOI] [PubMed] [Google Scholar]
- 225. Zanusso G, Vattemi G, Ferrari S, et al. Increased expression of the normal cellular isoform of prion protein in inclusion‐body myositis, inflammatory myopathies and denervation atrophy. Brain Pathol 2001;11:182–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Ahmed M, Machado PM, Miller A, et al. Targeting protein homeostasis in sporadic inclusion body myositis. Sci Transl Med 2016;8:331ra41–331ra41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Serrano‐Pozo A, Qian J, Monsell SE, et al. Mild to moderate Alzheimer dementia with insufficient neuropathological changes. Ann Neurol 2014;75:597–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Beach TG, Monsell SE, Phillips LE, Kukull W. Accuracy of the clinical diagnosis of Alzheimer disease at National Institute on Aging Alzheimer Disease Centers, 2005‐2010. J Neuropathol Exp Neurol 2012;71:266–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Monsell SE, Kukull WA, Roher AE, et al. Characterizing Apolipoprotein E ε4 Carriers and Noncarriers With the Clinical Diagnosis of Mild to Moderate Alzheimer Dementia and Minimal β‐Amyloid Peptide Plaques. JAMA Neurol 2015;72:1124–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Kaeberlein M. Deciphering the role of natural variation in age‐related protein homeostasis. BMC Biol 2013;11:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Komatsu M, Waguri S, Ueno T, et al. Impairment of starvation‐induced and constitutive autophagy in Atg7‐deficient mice. J Cell Biol 2005;169:425–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Hara T, Nakamura K, Matsui M, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006;441:885–889. [DOI] [PubMed] [Google Scholar]
- 233. Komatsu M, Waguri S, Chiba T, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006;441:880–884. [DOI] [PubMed] [Google Scholar]
- 234. Nakai A, Yamaguchi O, Takeda T, et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med 2007;13:619–624. [DOI] [PubMed] [Google Scholar]
- 235. Vilchez D, Saez I, Dillin A. The role of protein clearance mechanisms in organismal ageing and age‐related diseases. Nat Commun 2014;5:5659. [DOI] [PubMed] [Google Scholar]
- 236. Martínez‐Vicente M, Cuervo AM. Autophagy and neurodegeneration: when the cleaning crew goes on strike. Lancet Neurol 2007;6:352–361. [DOI] [PubMed] [Google Scholar]
- 237. Kaushik S, Cuervo AM. Proteostasis and aging. Nat Med 2015;21:1406–1415. [DOI] [PubMed] [Google Scholar]
- 238. López‐Otín C, Blasco MA, Partridge L, et al. The hallmarks of aging. Cell 2013;153:1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Finley D. Recognition and processing of ubiquitin‐protein conjugates by the proteasome. Annu Rev Biochem 2009;78:477–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Stadtmueller BM, Hill CP. Proteasome activators. Mol Cell 2011;41:8–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Davies KJ. Degradation of oxidized proteins by the 20S proteasome. Biochimie 2001;83:301–310. [DOI] [PubMed] [Google Scholar]
- 242. Fratta P, Engel WK, McFerrin J, et al. Proteasome inhibition and aggresome formation in sporadic inclusion‐body myositis and in amyloid‐beta precursor protein‐overexpressing cultured human muscle fibers. Am J Pathol 2005;167:517–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Groettrup M, Kirk CJ, Basler M. Proteasomes in immune cells: more than peptide producers? Nature Publishing Group 2010;10:73–78. [DOI] [PubMed] [Google Scholar]
- 244. Boes B, Hengel H, Ruppert T, et al. Interferon gamma stimulation modulates the proteolytic activity and cleavage site preference of 20S mouse proteasomes. J Exp Med 1994;179:901–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Akiyama K, Yokota K, Kagawa S, et al. cDNA cloning and interferon gamma down‐regulation of proteasomal subunits X and Y. Science 1994;265:1231–1234. [DOI] [PubMed] [Google Scholar]
- 246. Bhattarai S, Ghannam K, Krause S, et al. The immunoproteasomes are key to regulate myokines and MHC class I expression in idiopathic inflammatory myopathies. J Autoimmun 2016;75:118–129. [DOI] [PubMed] [Google Scholar]
- 247. Mizushima N, Klionsky DJ. Protein turnover via autophagy: implications for metabolism. Annu Rev Nutr 2007;27:19–40. [DOI] [PubMed] [Google Scholar]
- 248. Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell 2011;147:728–741. [DOI] [PubMed] [Google Scholar]
- 249. Münz C. Autophagy Beyond Intracellular MHC Class II Antigen Presentation. Trends Immunol 2016;37:755–763. [DOI] [PubMed] [Google Scholar]
- 250. Münz C, Of LAP. CUPS, and DRibbles ‐ Unconventional Use of Autophagy Proteins for MHC Restricted Antigen Presentation. Front Immunol 2015;6:200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Münz C. Autophagy proteins in antigen processing for presentation on MHC molecules. Immunol Rev 2016;272:17–27. [DOI] [PubMed] [Google Scholar]
- 252. Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol 2010;12:823–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Kimura T, Mandell M, Deretic V. Precision autophagy directed by receptor regulators ‐ emerging examples within the TRIM family. J Cell Sci 2016;129:881–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Maejima I, Takahashi A, Omori H, et al. Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J 2013;32:2336–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Randow F, Youle RJ. Self and nonself: how autophagy targets mitochondria and bacteria. Cell Host Microbe 2014;15:403–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Honsho M, Yamashita S‐I, Fujiki Y. Peroxisome homeostasis: mechanisms of division and selective degradation of peroxisomes in mammals. Biochim Biophys Acta 2016;1863:984–991. [DOI] [PubMed] [Google Scholar]
- 257. Rubinsztein DC, Mariño G, Kroemer G. Autophagy and aging. Cell 2011;146:682–695. [DOI] [PubMed] [Google Scholar]
- 258. Fukuhara N, Kumamoto T, Tsubaki T. Rimmed vacuoles. Acta Neuropathol 1980;51:229–235. [DOI] [PubMed] [Google Scholar]
- 259. Kumamoto T, Ueyama H, Tsumura H, et al. Expression of lysosome‐related proteins and genes in the skeletal muscles of inclusion body myositis. Acta Neuropathol 2004;107:59–65. [DOI] [PubMed] [Google Scholar]
- 260. Lünemann JD, Schmidt J, Schmid D, et al. β‐Amyloid is a substrate of autophagy in sporadic inclusion body myositis. Ann Neurol 2007;61:476–483. [DOI] [PubMed] [Google Scholar]
- 261. Girolamo F, Lia A, Amati A, et al. Overexpression of autophagic proteins in the skeletal muscle of sporadic inclusion body myositis. Neuropathol Appl Neurobiol 2013;39:736–749. [DOI] [PubMed] [Google Scholar]
- 262. Cacciottolo M, Nogalska A, D'Agostino C, et al. Chaperone‐mediated autophagy components are upregulated in sporadic inclusion‐body myositis muscle fibres. Neuropathol Appl Neurobiol 2013;39:750–761. [DOI] [PubMed] [Google Scholar]
- 263. Güttsches A‐K, Brady S, Krause K, et al. Proteomics of rimmed vacuoles define new risk allele in inclusion body myositis. Ann Neurol 2017;81:227–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Weihl CC, Baloh RH, Lee Y, et al. Targeted sequencing and identification of genetic variants in sporadic inclusion body myositis. Neuromuscul Disord 2015;25:289–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Gang Q, Bettencourt C, Machado PM, et al. Rare variants in SQSTM1 and VCP genes and risk of sporadic inclusion body myositis. Neurobiol Aging 2016;47:218.e1–218.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266. Schmidt K, Wienken M, Keller CW, et al. IL‐1β‐Induced Accumulation of Amyloid: macroautophagy in Skeletal Muscle Depends on ERK. Mediators Inflamm 2017;2017:5470831–5470837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267. Dengjel J, Schoor O, Fischer R, et al. Autophagy promotes MHC class II presentation of peptides from intracellular source proteins. Proc Natl Acad Sci USA 2005;102:7922–7927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Loi M, Müller A, Steinbach K, et al. Macroautophagy Proteins Control MHC Class I Levels on Dendritic Cells and Shape Anti‐viral CD8(+) T Cell Responses. Cell Rep 2016;15:1076–1087. [DOI] [PubMed] [Google Scholar]
- 269. Hubbard‐Lucey VM, Shono Y, Maurer K, et al. Autophagy gene Atg16L1 prevents lethal T cell alloreactivity mediated by dendritic cells. Immunity 2014;41:579–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. Lévy J, Cacheux W, Bara MA, et al. Intestinal inhibition of Atg7 prevents tumour initiation through a microbiome‐influenced immune response and suppresses tumour growth. Nat Cell Biol 2015;17:1062–1073. [DOI] [PubMed] [Google Scholar]
- 271. Wenger T, Terawaki S, Camosseto V, et al. Autophagy inhibition promotes defective neosynthesized proteins storage in ALIS, and induces redirection toward proteasome processing and MHCI‐restricted presentation. Autophagy 2012;8:350–363. [DOI] [PubMed] [Google Scholar]
- 272. Yu WH, Cuervo AM, Kumar A, et al. Macroautophagy–a novel Beta‐amyloid peptide‐generating pathway activated in Alzheimer's disease. J Cell Biol 2005;171:87–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273. Keller CW, Schmitz M, Münz C, et al. TNF‐α upregulates macroautophagic processing of APP/β‐amyloid in a human rhabdomyosarcoma cell line. J Neurol Sci 2013;325:103–107. [DOI] [PubMed] [Google Scholar]
- 274. Yang Y‐P, Hu L‐F, Zheng H‐F, et al. Application and interpretation of current autophagy inhibitors and activators. Acta Pharmacol Sin 2013;34:625–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275. Klionsky DJ, Elazar Z, Seglen PO, Rubinsztein DC. Does bafilomycin A1 block the fusion of autophagosomes with lysosomes?. Autophagy 2008;4:849–850. [DOI] [PubMed] [Google Scholar]
- 276. Chen G, Ke Z, Xu M, et al. Autophagy is a protective response to ethanol neurotoxicity. Autophagy 2012;8:1577–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277. Zhang J, Zhang Y, Li J, et al. Autophagosomes accumulation is associated with β‐amyloid deposits and secondary damage in the thalamus after focal cortical infarction in hypertensive rats. J Neurochem 2012;120:564–573. [DOI] [PubMed] [Google Scholar]
- 278. Xing S, Zhang Y, Li J, et al. Beclin 1 knockdown inhibits autophagic activation and prevents the secondary neurodegenerative damage in the ipsilateral thalamus following focal cerebral infarction. Autophagy 2012;8:63–76. [DOI] [PubMed] [Google Scholar]
- 279. Zheng L, Terman A, Hallbeck M, et al. Macroautophagy‐generated increase of lysosomal amyloid β‐protein mediates oxidant‐induced apoptosis of cultured neuroblastoma cells. Autophagy 2011;7:1528–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280. Bjørkøy G, Lamark T, Brech A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin‐induced cell death. J Cell Biol 2005;171:603–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281. Pankiv S, Clausen TH, Lamark T, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007;282:24131–24145. [DOI] [PubMed] [Google Scholar]
- 282. Seibenhener ML, Babu JR, Geetha T, et al. Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol Cell Biol 2004;24:8055–8068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283. Kirkin V, Lamark T, Sou Y‐S, et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell 2009;33:505–516. [DOI] [PubMed] [Google Scholar]
- 284. D'Agostino C, Nogalska A, Cacciottolo M, et al. Abnormalities of NBR1, a novel autophagy‐associated protein, in muscle fibers of sporadic inclusion‐body myositis. Acta Neuropathol 2011;122:627–636. [DOI] [PubMed] [Google Scholar]
- 285. Matsumoto G, Wada K, Okuno M, et al. Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol Cell 2011;44:279–289. [DOI] [PubMed] [Google Scholar]
- 286. Nakano S, Oki M, Kusaka H. The role of p62/SQSTM1 in sporadic inclusion body myositis. Neuromuscul Disord 2017;27:363–369. [DOI] [PubMed] [Google Scholar]
- 287. Yewdell JW, Antón LC, Bennink JR. Defective ribosomal products (DRiPs): a major source of antigenic peptides for MHC class I molecules? J Immunol 1996;157:1823–1826. [PubMed] [Google Scholar]
- 288. Hiniker A, Daniels BH, Lee HS, Margeta M. Comparative utility of LC3, p62 and TDP‐43 immunohistochemistry in differentiation of inclusion body myositis from polymyositis and related inflammatory myopathies. Acta Neuropathol Commun 2013;1:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289. Dalakas MC. Molecular immunology and genetics of inflammatory muscle diseases. Arch Neurol 1998;55:1509–1512. [DOI] [PubMed] [Google Scholar]
- 290. Schmidt J, Barthel K, Wrede A, et al. Interrelation of inflammation and APP in sIBM: IL‐1 beta induces accumulation of beta‐amyloid in skeletal muscle. Brain 2008;131(Pt 5):1228–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291. Adams V, Nehrhoff B, Späte U, et al. Induction of iNOS expression in skeletal muscle by IL‐1beta and NFkappaB activation: an in vitro and in vivo study. Cardiovasc Res 2002;54:95–104. [DOI] [PubMed] [Google Scholar]
- 292. Williams G, Brown T, Becker L, et al. Cytokine‐induced expression of nitric oxide synthase in C2C12 skeletal muscle myocytes. Am J Physiol 1994;267(4 Pt 2):R1020–R1025. [DOI] [PubMed] [Google Scholar]
- 293. Baron P, Galimberti D, Meda L, et al. Synergistic effect of beta‐amyloid protein and interferon gamma on nitric oxide production by C2C12 muscle cells. Brain 2000;123(Pt 2):374–379. [DOI] [PubMed] [Google Scholar]
- 294. Yang CC, Alvarez RB, Engel WK, Askanas V. Increase of nitric oxide synthases and nitrotyrosine in inclusion‐body myositis. NeuroReport 1996;8:153–158. [DOI] [PubMed] [Google Scholar]
- 295. Schmidt J, Barthel K, Zschüntzsch J, et al. Nitric oxide stress in sporadic inclusion body myositis muscle fibres: inhibition of inducible nitric oxide synthase prevents interleukin‐1β‐induced accumulation of β‐amyloid and cell death. Brain 2012;135(Pt 4):1102–1114. [DOI] [PubMed] [Google Scholar]
- 296. Kato K, Shinohara H, Goto S, et al. Copurification of small heat shock protein with alpha B crystallin from human skeletal muscle. J Biol Chem 1992;267:7718–7725. [PubMed] [Google Scholar]
- 297. Klemenz R, Fröhli E, Steiger RH, et al. Alpha B‐crystallin is a small heat shock protein. Proc Natl Acad Sci 1991;88:3652–3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298. Head MW, Corbin E, Goldman JE. Coordinate and independent regulation of alpha B‐crystallin and hsp27 expression in response to physiological stress. J Cell Physiol 1994;159:41–50. [DOI] [PubMed] [Google Scholar]
- 299. Renkawek K, Voorter CE, Bosman GJ, et al. Expression of alpha B‐crystallin in Alzheimer's disease. Acta Neuropathol 1994;87:155–160. [DOI] [PubMed] [Google Scholar]
- 300. Muth IE, Barthel K, Bähr M, et al. Proinflammatory cell stress in sporadic inclusion body myositis muscle: overexpression of alphaB‐crystallin is associated with amyloid precursor protein and accumulation of beta‐amyloid. J Neurol Neurosurg Psychiatr 2009;80:1344–1349. [DOI] [PubMed] [Google Scholar]
- 301. Banwell BL, Engel AG. AlphaB‐crystallin immunolocalization yields new insights into inclusion body myositis. Neurology 2000;54:1033–1041. [DOI] [PubMed] [Google Scholar]
- 302. Du Yan S, Zhu H, Fu J, et al. Amyloid‐beta peptide‐receptor for advanced glycation endproduct interaction elicits neuronal expression of macrophage‐colony stimulating factor: a proinflammatory pathway in Alzheimer disease. Proc Natl Acad Sci 1997;94:5296–5301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303. Andersson U, Wang H, Palmblad K, et al. High mobility group 1 protein (HMG‐1) stimulates proinflammatory cytokine synthesis in human monocytes. J Exp Med 2000;192:565–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304. Gardella S, Andrei C, Ferrera D, et al. The nuclear protein HMGB1 is secreted by monocytes via a non‐classical, vesicle‐mediated secretory pathway. EMBO Rep 2002;3:995–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305. Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002;418:191–195. [DOI] [PubMed] [Google Scholar]
- 306. Dormoy‐Raclet V, Cammas A, Celona B, et al. HuR and miR‐1192 regulate myogenesis by modulating the translation of HMGB1 mRNA. Nat Commun 2013;4:2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307. Riuzzi F, Sorci G, Sagheddu R, Donato R. HMGB1‐RAGE regulates muscle satellite cell homeostasis through p38‐MAPK‐ and myogenin‐dependent repression of Pax7 transcription. J Cell Sci 2012;125(Pt 6):1440–1454. [DOI] [PubMed] [Google Scholar]
- 308. Haslbeck KM, Friess U, Schleicher ED, et al. The RAGE pathway in inflammatory myopathies and limb girdle muscular dystrophy. Acta Neuropathol 2005;110:247–254. [DOI] [PubMed] [Google Scholar]
- 309. Ulfgren A‐K, Grundtman C, Borg K, et al. Down‐regulation of the aberrant expression of the inflammation mediator high mobility group box chromosomal protein 1 in muscle tissue of patients with polymyositis and dermatomyositis treated with corticosteroids. Arthritis Rheum 2004;50:1586–1594. [DOI] [PubMed] [Google Scholar]
- 310. Muth IE, Zschüntzsch J, Kleinschnitz K, et al. HMGB1 and RAGE in skeletal muscle inflammation: Implications for protein accumulation in inclusion body myositis. Exp Neurol 2015;271:189–197. [DOI] [PubMed] [Google Scholar]