

Abstract
Objective
To describe the rationale for a novel study design and baseline characteristics of a disease‐modifying trial of isradipine 10 mg daily in early Parkinson disease (PD).
Methods
STEADY‐PDIII is a 36‐month, Phase 3, parallel group, placebo‐controlled study of the efficacy of isradipine 10 mg daily in 336 participants with early PD as measured by the change in the Unified Parkinson Disease Rating Scale (UPDRS) Part I‐III score in the practically defined ON state. Secondary outcome measures include clinically meaningful measures of disability progression in early PD: (1) Time to initiation and utilization of dopaminergic therapy; (2) Time to onset of motor complications; (3) Change in nonmotor disability. Exploratory measures include global measures of functional disability, quality of life, change in the ambulatory capacity, cognitive function, and pharmacokinetic analysis. Rationale for the current design and alternative design approaches are discussed.
Results
The entire cohort of 336 participants was enrolled at 55 Parkinson Study Group sites in North America. The percentage of male participants were 68.5% with a mean age of 61.9 years (sd 9.0), mean Hoehn and Yahr stage of 1.7 (sd 0.5), mean UPDRS total of 23.1 (sd 8.6), and MoCA of 28.1 (sd 1.4).
Interpretation
STEADY‐PD III has a novel and innovative design allowing for the determination of longer duration benefits on clinically relevant outcomes in a relatively small cohort on top of the benefit derived from symptomatic therapy. Baseline characteristics are similar to those in previously enrolled de novo PD trials. This study represents a unique opportunity to evaluate the potential impact of a novel therapy to slow progression of PD disability and provide clinically meaningful benefits.
Introduction
Parkinson disease (PD) is a significant and increasing public health issue. PD is the second most common chronic neurodegenerative disease, after Alzheimer's disease, affecting nearly 1% of the population over the age of 65.1 The prevalence of PD is expected to double in the next 20 years in the world's most populous nations.2 The economic burden of PD is estimated to be $23 billion annually in US and projected to increase to $50 billion by year 2040.3 Current therapy is limited to symptomatic treatment; however, the disease continues to progress with accumulation of significant disability, worsening quality of life, reduced productivity, nursing home placement, and increased mortality.4 Attempts to slow or modify disease progression in PD have resulted in mixed outcomes and no treatment has yet to definitively demonstrate disease modification (see Table 1 for recent disease‐modifying trials).5, 6, 7, 8 Therefore, treatments that slow disease progression remain a major unmet therapeutic need in PD.
Table 1.
Representative Phase III disease‐modifying trials in Parkinson disease
| Trial | Intervention | N | Design | PD population | Duration | Primary outcome(s) | Results |
|---|---|---|---|---|---|---|---|
| DATATOP | Deprenyl and centertocopherol | 800 | 2 × 2 factorial | Early untreated | 24 months | Time to development of disability requiring levodopa therapy | Deprenyl resulted in reduced hazard of requiring levodopa therapy |
| PRECEPT |
CEP‐1347 10 mg BID 25 mg BID 50 mg BID |
806 | Parallel group | Early untreated | 24 months | Time to development of disability requiring dopaminergic therapy | Terminated early for futility |
| QE3 |
Coenzyme Q10 1200 mg/day 2400 mg/day |
600 | Parallel group | Early untreated | 16 months | UPDRS change | Terminated for prespecified futility |
| ADAGIO |
Rasagiline 1 mg/day 2 mg/day |
1176 | Delayed start | Early untreated | 18 months |
|
1 mg/day but not 2 mg/day met all criteria for efficacy |
| LS1 | Creatine | 1741 | Parallel group | Early stable treatment | 60 months | Global statistical test defined by 5 outcome measures: Modified Rankin Scale, Symbol Digit Modalities Test, PDQ‐39 Summary Index, Schwab and England Activities of Daily Living scale, and ambulatory capacity (UPDRS) | Terminated due to futility in an interim analysis of 955 subjects followed up for 5 years |
Isradipine, a dihydropyridine calcium channel antagonist (DHP) that is approved for the treatment of hypertension, is being tested as a potential disease‐modifying intervention in early PD. Isradipine was shown to be neuroprotective in in vitro and in vivo models of parkinsonism.9, 10 The mechanism of neuroprotection is linked to selective vulnerability of substantia nigra pars compacta neurons that preferentially express L‐type calcium channels. Neuroprotective effects of isradipine are achieved at the plasma concentration that is obtained within the safe dose range for human administration10, 11 and consistent with the tolerable dosage identified in our phase II study of isradipine in PD (STEADY‐PDII).12 Isradipine is the most potent of the clinically available Cav1.3 DHPs and has excellent central nervous system penetration suggesting it is the optimal DHP to target this novel mechanism of neuroprotection.13, 14
Importantly, multiple epidemiological studies have demonstrated a reduced risk of development of PD in individuals treated with DHPs compared with other antihypertensive agents15, 16, 17 In addition, 4733 hypertensive individuals with parkinsonism treated with DHPs had a decreased risk of requiring symptomatic therapy (ST), admission to a nursing home, and death compared with those treated with other antihypertensive agents.18 Although select epidemiological studies have failed to demonstrate this effect, these studies have been limited by small sample sizes and nonrepresentative cohorts.19, 20 Therefore, convergent data from in vivo, in vitro, and epidemiological studies strongly support the potential ability of isradipine to slow progression of disability in PD; representing the strongest preclinical and clinical rationale of any past or current putative disease‐modifying agent for PD.
In addition to sufficient preclinical and early clinical data, it is critical to use a trial design and outcomes that will be sensitive to clinically meaningful impacts of an intervention above and beyond current ST. Most previous trials (Table 1) have used designs that rely on assessments prior to the initiation of ST or have used the time to initiation of ST as the primary outcome. These studies may be affected by differential drop out and differential use of symptomatic therapies and relatively short duration of follow‐up.
STEADY‐PDIII (clinicaltrials.gov NCT02168842) is a 36–month, parallel group, double‐blind, placebo‐controlled trial that will evaluate the effect of isradipine on the progression of PD disability in untreated individuals with early PD.
Methods
Trial design
STEADY‐PD III is an ongoing 36‐month, double‐blind, randomized, placebo‐controlled study of isradipine in 336 participants with early PD at baseline not receiving or requiring ST (Fig. 1). This design will test the hypothesis that individuals treated with isradipine will have slower progression of PD disability as determined by the change in the total Unified Parkinson Disease Rating Scale (UPDRS) score21 in the active treatment arm versus placebo between baseline and 36 months. Eligible participants are randomized to isradipine 5 mg twice daily or matching placebo. Participants are titrated to the treatment dosage over a period of 4–12 weeks and then followed prospectively and systematically during a maintenance period over the remaining 36 months followed by a 3‐day titration off the study drug and 2 week post‐titration safety visit. Temporary study drug suspensions are allowed at the discretion of the investigator and participants who permanently discontinue the study drug are encouraged to remain in the study.
Figure 1.
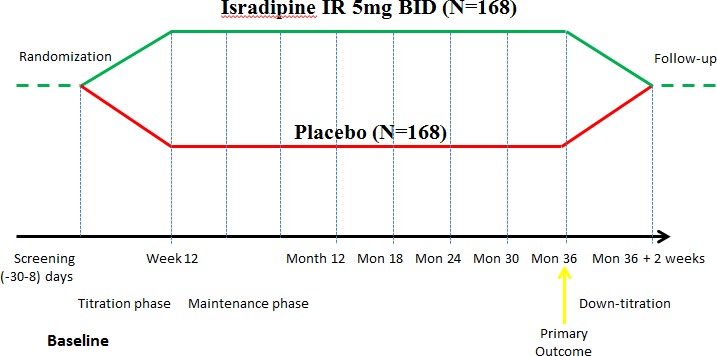
Phase III Study Design.
Setting
The study is being conducted at 57 Parkinson Study Group (PSG) sites in North America and is funded by the National Institute of Neurological Disorders and Stroke (NINDS) and the Michael J. Fox Foundation. The PSG is an independent consortium of scientific investigators committed to the cooperative planning, implementation, analysis, and reporting of controlled clinical trials and other research in PD and has successfully completed over 35 multi‐center cooperative therapeutic studies including STEADY‐PDII.
Participants
Eligible participants have early idiopathic PD (presence of two out of three cardinal manifestations of PD)22; Age greater than or equal to 30 years at the time of diagnosis; Hoehn and Yahr stage23 less than or equal to 2; Diagnosis of PD less than 3 years, currently NOT receiving ST (levodopa, dopamine agonist or MAO‐B inhibitors) and NOT projected to require ST for at least 3 months from the baseline visit. Use of amantadine and/or anticholinergics is allowed at stable dosages prior to enrollment. The key exclusion criteria include a diagnosis of an atypical parkinsonism; prior exposure to ST, history of orthostatic hypotension (based on standard definitions), bradycardia, congestive heart failure or other cardiac and other systemic diseases, abnormalities on the screening laboratories or ECG that might preclude safe participation in the study; presence of cognitive dysfunction defined by a Montreal Cognitive assessment (MOCA)(104) score < 26;24 clinically significant depression as determined by a Beck Depression Inventory II (BDI‐II) score > 15.25 Participants may take up to two other antihypertensives with the exception of calcium channel blockers which are exclusionary.
In addition, participants must meet blood pressure criteria during home blood pressure monitoring prior to initiating study drug.26
Outcome measures
Figure 2 outlines the primary and major secondary outcomes in this study.
Figure 2.
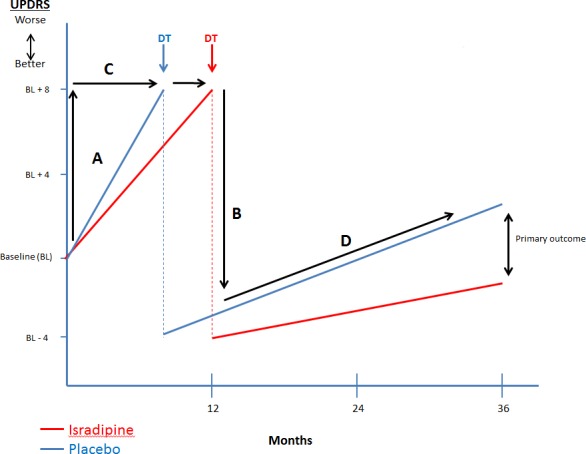
Overview of Efficacy Analyses. Primary outcomes – change in UPDRS from baseline to 36 months. A‐change in UPDRS prior to initiation of dopaminergic therapy (DT). B‐change in UPDRS due to ST initiation. C‐time to initiation of ST. D‐trajectory of UPDRS change over time.
The primary outcome is the change in total UPDRS (sum of mental, ADL and motor components) from baseline to 36 months in the medications “ON” state approximately 1 h after dose of ST for those receiving symptomatic treatment. The UPDRS is a valid and reliable measure of PD disability that has been effectively used in a number of PD trials.21, 27, 28
Key secondary outcomes of clinical importance have been identified and include: (1) Time to initiation of ST has been used as a primary outcome measure in several previous studies of putative disease‐modifying agents5, 29 and reflects progression early in disease not obscured by symptomatic therapy; (2) Time to and severity of motor complications may reflect a secondary measure of progression once type of initial symptomatic treatment is accounted for30, 31; (3) A potential beneficial effect of isradipine on disease progression could be masked by differential usage of ST. To account for this factor, we will evaluate differential use of ST by calculating the levodopa equivalent dosages between treatment groups32; (4) Incidence and severity of nonmotor symptoms, as these contribute disproportionately to quality of life and reflect clinically relevant outcomes in PD.33, 34, 35
A variety of exploratory outcome measures will be evaluated including global measures of functional disability measured by the modified Rankin scale,36 quality of life measured by PDQ‐3937 and NeuroQOL,38 the change in the ambulatory capacity (sum of 5 UPDRS items: falling, freezing, walking, gait, postural stability)39, and cognitive function as measured by MOCA.24 Finally, we will model the trajectory of UPDRS change before and after initiation of ST (D in Fig. 2).
Plasma pharmacokinetic (PK) samples will be collected at the screening, 3 month and 6 month visits. The objective of collecting blood PK samples is to confirm isradipine trough concentrations and to establish a sparse PK profile of isradipine in this population. In addition, blood samples to extract DNA will be collected at screening and plasma will be collected at screening and at the end of the study and stored for future unspecified research.
Statistical analyses
Efficacy analyses will use the intent‐to‐treat principle. The primary analysis will compare the active treatment group (all participants randomized to receive active isradipine) with the placebo group. All P‐values for efficacy outcomes will be two‐sided.
The primary analysis will use analysis of covariance applied to the change from baseline in the total UPDRS score. The baseline value will also be entered into the model as a continuous variable, the assigned treatment and enrolling site will be entered as categorical variables. A two‐tailed test with α = 0.05 will be used to declare statistical significance.
Secondary efficacy analyses of continuous outcome measures will be performed similar to the primary analysis. The time to initiation of ST and the time to onset of motor complications will be analyzed using Kaplan–Meier plots and Cox Regression.
Supplementary analyses of the final study outcomes will be conducted with current use of symptomatic medication (levodopa equivalents) added as an additional predictor variable. The purpose of this analysis is to assess whether any differences seen in the primary outcome variable could be attributed to differential use of ST between the treatment groups. We aim to demonstrate that at 36 months participants on isradipine will have less functional decline than participants on placebo, without requiring more ST. We will also perform exploratory analyses of the primary outcome, using quantitative modeling along the lines suggested by Holford and Nutt,40 which permits exploration of both short‐term symptomatic effects and long‐term disease‐modifying effects of treatment in order to further evaluate the differential impact of isradipine and ST.
Recognizing that the study does not have high power to detect treatment effects among subgroups, we will conduct exploratory analyses to check the consistency of treatment effects on the primary and secondary efficacy measures in relation to selected baseline characteristics, including gender and race/ethnicity.
Power and sample size considerations
Previous studies29, 30, 41 support a standard deviation of 12.0 units for the change in the primary outcome, total UPDRS from baseline to 36 months. The same data suggest an average change in total UPDRS of around 4.0 points over this same time period. However, this change is deceiving, as the change would likely be much greater in the absence of symptomatic treatment. If we assume that treatment with levodopa or a dopaminergic agonist provides a “bonus” of 12 points, then the underlying true decline in function over this period would be approximately 16 points, a value broadly consistent with the rate of change in total UPDRS in participants prior to treatment. We have chosen to power our study to detect a 4‐point effect, representing an overall 25% reduction in the underlying rate of progression. Using the above assumptions, a two‐sided test with α = 0.05 and β = 0.8 and making allowance for 15% dropouts, the required sample size is 168 participants per group or a total 336 participants.
We are also sufficiently powered to address our key secondary outcomes. Given the sample size above, we will be able to detect a 29% reduction in the risk of initiating ST; a reduction of 40% in the risk of developing motor complications; an approximately 25% reduction in the dosage of ST; and an effect size of 1.5 points on the nonmotor experiences of daily living between treatment groups.
Interim analyses
An interim analysis for futility and efficacy will be performed after primary outcome data are available for the first 168 participants (50%) to enroll. The study will be terminated for futility if the interim analysis shows that the conditional power of rejecting the null hypothesis in favor of a beneficial effect of isradipine is lower than 20% under any scenario that is consistent with the data accrued at that time. A two‐sided P‐value in favor of isradipine of less than 0.001 will be required to stop for efficacy at the interim analysis. The stringent alpha level for efficacy was chosen so as to have minimal effect on the final P‐value, should the study run to completion. In addressing futility, the DSMB will examine a range of possible treatment effects consistent with the data obtained in the study at the time of analysis.
Results
Enrollment of the 336 participants began in November 2014 and was completed in October 2015 at 55 of the 57 active PSG sites. The final subject is expected to complete the study in November 2018. Baseline characteristics of the enrolled cohort are detailed in Table 2. At the time of this report, 330 participants remain active in the study with 322 participants on active drug.
Table 2.
Baseline characteristics of the enrolled cohort
| Enrolled cohort (n = 336) | Valuea |
|---|---|
| Age | 61.9 (9.0) |
| Male gender, n (%) | 230 (68.5) |
| White, non‐Hispanic, n (%) | 300 (89.3) |
| Years from diagnosis | 0.9 (0.7) |
| Hoehn and Yahr Stage | 1.7 (0.5) |
| Schwab and England ADL score | 94.0 (7.9) |
| Total UPDRS | 23.1 (8.6) |
| Mental UPDRS | 0.7 (1.1) |
| ADL UPDRS | 5.2 (3.1) |
| Motor UPDRS | 17.2 (7.0) |
| MDS‐UPDRS Total | 32.4 (11.6) |
| MoCA | 28.1 (1.4) |
| Amantadine use at baseline, n (%) | 20 (6.0) |
| Anticholinergic use at baseline, n (%) | 5 (1.5) |
ADL, activities of daily living; UPDRS, Unified Parkinson Disease Rating Scale; MDS, Movement Disorders Society; MoCA, Montreal Cognitive Assessment.
Values represent mean (standard deviation) unless otherwise specified.
Discussion
STEADY‐PDIII is a novel PD disease‐modifying trial evaluating efficacy of isradipine compared with placebo over 36 months. Several novel aspects of study design can be highlighted. It is the longest duration disease modifying trial ever conducted in de novo PD. In addition, the primary outcome (UPDRS change in the practically defined ON state) is powered to detect a 25% slowing of functional decline with isradipine above the benefit from ST, a difference that would be sufficient to influence clinical practice and may suggest the likelihood of longer term benefit. Finally, we are looking at a variety of relevant motor and nonmotor outcomes that will serve to support the effect of isradipine on outcomes that are clinically relevant to patients and clinicians.
PD is a slowly progressive neurodegenerative disease. Most of the previously conducted disease‐modifying studies enrolled participants with newly diagnosed PD not yet requiring ST and followed them for a relatively short period of time (12–24 months) assuming that if benefit is shown it will persist long term.42 In case the participant required initiation of ST, the last observation prior to symptomatic treatment was carried forward. Such design is driven by lack of objective biomarkers of PD progression and the significant impact of ST on standard clinical outcome measures. However, this design is artificial and does not address “real life scenarios” where all patients are ultimately treated with ST. Indeed, on average 50% of de novo PD patients initiate ST within 1 year43 with nearly 100% requiring therapy by 3 years.44, 45 If the effect of isradipine on progression influences the rates of initiation of therapy, then we will be able to evaluate this through our key secondary outcome measures looking at time to initiation of ST and differential use of ST. Even if an intervention is effective early in the course of the disease, it remains to be proven that the benefit will persist longer term and specifically after initiation of ST. The interpretation of previous disease‐modifying therapies has been obscured by this lack of long‐term follow‐up.
STEADY‐PDIII attempts to address these limitations through 36‐month follow‐up of a randomized and blinded cohort. At 36 months, nearly all participants are expected to be treated with symptomatic therapies; therefore, this study is powered to demonstrate a disease‐modifying effect, if such exists, “on top of” the symptomatic benefit of existing treatments, making the results more clinically relevant and reflecting a “real life scenario” in a relatively small cohort of patients. Although we recognize that 36 months is not a substantially long period to see the emergences of long‐term complications such as postural instability and dementia, it is the longest duration ever proposed for a study in a de novo untreated PD population and is likely long enough to provide insight into the effect of isradipine on relevant motor and nonmotor outcomes. It is also a practically feasible time to maximize participant retention. Thus, the study design is novel in that it allows us to take advantage of a relatively small cohort to address the longer duration benefits of isradipine on top of the benefit derived from ST and to address clinically relevant longer duration motor and nonmotor outcomes.
We considered alternative study designs including a “simple long duration study” design (LS‐1), but this design would require in excess of 1500 participants and 7–8 years to complete.39 Another design used in PD neuroprotective trials is the delayed‐start design.7 The arguments against a delayed‐start design are the lack of demonstrable symptomatic benefit of isradipine, the requirement of >1000 participants for sufficient power, and controversy on its ability to demonstrate disease modification in PD. Another consideration would be to enroll individuals at the time of initiation of symptomatic therapy (e.g., CALM‐PD),30 however, this would not allow us to evaluate the impact of isradipine on progression early in disease not confounded by symptomatic therapy, would not allow us to assess the impact of isradipine on the timing of initiation of ST and would be unlikely to add value as both scenarios would assess baseline UPDRS prior to the initiation of symptomatic therapy. In addition, enrolling participants as early as possible in the disease process would allow us to maximize the neuroprotective benefit of isradipine if such an effect exists. We also considered a prolonged wash out at the end of study or at the time of initiation of ST to reassess for the evidence of symptomatic benefit, but there are strong arguments against such design, including lack of obvious symptomatic effect of isradipine in our Phase II STEADY‐PD2 study and participant burden. In addition, there is no consensus regarding the necessary duration of the washout that would be required for isradipine.5, 28 Therefore, our design represents the most rational approach to study the efficacy of isradipine on disability in PD.
Our primary outcome is the change in UPDRS in the practically defined “ON” state from baseline to 36 months. Despite its limitations, UPDRS remains the best characterized outcome measure in PD and motor UPDRS correlates with neuronal loss in the substantia nigra.46 In addition, substantial data exist on the rate of change in the UPDRS in the de novo PD population and on the clinical meaningfulness of this outcome to allow us to adequately power this study. We have chosen to evaluate UPDRS in the medication “ON” state once ST is initiated, which will allow us to identify the benefit of isradipine “on top of” the benefit conferred by ST, an outcome with “real world” relevance to patients and clinicians. We have carefully considered UPDRS “OFF” as an alternative primary outcome. While it may be argued that the “OFF” assessment is a better representation of dopaminergic deficit this is not supported by the clinical data (31–33).47, 48, 49 Both levodopa and dopamine agonists have shown long duration effects on UPDRS lasting days and even weeks, so that traditional 12 h off medication assessment does not reflect true dopaminergic deficiency. Despite these limitations, we recognize the potential value of “OFF” assessments and the motor UPDRS will be assessed as an exploratory outcome in the defined medication “OFF” state (at least 12 h after last dose of ST) once ST has been initiated.
In addition, we have identified a number of key secondary outcomes to corroborate the findings from the primary analysis. These outcomes include time to initiation of ST, time to the development of motor complications, use of ST and nonmotor disability. Time to initiation of ST has been a primary outcome in several completed studies that examined the efficacy of putative disease‐modifying interventions.5, 29 Although it has been criticized for the subjective nature of the measure and being impacted by the change in the treatment algorithms that overall lead to the earlier initiation of ST, nevertheless it can be considered a surrogate measure of the disease progression and allows us to compare our findings with previous trials. The differential use of ST has the potential to obscure the results and may represent a surrogate of disease severity. For instance, individuals with greater progression may be on higher dosages of ST which may offset the benefit of slower progression as measured by the UPDRS. Not only will this serve as a surrogate of disease progression and severity, it will allow us to conduct exploratory analyses accounting for ST effects.
The development of motor complications represents a significant milestone in PD progression and results in impaired quality of life, function, and social isolation.50 Therapies aimed at preventing or ameliorating motor complications represent a major unmet therapeutic need in PD. If isradipine only resulted in a difference in the rate of motor complications, this would represent a clinically meaningful outcome that would likely influence care. Nonmotor symptoms can be challenging to treat and have a disproportionate impact on quality of life.51, 52, 53 Therefore, therapy that influences these outcomes will likely have a significant impact on PD quality of life. The MDS‐UPDRS evaluated a variety of nonmotor outcomes not assessed in the traditional UPDRS. STEADY‐PD III represents the first interventional trial in de novo population to systematically evaluate the MDS‐UPDRS and will allow for further validation of this scale. We have chosen not to use it as the primary outcome as there were limited data on the change in MDS‐UPDRS in de novo PD to power the study. We have also chosen a number of exploratory measures that take advantage of the longer duration of follow‐up in this study compared with previous de novo studies and represent clinically valuable and complementary outcomes in PD. These measures represent components of the NINDS Common Data Elements and have largely been validated in PD and include measures of function, quality of life, gait, and cognition.
The study design has some limitations. A biomarker to validate target engagement of isradipine at the CAV1.3 channel does not exist and therefore a negative study may reflect a failure of target engagement. We have considered collecting biomarkers of oxidative stress and mitochondrial function as potential down steam effects of isradipine but these would only be indirect measures and have not been validated with isradipine in in vivo or in vitro models. We are collecting DNA and plasma for future analyses of novel biomarkers that may assist in interpretation of the study. We will analyze PK data to ensure that a minimum concentration necessary for neuroprotection is achieved and to address whether variability in clinical response is related to variations in serum concentrations. A final limitation is that participants are enrolled based on the clinical diagnosis of PD, raising the possibility that approximately 10% of participants might not have a presynaptic dopaminergic deficit. The use of DAT scan at enrollment could obviate this concern but would be associated with increased costs and time and would not definitively exclude individuals without PD. All investigators are credentialed by the PSG, experienced in the diagnosis and care of PD and the conduct of PD‐related trials and therefore, we anticipate a low false‐positive diagnosis rate. Regardless, we are collecting data on the change in diagnosis and will conduct post hoc analyses that incorporate this information.
In conclusion, this study is testing isradipine as a potential novel neuroprotective agent in PD based on robust preclinical and strong epidemiological data. The STEADY‐PDIII study design is unique in assessing the impact of isradipine over 36 months, at a time point where all participants will likely be on ST allowing us to determine if the benefit is sustained “on top of” traditional ST. In addition, the study design allows us to determine if the effects on motor function are corroborated by important secondary outcomes assessing clinically relevant measures of ST use, motor complications, nonmotor function, global disability, quality of life, ambulatory capacity, and cognition. This design is novel and innovative and allows for the determination of longer duration benefits on several clinically relevant outcomes in a relatively small cohort on top of the benefit derived from ST.
Conflict of Interest
None of the authors have relevant conflicts of interest.
References
- 1. de Lau LM, Breteler MM. Epidemiology of Parkinson's disease. Lancet Neurol 2006;5:525–535. [DOI] [PubMed] [Google Scholar]
- 2. Dorsey ER, Constantinescu R, Thompson JP, et al. Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology 2007;68:384–386. [DOI] [PubMed] [Google Scholar]
- 3. Dodel RC, Singer M, Kohne‐Volland R, et al. The economic impact of Parkinson's disease. An estimation based on a 3‐month prospective analysis. Pharmacoeconomics 1998;14:299–312. [DOI] [PubMed] [Google Scholar]
- 4. Huse DM, Schulman K, Orsini L, et al. Burden of illness in Parkinson's disease. Mov Disord 2005;20:1449–1454. [DOI] [PubMed] [Google Scholar]
- 5. Parkinson Study Group . Effects of tocopherol and deprenyl on the progression of disability in early Parkinson's disease. N Engl J Med 1993;328:176–183. [DOI] [PubMed] [Google Scholar]
- 6. The NINDS NET‐PD Investigators . A randomized, double‐blind, futility clinical trial of creatine and minocycline in early Parkinson disease. Neurology 2006;66:664–671. [DOI] [PubMed] [Google Scholar]
- 7. Rascol O, Fitzer‐Attas CJ, Hauser R, et al. A double‐blind, delayed‐start trial of rasagiline in Parkinson's disease (the ADAGIO study): prespecified and post‐hoc analyses of the need for additional therapies, changes in UPDRS scores, and non‐motor outcomes. Lancet Neurol 2011;10:415–423. [DOI] [PubMed] [Google Scholar]
- 8. Beal MF, Oakes D, Shoulson I, et al. A randomized clinical trial of high‐dosage coenzyme Q10 in early Parkinson disease: no evidence of benefit. JAMA Neurol 2014;71:543–552. [DOI] [PubMed] [Google Scholar]
- 9. Chan CS, Guzman JN, Ilijic E, et al. ‘Rejuvenation’ protects neurons in mouse models of Parkinson's disease. Nature 2007;447:1081–1086. [DOI] [PubMed] [Google Scholar]
- 10. Ilijic E, Guzman JN, Surmeier DJ. The L‐type channel antagonist isradipine is neuroprotective in a mouse model of Parkinson's disease. Neurobiol Dis 2011;43:364–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Anekonda TS, Quinn JF. Calcium channel blocking as a therapeutic strategy for Alzheimer's disease: the case for isradipine. Biochim Biophys Acta 2011;1812:1584–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Parkinson Study Group . Phase II safety, tolerability, and dose selection study of isradipine as a potential disease‐modifying intervention in early Parkinson's disease (STEADY‐PD). Mov Disord 2013;28:1823–1831. [DOI] [PubMed] [Google Scholar]
- 13. Sinnegger‐Brauns MJ, Huber IG, Koschak A, et al. Expression and 1,4‐dihydropyridine‐binding properties of brain L‐type calcium channel isoforms. Mol Pharmacol 2009;75:407–414. [DOI] [PubMed] [Google Scholar]
- 14. Puopolo M, Raviola E, Bean BP. Roles of subthreshold calcium current and sodium current in spontaneous firing of mouse midbrain dopamine neurons. J Neurosci 2007;27:645–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Becker C, Jick SS, Meier CR. Use of antihypertensives and the risk of Parkinson disease. Neurology 2008;70:1438–1444. [DOI] [PubMed] [Google Scholar]
- 16. Ritz B, Rhodes SL, Qian L, et al. L‐type calcium channel blockers and Parkinson disease in Denmark. Ann Neurol 2010;67:600–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pasternak B, Svanstrom H, Nielsen NM, et al. Use of calcium channel blockers and Parkinson's disease. Am J Epidemiol 2012;175:627–635. [DOI] [PubMed] [Google Scholar]
- 18. Marras C, Gruneir A, Rochon P, et al. Dihydropyridine calcium channel blockers and the progression of parkinsonism. Ann Neurol 2012;71:362–369. [DOI] [PubMed] [Google Scholar]
- 19. Ton TG, Heckbert SR, Longstreth WT Jr, et al. Calcium channel blockers and beta‐blockers in relation to Parkinson's disease. Parkinsonism Relat Disord 2007;13:165–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Simon KC, Gao X, Chen H, et al. Calcium channel blocker use and risk of Parkinson's disease. Mov Disord 2010;25:1818–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fahn S, Elton RL, UPDRS Development Committee. Unified Parkinson's Disease Rating Scale In: Fahn S. M. C., Calne D. B., and Lieberman A., Eds., Recent Developments in Parkinson's Disease. Florman Park, NJ: Macmillan; 1987. Pp. 153–163. [Google Scholar]
- 22. Hughes AJ, Ben‐Shlomo Y, Daniel SE, Lees AJ. What features improve the accuracy of clinical diagnosis in Parkinson's disease: a clinicopathologic study. Neurology 1992;42:1142–1146. [DOI] [PubMed] [Google Scholar]
- 23. Hoehn MM, Yahr MD. Parkinsonism: onset, progression and mortality. Neurology 1967;17:427–442. [DOI] [PubMed] [Google Scholar]
- 24. Nasreddine ZS, Phillips NA, Bedirian V, et al. The Montreal cognitive assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc 2005;53:695–699. [DOI] [PubMed] [Google Scholar]
- 25. Beck Beck AT, Inventory Depression. San Antonio. Texas: The Psychological Corporation, 1987.Ref Type: Pamphlet [Google Scholar]
- 26. Adams JL, Biemiller RA, Andrzejewski KL, et al. Utilizing remote blood pressure monitoring in a Phase III clinical drug trial for Parkinson's disease [abstract]. Mov Disord 2016;31 (suppl 2). http://www.mdsabstracts.org/abstract/utilizing-remote-blood-pressure-monitoring-in-a-phase-iii-clinical-drug-trial-for-parkinsons-disease/. Accessed April 25, 2017. [Google Scholar]
- 27. Parkinson Study Group . A controlled trial of rasagiline in early Parkinson disease. Arch Neurol 2002;59:1937–1943. [DOI] [PubMed] [Google Scholar]
- 28. Fahn S. Parkinson Study Group . Results of the ELLDOPA (earlier vs. later levodopa) study. Mov Disord 2002;17:S13–S14.12211136 [Google Scholar]
- 29. Mixed lineage kinase inhibitor . CEP‐1347 fails to delay disability in early Parkinson disease. Neurology 2007;69:1480–1490. [DOI] [PubMed] [Google Scholar]
- 30. Parkinson Study Group . Pramipexole vs levodopa as initial treatment for Parkinson disease: a randomized controlled trial. JAMA 2000;284:1931–1938. [DOI] [PubMed] [Google Scholar]
- 31. Rascol O, Brooks DJ, Korczyn AD, et al. A five‐year study of the incidence of dyskinesia in patients with early Parkinson's disease who were treated with ropinirole or levodopa. N Engl J Med 2000;342:1484–1491. [DOI] [PubMed] [Google Scholar]
- 32. Tomlinson CL, Stowe R, Patel S, et al. Systematic review of levodopa dose equivalency reporting in Parkinson's disease. Mov Disord 2010;25:2649–2653. [DOI] [PubMed] [Google Scholar]
- 33. Allain H, Schuck S, Mauduit N. Depression in Parkinson's disease. BMJ 2000;2000:1287–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cahn DA, Sullivan EV, Shear PK, et al. Differential contributions of cognitive and motor component processes to physical and instrumental activities of daily living in Parkinson's Disease. Arch Clin Neuropsychol 1998;13:575–583. [PubMed] [Google Scholar]
- 35. Gallagher DA, Schrag A. Psychosis, apathy, depression and anxiety in Parkinson's disease. Neurobiol Dis 2012;46:581–589. [DOI] [PubMed] [Google Scholar]
- 36. Weisscher N, Vermeulen M, Roos YB, de Haan RJ. What should be defined as good outcome in stroke trials; a modified Rankin score of 0‐1 or 0‐2? J Neurol 2008;255:867–874. [DOI] [PubMed] [Google Scholar]
- 37. Peto V, Jenkinson C, Fitzpatrick R, Greenhall R. The development and validation of a short measure of functioning and well being for individuals with Parkinson's disease. Qual Life Res 1995;4:241–248. [DOI] [PubMed] [Google Scholar]
- 38. Nowinski CJ, Siderowf A, Simuni T, et al. Neuro‐QoL health‐related quality of life measurement system: validation in Parkinson's disease. Mov Disord 2016;31:725–733. [DOI] [PubMed] [Google Scholar]
- 39. Elm JJ. Design innovations and baseline findings in a long‐term Parkinson's trial: the National institute of neurological disorders and stroke exploratory trials in Parkinson's disease long‐term study‐1. Mov Disord 2012;27:1513–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Holford NH, Nutt JG. Interpreting the results of Parkinson's disease clinical trials: time for a change. Mov Disord 2011;26:569–577. [DOI] [PubMed] [Google Scholar]
- 41. Palhagen S, Heinonen EH, Hagglund J, et al. Selegiline delays the onset of disability in de novo parkinsonian patients. Swedish Parkinson Study Group. Neurology 1998;51:520–525. [DOI] [PubMed] [Google Scholar]
- 42. Hart RG, Pearce LA, Ravina BM, et al. Neuroprotection trials in Parkinson's disease: systematic review. Mov Disord 2009;24:647–654. [DOI] [PubMed] [Google Scholar]
- 43. Parashos SA, Swearingen CJ, Biglan KM, et al. Determinants of the timing of symptomatic treatment in early Parkinson disease: The National Institutes of health exploratory trials in Parkinson disease (NET‐PD) experience. Arch Neurol 2009;66:1099–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shoulson I. DATATOP: a decade of neuroprotective inquiry. Parkinson Study Group. Deprenyl and tocopherol antioxidative therapy of parkinsonism. Ann Neurol 1998;44:S160–S166. [PubMed] [Google Scholar]
- 45. Ravina B, Tanner C, Dieuliis D, et al. A longitudinal program for biomarker development in Parkinson's disease: a feasibility study. Mov Disord 2009;24:2081–2090. [DOI] [PubMed] [Google Scholar]
- 46. Greffard S, Verny M, Bonnet AM, et al. Motor score of the Unified Parkinson Disease Rating Scale as a good predictor of Lewy body‐associated neuronal loss in the substantia nigra. Arch Neurol 2006;63:584–588. [DOI] [PubMed] [Google Scholar]
- 47. Fahn S, Oakes D, Shoulson I, et al. Levodopa and the progression of Parkinson's disease. N Engl J Med 2004;351:2498–2508. [DOI] [PubMed] [Google Scholar]
- 48. Stocchi F, Vacca L, Berardelli A, et al. Long‐duration effect and the postsynaptic compartment: study using a dopamine agonist with a short half‐life. Mov Disord 2001;16:301–305. [DOI] [PubMed] [Google Scholar]
- 49. Barbato L, Stocchi F, Monge A, et al. The long‐duration action of levodopa may be due to a postsynaptic effect. Clin Neuropharmacol 1997;20:394–401. [DOI] [PubMed] [Google Scholar]
- 50. Hechtner MC, Vogt T, Zollner Y, et al. Quality of life in Parkinson's disease patients with motor fluctuations and dyskinesias in five European countries. Parkinsonism Relat Disord 2014;20:969–974. [DOI] [PubMed] [Google Scholar]
- 51. Hobson P, Holden A, Meara J. Measuring the impact of Parkinson's disease with the Parkinson's Disease Quality of Life questionnaire. Age Ageing 1999;28:341–346. [DOI] [PubMed] [Google Scholar]
- 52. Martinez‐Martin P, Rodriguez‐Blazquez C, Kurtis MM, Chaudhuri KR. The impact of non‐motor symptoms on health‐related quality of life of patients with Parkinson's disease. Mov Disord 2011;26:399–406. [DOI] [PubMed] [Google Scholar]
- 53. Leroi I, McDonald K, Pantula H, Harbishettar V. Cognitive impairment in Parkinson disease: impact on quality of life, disability, and caregiver burden. J Geriatr Psychiatry Neurol 2012;25:208–214. [DOI] [PubMed] [Google Scholar]