

The idea that activity-dependent homeostatic plasticity is driven solely by firing has wide credence. In this report we show that homeostatic compensation after loss of an ion channel conductance is tailored to identity of the channel lost, not its properties.
Keywords: Drosophila, motor neuron, ion channel, homeostasis, excitability
Abstract
Homeostatic control of intrinsic excitability is important for long-term regulation of neuronal activity. In conjunction with many other forms of plasticity, intrinsic homeostasis helps neurons maintain stable activity regimes in the face of external input variability and destabilizing genetic mutations. In this study, we report a mechanism by which Drosophila melanogaster larval motor neurons stabilize hyperactivity induced by the loss of the delayed rectifying K+ channel Shaker cognate B (Shab), by upregulating the Ca2+-dependent K+ channel encoded by the slowpoke (slo) gene. We also show that loss of SLO does not trigger a reciprocal compensatory upregulation of SHAB, implying that homeostatic signaling pathways utilize compensatory pathways unique to the channel that was mutated. SLO upregulation due to loss of SHAB involves nuclear Ca2+ signaling and dCREB, suggesting that the slo homeostatic response is transcriptionally mediated. Examination of the changes in gene expression induced by these mutations suggests that there is not a generic transcriptional response to increased excitability in motor neurons, but that homeostatic compensations are influenced by the identity of the lost conductance.
NEW & NOTEWORTHY The idea that activity-dependent homeostatic plasticity is driven solely by firing has wide credence. In this report we show that homeostatic compensation after loss of an ion channel conductance is tailored to identity of the channel lost, not its properties.
one of the major determinants of neuronal electrical activity is the distribution and density of voltage-gated ion channels. These ion channels, along with ion pumps and the signaling molecules that modulate them, determine the intrinsic firing behavior of neurons (Neverisky and Abbott 2015). Regulation of excitability must be both plastic, to respond to developmental and learning-induced changes in activity, and robust, to provide stability in the face of environmental changes (Ghezzi et al. 2004; Nataraj et al. 2010; Turrigiano et al. 1994). Remarkable homeostatic compensations that protect against potentially debilitating electrical activity have been observed in vertebrates (Turrigiano 2012) and in electrophysiologically accessible invertebrate systems, such as the crustacean stomatogastric ganglion (Marder and Bucher 2007), the Drosophila neuromuscular junction (Davis and Müller 2015), and cultured Drosophila neurons (Peng and Wu 2007b; Ping and Tsunoda 2012).
The regulation of neuronal excitability can be disrupted in many ways, and the nature of the disruption (Parrish et al. 2014) and its time course (Nerbonne et al. 2008; Swensen and Bean 2005) can affect the cell’s response. For an animal to survive the long-term loss of an important ion conductance, it has to activate processes that restore cell function. Mutations in ion channel genes impose long-term changes in excitability that engage these homeostatic compensation mechanisms, but the nature of the homeostatic changes and the underlying principles that govern them are poorly understood (Davis 2006).
Our initial molecular understanding of ion channels came from genetic studies performed in Drosophila (Bellen et al. 2010; Salkoff and Tanouye 1986), where it was quickly recognized that there was substantial conservation between insects and mammals (Wei et al. 1990). There is also similarity in the ways that nervous systems deal with dysregulation, at both the molecular and cellular levels (Davis and Müller 2015; Giachello and Baines 2017). In this study, we utilize the well-characterized larval motor neuron preparation to investigate the consequences of long-term global and local perturbations of excitability. We manipulate two potassium channel genes, Shab and slo, and identify somatic current compensations and global transcriptional changes that occur in response to their disruption. Despite having similar somatic amplitude and kinetics, loss of the two conductances does not generate the same homeostatic response.
MATERIALS AND METHODS
Stocks.
Canton Special (Canton S, or CS) was used as the wild-type control for the homozygous Shab null mutant line Shab3 (Hegde et al. 1999) and the slo1 homozygous line (obtained from Barry Ganetzky, University of Wisconsin). OK371-GAL4, which expresses GAL4 in all motor and glutamatergic neurons (Mahr and Aberle 2006), was used as the driver for all upstream activation sequence (UAS) lines. UAS-Shab RNAi (transformant ID 108861) and UAS-slo RNAi (transformant ID 1044211) used for mRNA knockdown experiments were obtained from the Vienna Drosophila RNAi Center. The genotypes in these experiments were w;OK371-GAL4/UAS-ShabRNAi;+;+ (“Shab RNAi”) and w;OK371-GAL4/UAS-sloRNAi;+;+ (“slo RNAi”). Controls were heterozygotes of the associated UAS lines for each experiment and the GAL4 line. The double knockdown experiment required the recombination of OK371-GAL4 with either UAS-ShabRNAi or UAS-sloRNAi, and was confirmed by PCR genotyping. These lines were then crossed to produce the experimental line w;OK371-GAL4,UAS-ShabRNAi/OK371-GAL4,UAS-sloRNAi;+;+ (“Double KD” or “Shab RNAi + slo RNAi”). UAS-CaMBP4 (“CaM Sponge”) was a gift from Dr. Hilmar Bading (Weislogel et al. 2013) and was crossed to OK371-GAL4 and the recombinant OK371-GAL4,UAS-ShabRNAi line we generated to examine the role of nuclear calcium in the Shab knockdown. UAS-dCREB26 Blocker (“dnCREB”) was a gift from Dr. Jerry Yin and was crossed to OK371-GAL4 and the recombinant OK371-GAL4,UAS-ShabRNAi to examine the role of CREB-mediated transcription in the Shab knockdown.
Dissection.
All animals used were female larvae picked at the wandering third instar stage to eliminate potential sex-specific differences. The initial dissection to expose the ventral ganglia was done under a Leica Wild M3Z (Leica Microsystems, Buffalo Grove, IL) in chilled 0 mM Ca2+ “A” solution (containing in mM: 118 NaCl, 2 NaOH, 2 KCl, 4 MgCl2, 22.3 sucrose, 5 trehalose, 5 HEPES, and 1.8 CaCl2, pH 7.1–7.2) (Jan and Jan 1976) to reduce muscle contraction. A secondary protease dissection to expose motor neuron soma clusters was performed under a BX50WI compound microscope (Olympus, Center Valley, PA) with a ×40 water-immersion lens with a continual flow of 0 mM Ca2+ “A” solution. Glass micropipettes (BF120-90-100; Friedrich and Dimmoch, Milville, NJ) were used to deliver the protease solution, pulled to a relatively wide diameter (with tip resistance <2–4 MΩ) with a Flaming Brown P-97 micropipette puller (Sutter Instruments, Novato, CA). The protease solution contained protease XIV (Sigma-Aldrich, St. Louis, MO) diluted to 0.1% concentration in internal solution (containing in mM: 20 KCl, 0.1 CaCl2, 2 MgCl2, 1.1 EGTA, 120 K-gluconate, and 10 HEPES, pH 7.2). Application of the solution was done with a MP285 micromanipulator (Sutter Instruments) until soma clusters became distinct from the outer ventral ganglia sheath.
Electrophysiology.
The MN1-Ib neuron was selected for all recordings on the basis of its readily identifiable position in the motor neuron cluster (Fig. 1A and Choi et al. 2004). Fresh micropipettes were pulled to 2–5 MΩs, fire polished with a custom-made device, and filled with internal solution. Slight outward pressure was applied through mouth suction before cell contact, and gigaohm seals (>1 GΩ) were formed through relief of mouth suction. Whole cell configuration was then achieved through sudden inward pressure via suction. All protocols were preceded by a measurement of several cell properties using the membrane test function in pCLAMP 10: cell capacitance, membrane resistance, and series resistance (shown for all cells in Table 1). The external bath was “A” solution, first without and then with the addition of 1.8 mM Ca2+ to distinguish the contributions of Ca2+ and Ca2+-dependent currents. All cells were recorded under both conditions, and only data sets where both conditions had stable cell properties were used.
Fig. 1.
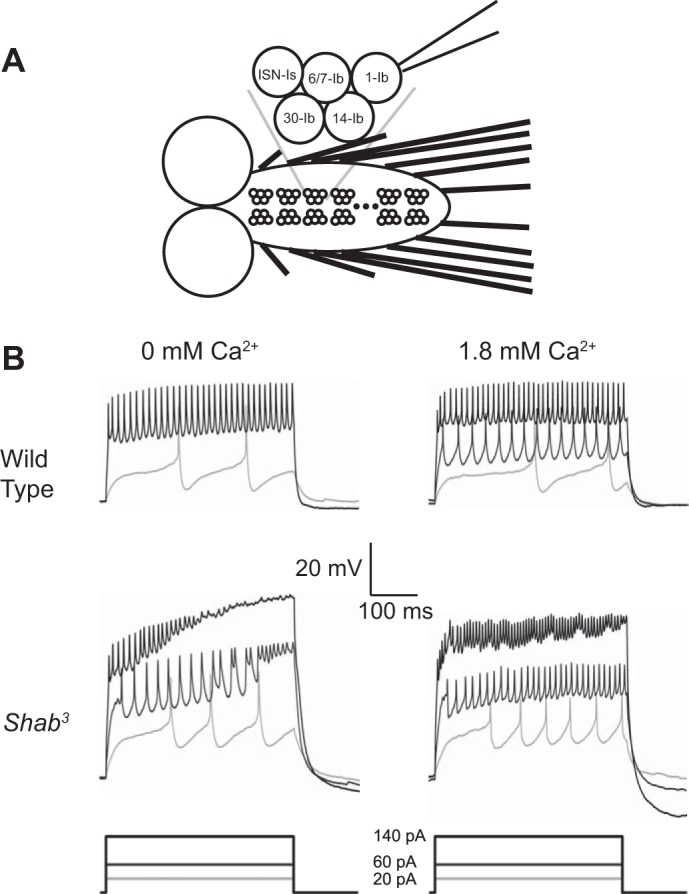
Shab3 mutants exhibit Ca2+-suppressed firing instability. A: schematic of the third instar larval brain. Ventral ganglion is denoted by bracket. Individual motor neuron clusters are arranged in a stereotyped repeating pattern. The MN1-Ib neuron, shown attached to a patch electrode, is distinguishable from other identified neurons in each cluster by position. B: representative voltage traces from current-clamp experiments. Wild type (Canton S) is represented in top row. Traces from Shab3 recordings are shown in bottom row. Recordings taken in 0 mM external Ca2+ are shown at left, with 1.8 mM Ca2+ recordings shown at right. Removal of SHAB results in a destabilization of firing behavior at higher current injection levels in 0 mM Ca2+. This instability was seen in 12/13 cells recorded and is suppressed by addition of external Ca2+.
Table 1.
Cell properties across all voltage-clamp experiments
| Genotype | No. of Cells | Cell Capacitance, pA/pF |
Membrane Resistance, GΩ |
Series Resistance, MΩ |
|||
|---|---|---|---|---|---|---|---|
| Initial | Drift | Initial | Drift | Initial | Drift | ||
| Canton S wild type | 5 | 24.1 ± 1.0 | −1.9 ± 0.2 | 0.8 ± 0.1 | 0.0 ± 0.0 | 25.4 ± 6.1 | 3.0 ± 2.3 |
| Shab3 | 5 | 27.3 ± 2.1 | −2.0 ± 0.5 | 0.8 ± 0.1 | 0.0 ± 0.1 | 14.5 ± 5.9 | −1.3 ± 2.8 |
| OK371-GAL4 | 6 | 26.2 ± 0.9 | −2.8 ± 0.5 | 0.7 ± 0.0 | 0.1 ± 0.0 | 13.3 ± 2.5 | 1.4 ± 1.6 |
| UAS-ShabRNAi | 14 | 27.2 ± 1.4 | −2.4 ± 0.3 | 0.8 ± 0.0 | 0.0 ± 0.0 | 19.7 ± 2.1 | 2.8 ± 0.8 |
| UAS-SloRNAi | 17 | 27.4 ± 0.6 | −2.2 ± 0.4 | 0.7 ± 0.0 | 0.1 ± 0.0 | 17.1 ± 1.1 | 1.9 ± 1.8 |
| OK371>UAS-ShabRNAi | 14 | 23.7 ± 0.9 | −1.6 ± 0.2 | 0.7 ± 0.0 | 0.1 ± 0.0 | 13.5 ± 1.2 | 1.3 ± 1.7 |
| OK371>UAS-SloRNAi | 19 | 23.8 ± 0.5 | −2.1 ± 0.3 | 0.7 ± 0.1 | 0.1 ± 0.0 | 20.9 ± 1.8 | 3.3 ± 2.1 |
| OK371>UAS-ShabRNAi+UAS-SloRNAi | 10 | 23.6 ± 1.2 | −1.8 ± 0.3 | 0.7 ± 0.1 | 0.1 ± 0.0 | 17.3 ± 1.9 | 2.2 ± 0.1 |
| OK371>dnCREB | 4 | 26.6 ± 3.0 | −1.5 ± 0.1 | 0.8 ± 0.1 | 0.1 ± 0.0 | 22.3 ± 5.5 | 1.7 ± 2.2 |
| OK371>CaM Sponge | 4 | 24.3 ± 0.6 | −1.5 ± 0.6 | 0.8 ± 0.1 | 0.1 ± 0.0 | 20.3 ± 2.8 | 1.3 ± 1.1 |
| OK371>dnCREB+ShabRNAi | 3 | 24.8 ± 2.2 | 0.0 ± 2.9 | 0.6 ± 0.1 | 0.1 ± 0.1 | 10.2 ± 1.5 | 2.7 ± 2.5 |
| OK371>CaM Sponge+ShabRNAi | 5 | 23.6 ± 0.8 | −1.9 ± 0.2 | 0.9 ± 0.1 | 0.1 ± 0.0 | 20.8 ± 1.6 | 3.7 ± 1.2 |
In all experiments, cells were patched and initially recorded in 0 mM Ca2+ and then switched to 1.8 mM Ca2+ by bath exchange. Values are means ± SE for initial cell properties and the magnitude of drift after bath exchange. One-way ANOVAs were performed across all groups. For capacitance (P = 0.0056) and series resistance (P = 0.0075), the ANOVA was significant, but Tukey-Kramer post hoc tests did not reveal any specific differences. For membrane resistance, the ANOVA failed to reach significance (P = 0.1310). Only cells that survived the entire protocol were used for analysis.
Current-clamp and voltage-clamp experiments were performed with an Axopatch 200B amplifier, Digidata 1332A digitizer, and Clampex 8 software (Molecular Devices, Sunnyvale, CA). Current injections for current-clamp experiments were applied from 0 to 140 pA in 10-pA steps, with 10-s intermissions to ensure baseline recovery. Patch-clamp experiments followed a repeating series of 4 protocols: 1) 500-ms square-pulsed holding potentials from −80 to 60 mV in 10-mV steps; 2) a P/N (n = 10) protocol to estimate the leak current; 3) 500-ms squared-pulsed holding potentials as in protocol 1, but preceded by a −20-mV prepulse potential; and 4) a P/N (n = 10) protocol of protocol 3 to estimate the leak current. We did not compensate for voltage drops due to the series resistance. TTX (1 μM; Tocris, Ellisville, MO) was added to the bath solution in voltage-clamp experiments to eliminate the Na+ inward current.
Data analysis for electrophysiology.
Offline leak current subtraction was performed by first linearly fitting the currents in protocols 2 and 4 to the equation i = g(V − Ve), where i is the current, g is the leak conductance, V is the membrane potential, and Ve is the leak reversal potential. This modeled leak current was then subtracted from protocols 1 and 3, respectively. Steady-state noninactivating currents were estimated by averaging the late current in the leak-subtracted prepulse protocol. All operations except an import function were performed with custom scripts written in MATLAB (The MathWorks, Natick, MA). Currents were normalized to capacitance for each cell to reduce variance and are reported as current density.
Two-way ANOVAs of total, Ca2+-dependent (IKCa), and delayed K+ (IKd) current densities were used to determine significant differences in voltage-current density relationships with holding potential and genotype as variables. Significance reflects comparisons done with the Tukey-Kramer post hoc test. For comparison of current densities at a single holding potential, one-way ANOVA with a Tukey-Kramer post hoc test was used to determine statistically significant differences. Significance groups are indicated by letters in Fig. 2C. For Fig. 4, data from GAL4 and UAS RNA interference (RNAi) genetic background controls were found to be statistically indistinguishable by one-way ANOVA and were pooled for comparisons to experimental data; P values for these data are presented in Table 2. Statistical analysis was done in StatPlus (AnalystSoft, Walnut, CA) and MATLAB.
Fig. 2.

Mutation of Shab reduces total outward current and alters the balance of Ca2+-dependent and Ca2+-independent steady-state currents. A: current traces from voltage-clamp experiments on cells shown in Fig. 1B. Wild type (Canton S) is represented in the top row. Traces from Shab3 recordings are shown in bottom row. Recordings taken in 0 mM external Ca2+ are shown at left, with 1.8 mM Ca2+ recordings shown at right. B: total current density from voltage-clamp experiments done in 0 or 1.8 mM Ca2+ are shown for both genotypes. No significant difference in the outward currents can be seen in wild type when Ca2+ is added. In contrast, outward currents in Shab3 are substantially increased in Ca2+ (P < 0.0005). C: gene dosage effect on Ca2+-dependent currents. The percentage of currents that are Ca2+ dependent is significantly higher in Shab3 motor neurons than in wild type (P < 0.05), whereas the Shab3/+ heterozygote falls midway between the homozygote and wild type and is not significantly different from either wild type or Shab3. A,BDifferent letters indicate groups with a significant difference. D: IKd and IKCa currents in wild type and Shab3 mutants. IKd and IKCa for wild type and Shab3 were significantly different (P < 0.0005). E: difference plot for Shab3 mutant and wild type. The steady-state current at each holding potential for wild type was subtracted from currents produced in Shab3 neurons at the same potential. The downward deflection of the IKd curve demonstrates the loss of SHAB-mediated currents, whereas the upward deflection of the IKCa line indicates that there is an increase in this current in Shab3. The shape of the curve shows the voltage-dependence profile of the difference current. All error bars are SE; n = 5 for both wild type and Shab3. For data in B and D, 2-way ANOVAs with holding potential and genotype were performed and the Tukey-Kramer post hoc test was used to determine significant differences between genotypes. For data in C, significance was determined by 1-way ANOVA with the Tukey-Kramer post hoc test.
Fig. 4.

slowpoke expression is required for upregulation of IKCa in response to reduction of SHAB. A: IKd and IKCa current densities for control and experimental genotypes at 20-mV holding potential. slo RNAi significantly decreases IKCa compared with controls. Shab RNAi decreases IKd but significantly increases IKCa. The presence of both RNAis does not further decrease IKd beyond the level present in Shab RNAi, but it blocks the increase in IKCa seen with Shab RNAi alone. P values for comparisons are shown in Table 2. *P < 0.05 compared with controls; ANOVA with Tukey-Kramer post hoc test. Data are means ± SE; n = 6 for OK371/+, n = 14 for UAS-ShabRNAi/+, n = 17 for UAS-sloRNAi/+, n = 14 for OK371/UAS-ShabRNAi, n = 19 for OK371/UAS-sloRNAi, and n = 10 for OK371,UAS-ShabRNAi/OK371-GAL4,UAS-sloRNAi. B: total currents in 1.8 mM Ca2+ are compared for animals expressing Shab, slo, or Shab+slo RNAi in motor neurons and for the OK371/+ control. slo RNAi ± Shab RNAi reduces total currents (P < 0.005), whereas Shab RNAi alone does not. C: IKd for animals expressing Shab, slo, or Shab+slo RNAi in motor neurons (format as in B). Shab RNAi reduces IKd compared with both GAL4 and UAS controls and slo RNAi (P < 0.0005). D: IKCa for animals expressing Shab, slo, or Shab+slo RNAi in motor neurons (format as in B). The presence of slo RNAi blocks the increase in IKCa produced by Shab RNAi (P < 0.0005). E: difference plot for Shab+slo RNAi and Shab RNAi. The steady-state current at each holding potential for Shab RNAi was subtracted from currents produced in neurons expressing both Shab RNAi and slo RNAi at the same potential. The flat IKd curve demonstrates that slo RNAi does not change the ability of Shab RNAi to reduce IKd. The downward deflection of the IKCa line indicates that slo RNAi decreases the ability of Shab RNAi to produce an increase in IKCa. For data in B–D, two-way ANOVAs with holding potential and genotype were performed and the Tukey-Kramer post hoc test was used to determine significant differences between genotypes. Data are means ± SE.
Table 2.
P values for pairwise comparisons at 20 mV in Fig. 4A
| Genotype 1 | Genotype 2 | IKCa | IKd |
|---|---|---|---|
| Shab RNAi | GAL4+UAS control | <0.0001 | 0.0001 |
| slo RNAi | <0.0001 | 0.0426 | |
| Double KD | <0.0001 | 0.9814 | |
| slo RNAi | GAL4+UAS control | 0.0053 | 0.3614 |
| Shab RNAi | <0.0001 | 0.0426 | |
| Double KD | 0.5211 | 0.1879 | |
| Double KD | GAL4+UAS control | 0.6209 | 0.0040 |
| Shab RNAi | <0.0001 | 0.9814 | |
| slo RNAi | 0.5211 | 0.1879 |
Data are P values from one-way ANOVA performed with Tukey-Kramer post hoc tests to examine differences in currents due to genotype. P values <0.05 are in bold type. Double KD, double knockdown.
PCR analysis.
The primers used in PCR amplification to confirm the identity of recombinant double knockdown lines were: ShabRNAi left (TAG CCT CCC TAG CGC GAA CAC CGC AT), ShabRNAi right (TGG CGC CCC TAG ATG TGA CGT TGA AC), sloRNAi left (CGC ATG TAG CCT GCC TGT TAT CAT GG), sloRNAi right (TGG CGC CCC TAG ATG GGG CTC GGT G), UAS left (TGC AGT TGA TTT ACT TGG TTG C), and UAS right (CTC CGA GCG GAG ACT CTA) (IDT, Coralville, IA).
RNA sequencing.
Dissection of 8–10 third instar larval brains of each genotype (Canton S wild type, slo1 and Shab3 mutants) was done in PBS, and total RNA was extracted using an Arcturus PicoPure RNA isolation kit (Applied Biosystems/Life Technologies, Carlsbad, CA) with an on-column DNase I digestion following elution. Extracted RNA was assessed and quantified by NanoDrop (Thermo Scientific/Nanodrop, Wilmington, DE). RNA (500 ng) was used to make mRNA libraries by using the TruSeq RNA kit (v2; Illumina, San Diego, CA) with one-third recommended reaction volumes to be more cost effective. Libraries were evaluated and quantified using the Agilent RNA 6000 Nano kit (Agilent Technologies, Palo Alto, CA) and Bioanalyzer (Agilent Technologies). A 2 nM mixture of 10 different barcoded libraries was mixed together, and 8 pmol of this sample were sequenced in a single lane (Illumina HiSeq 2000). The average size of a library was 15 million reads. Three independent replicates of wild type, slo1, and Shab3 larval brains were performed.
The resulting data were parsed to separate the barcoded reads, and the sequence files (fastq format) were aligned to the Drosophila genome (version dm3) via TopHat 2.0.10 (Kim et al. 2013) using the following criteria: m 1 -F 0 -p 6 -g 2 -I 50000 –microexon-search –no-no-coverage-search. Only the sequencing reads that mapped uniquely to the Drosophila genome were used for further analysis.
Differential gene expression analysis.
Gene expression levels and confidence intervals were calculated by running Cufflinks package 2.1.1 (Trapnell et al. 2012) on the alignments from TopHat. Cufflinks commands were the following:
cufflinks -o/output -G dm3.gtf tophat_aligned_reads.sam & cuffmerge assembly_gtf_file.txt & (merge of all cufflinks output files into a gtf file).
cuffdiff -L genoptype1,genotype2 -o/output assembly_file.gtf -T replicate1.sam, replicate2.sam, replicate3.sam replicate1.sam, replicate2,replicate3.sam &
For Table 3, genes that had significantly different expression (P < 0.05; see Supplemental Table S1) by pairwise comparison were searched against the FlyBase (http://Flybase.org) list of ion channel genes. (Supplemental material for this article is available online at the Journal of Neurophysiology website.) For gene ontology (GO) analysis, the lists of genes for which pairwise comparisons showed significant changes corrected for false discovery rate (Q < 0.05; see Supplemental Table 1) were collected using PANTHER (Mi et al. 2016) and sorted using Excel search functions. Functional annotation and enrichment analysis was done using DAVID (https://david.ncifcrf.gov; Huang da et al. 2009a; Huang da et al. 2009b).
Table 3.
Ion channel gene expression changes in slo and Shab
|
Shab vs. Wild Type |
slo vs. Wild Type |
||||
|---|---|---|---|---|---|
| Ion channel | Change | FPKM (CI) | Ion channel | Change | FPKM with CI |
| CG42260, CNG family cation channel | 2.64-fold increase | CS = 4.58 (2.60–6.56) | CG42260, CNG family cation channel | 2.67-fold increase | CS = 4.58 (2.60–6.56) |
| Shab = 12.09 (7.95–16.22) | slo = 12.21 (8.05–16.38) | ||||
| slo, Ca2+-activated potassium channel | 1.68-fold increase | CS = 7.21 (4.56–9.85) | CG8713, two-pore potassium channel | 13.94-fold increase | CS = 0.92 (0–2.51) |
| Shab = 12.07 (7.83–16.31) | slo = 12.80 (5.67–19.92) | ||||
| cngl, CNG family cation channel | 1.57-fold increase | CS = 11.06 (7.35–14.76) | CG42732, Ca2+-activated potassium channel | 1.61-fold increase | CS = 9.38 (5.96–12.79) |
| Shab = 16.14 (10.79–21.48) | slo = 15.12 (10.19–20.05) | ||||
| Ih, hyperpolarization-activated cation channel | 1.46-fold increase | CS = 25.88 (16.59–35.18) | CG7589, ligand-gated chloride channel | 5.96-fold decrease | CS = 1.56 (0–3.36) |
| Shab = 38.04 (24.53–51.55) | slo = 0.26 (0–0.62) | ||||
| cac, voltage-gated calcium channel, Cav2 homolog | 1.40-fold increase | CS = 15.30 (10.28–20.31) | rpk, amiloride-sensitive sodium channel | 2.04-fold decrease | CS = 2.32 (0.37–4.30) |
| Shab = 21.40 (14.62–28.19) | slo = 1.02 (0–2.21) | ||||
| CG11155, ligand-gated cation channel | 1.32-fold increase | CS = 84.21 (49.07–119.21) | |||
| slo = 109.19 (66.52–151.85) | |||||
| NtR, ligand-gated cation channel | 1.74-fold decrease | CS = 7.19 (2.96–11.42) | |||
| slo = 4.10 (1.22–6.99) | |||||
Libraries were prepared from third instar larval brains of Canton S (CS) wild type, slo1, and Shab3, and resulting profiles were compared using Cuffdiff (Cufflinks 2.1.1) to extract statistically significant (P < 0.05) differences to identify ion channel genes. Three replicates were done for each genotype. Fragments per kilobase of transcript per million mapped reads (FPKM) are shown with the confidence intervals (CI) used for differential expression testing to give an estimate of expression levels and variability. Note that only CG42260, slo, and CG42732 met the more stringent Q < 0.05 cutoff.
RESULTS
Shab3 null neurons exhibit unstable firing in 0 mM external Ca2+.
To examine the nature of long-term neuronal compensation after a disturbance of excitability, we looked at the effects of mutations in Drosophila potassium channels. The Shab K+ channel was an attractive starting point because it is the major delayed rectifier in Drosophila motor neurons (Tsunoda and Salkoff 1995) and because its expression is localized to the soma and primary neurite in other arthropod species (French et al. 2004), making it readily measurable in whole cell patch-clamp mode. The effects of Shab mutations have been examined in the larval body-wall muscle (Chopra et al. 2000; Singh and Singh 1999), giant neuron (Peng and Wu 2007b), and embryonic culture preparations (Tsunoda and Salkoff 1995). To assess the effects of chronic loss of this conductance on central neuron function, we recorded from third instar larval motor neurons. These neurons are well characterized, and individual identified motor neurons can be accessed reproducibly due to their position (Choi et al. 2004). In this study we record exclusively from MN-1b (Fig. 1A).
To determine the overall effect of loss of this conductance on firing, we employed whole cell patch clamp in current-clamp mode. Comparison of recordings taken in 0 mM external Ca2+ from Shab3, a null allele with a small deletion in the Shab gene (Hegde et al. 1999), and Canton S wild type revealed a dramatic instability of current injection-induced neuronal firing that was exclusive to Shab3 and present in 12 of the 13 cells recorded (Fig. 1B, left). This phenotype was similar to that observed in the giant neuron preparation (Peng and Wu 2007a). This instability manifested after long periods of depolarization and never appeared near stimulus onset, suggesting that deficit in a slow sustained current rather than loss of a fast transient current was responsible for the phenotype (Ford and Davis 2014; Ma and Koester 1996).
Addition of Ca2+ to Shab3 neurons suppresses firing instability.
Under physiological conditions, Ca2+ channels and Ca2+-dependent channels play a critical role in determining neuronal excitability. Because our initial recordings were done in 0 mM external Ca2+, we sought to determine the effects of Ca2+-dependent processes on the observed instability. Addition of 1.8 mM Ca2+ into the bath solution normalized firing in current-clamp in Shab3, with wild type experiencing a small increase in firing rate (Fig. 1B, right). Whereas Ca2+channels such as DMCA1D conduct an excitatory fast-inactivating inward current (Worrell and Levine 2008), excitability-suppressing Ca2+-dependent outward currents (IKCa) can display either inactivating or noninactivating kinetics (Elkins et al. 1986; Singh and Wu 1989), similar to Shab. It therefore seemed likely that a change in outward currents was responsible for the normalization of firing.
Shab mutants have decreased outward currents in 0 mM Ca2+.
To investigate the mechanisms underlying these observations, we recorded in voltage-clamp mode to isolate outward currents. Recordings from the cells shown in Fig. 1 are presented in Fig. 2A. Examination of current-voltage relationships in the presence and absence of external Ca2+ revealed that the total outward current was different between genotypes (Fig. 2B). The addition of Ca2+ did not significantly increase the steady-state outward current in wild type but did induce a large increase in Shab3 (P < 0.0005; Fig. 2B). Although the total outward currents in Shab3 did not reach wild-type levels (P < 0.0005) in the presence of Ca2+, a much higher proportion of the total current in Shab3 than in wild type was due to a Ca2+-dependent component (Fig. 2C; P < 0.05 for comparison between wild type and Shab3). This increase in the amount of Ca2+-dependent outward current was dependent on genetic dosage: Shab3/+ heterozygotes had an intermediate level of Ca2+-dependent outward current (Fig. 2C). Interestingly, none of the heterozygote neurons showed firing instability (0/8 in 0 mM Ca2+), indicating that property was also dose dependent.
Figure 2D shows isolated IKd and IKCa currents for wild type and Shab3. Reduction of the Ca2+-independent IKd in Shab3 was large, but it did not go to zero. This suggested the possibility that other noninactivating channels such as SHAW (Hodge et al. 2005; Tsunoda and Salkoff 1995) contribute to IKd, although we did not pursue this hypothesis further. As expected, IKCa was substantially increased (P < 0.0005) in the Shab3 mutant compared with wild type.
We hypothesized that the stabilization of Shab3 firing behavior we observed in 1.8 mM Ca2+ was due to a homeostatic increase in IKCa, which compensated for the loss of IKd. By plotting the difference between genotypes for the isolated IKd and IKCa currents as a function of holding potential, the redistribution of current balance and its voltage dependence is easily seen (Fig. 2E). In this type of plot, a flat line would indicate that there was no difference between genotypes. The downward deflection of the IKd curve reflects the loss of SHAB function in the Shab3 mutant and its voltage dependence. The upward deflection of the IKCa plot indicates that Shab3 has an increase in Ca2+-dependent current. Although the magnitude differences between IKd and IKCa change are not exactly matched, a general trend toward compensation can be seen.
RNAi knockdown of Shab mRNA in motor neurons also reduces IKd and increases IKCa currents.
Because Shab is likely to be expressed in many neurons in the central nervous system, it was possible that the changes in K+ conductances we observed were driven by circuit-level alterations in the brain rather than a cell-autonomous homeostatic process. To distinguish between these possibilities, and to confirm the mutant phenotype, we used RNAi knockdown. The GAL4/UAS system (Brand and Dormand 1995) was utilized to express UAS-Shab RNAi under control of OK371-GAL4, a driver that expresses GAL4 in glutamatergic motor neurons (Mahr and Aberle 2006). Control and knockdown had statistically indistinguishable total outward currents in the presence of Ca2+ (Fig. 3A), but the proportion of total current that was Ca2+ dependent was increased in the knockdown motor neurons, indicating compensation similar to that seen in the mutant.
Fig. 3.

RNAi knockdown of Shab alters the balance of Ca2+-dependent and Ca2+-independent steady-state currents in a cell-autonomous manner. A: total current density from voltage-clamp experiments done in 0 or 1.8 mM Ca2+ are shown for animals expressing Shab RNAi in motor neurons and for UAS-alone control animals. Shab RNAi reduces total current in 0 mM Ca2+ (P < 0.0005 compared with both GAL4 and UAS controls) but not in 1.8 mM Ca2+ (P > 0.05). B: IKd and IKCa currents in Shab RNAi and UAS control neurons. Compared with both UAS and GAL4 controls, loss of SHAB reduces IKd (P < 0.0005) but increases IKCa (P < 0.0005), similar to the effect of the Shab3 mutant. C: difference plot for Shab RNAi and GAL4 control. The steady-state current at each holding potential for control was subtracted from currents produced in Shab RNAi neurons at the same potential. The downward deflection of the IKd curve demonstrates the loss of SHAB-mediated currents, whereas the upward deflection of the IKCa line indicates that there is an increase in this current in Shab RNAi. The shape of the curve shows the voltage-dependence profile of the difference current. All error bars are SE; n = 6 for GAL4 control and n = 14 for both UAS control and Shab RNAi. For data in A and B, 2-way ANOVAs with holding potential and genotype were performed and the Tukey-Kramer post hoc test was used to determine significant differences between genotypes.
Separation of the currents revealed that knockdown of Shab in these cells produced a significant reduction in IKd similar to that observed in the Shab3 mutant (P < 0.0005 compared with both GAL4 and UAS controls; Fig. 3B). Not surprisingly, the magnitude of the knockdown was smaller than in the mutant (mean IKd reduction at 60 mV was 25.22 pA/pF in wild type vs. Shab3 and 18.28 pA/pF in RNAi control vs. knockdown). The reduction of IKd in the knockdown was accompanied by an increase in IKCa (average ± SE IKCa densities at 60 mV were 20.07 ± 7.57 pA/pF in control vs. 54.16 ± 10.27 pA/pF in knockdown; P < 0.0005 across genotypes; Fig. 3B). This was similar to that in the Shab3 mutant strain, providing further evidence that the increase in IKCa was due to a cell autonomous homeostatic process. IKd loss and IKCa gain were roughly current density matched, except at the highest holding potentials (Fig. 3C), indicating the neuron is able to stabilize its overall excitability at the most physiologically relevant operating voltages.
RNAi knockdown of slowpoke mRNA results in decreased IKCa currents.
With strong evidence for homeostatic regulation by IKCa currents, we sought to determine the identity of the gene(s) that encoded the enhanced IKCa current. Potential candidates for Ca2+-dependent K+ channel-encoding genes were ether-a-go-go (Srinivasan et al. 2012; Zhong and Wu 1991), the small-conductance Ca2+-dependent K+ channel SK (Abou Tayoun et al. 2012), and the large-conductance Ca2+-dependent K+ channel slowpoke (Atkinson et al. 1991; Elkins et al. 1986; Lee et al. 2008). We chose to test the role of slo first because of its known involvement in homeostatic compensation (Ghezzi et al. 2004; Lee et al. 2008). Knockdown of slo using GAL4/UAS in the same fashion as in the Shab knockdown produced a significant, but not complete, reduction of Ca2+-dependent outward current recorded in 1.8 mM Ca2+ (Fig. 4A and Table 2) compared with genetic controls. Curiously, we saw no evidence for a homeostatic compensation similar to that observed in Shab (i.e., total somatic currents were reduced by loss of SLO; Fig. 4, A and B). We conclude that SLO provides a significant component of the IKCa current we measure in soma, but that its loss elicits a different type of homeostatic response than loss of SHAB.
Knockdown of slowpoke blocks compensation by IKCa currents when Shab is reduced.
To determine if induction of slo contributed to the increase in IKCa seen in the Shab mutant and knockdown, we performed recordings in double knockdowns of Shab and slo mRNA in motor neurons (w;OK371-GAL4,UAS-ShabRNAi/OK371-GAL4,UAS-sloRNAi;+;+). A significant reduction of IKd was observed in the double knockdown, with currents nearly identical in magnitude to those in the Shab-only knockdown (P > 0.9; Fig. 4, A and C). IKCa currents were also significantly reduced, eliminating nearly all of the total outward current compensation seen in the Shab-only knockdown (Fig. 4, A and D; P < 0.0005 for comparison of Shab RNAi to double knockdown), i.e., bringing the level down to near that seen in controls. Interestingly, total current in the Shab-only knockdown was not significantly changed compared with the GAL4 or UAS control, indicating that the upregulation of IKCa was well matched to the loss of IKd.
The difference plot between these genotypes highlights the inability of the double knockdown to balance currents (Fig. 4E). The flat IKd curve between Shab RNAi and Shab + slo RNAi further demonstrates that the reduction in IKd is identical for both genotypes, indicating that addition of a second UAS transgene did not blunt the Shab RNAi response. The significant downward slope in IKCa, which largely accounts for the reduction in total current, shows that the failure to increase total current is due to lack of SLO.
Homeostatic increases in IKCa involve activity-dependent transcriptional pathways.
To determine if the increases in IKCa involve transcriptional changes, we examined the consequences of inhibiting the major activity-dependent transcriptional pathways to the homeostatic compensation. Nuclear Ca2+ signaling is a central, conserved component of the transcriptional response pathway (Hardingham et al. 2001; Weislogel et al. 2013). Nuclear Ca2+ binds to a distinct pool of calmodulin (CaM), which has then been shown to activate a CREB-binding protein transcriptional cascade. This form of nuclear signaling has been shown to mediate synaptic homeostatic plasticity in mammals (Ibata et al. 2008). In Drosophila, CREB is central to signaling pathways involved in memory formation and circadian rhythms (Belvin et al. 1999; Perazzona et al. 2004; Tubon et al. 2013) and has been shown to be involved in the regulation of slo in alcohol tolerance behavioral assays (Wang et al. 2009). In these studies, slo was shown to be upregulated in adult flies exposed to alcohol, likely as a response to the excitatory effects of benzyl alcohol. Because disruption of CREB in these assays eliminated the slo response to alcohol, we wondered if a disruption of CREB or nuclear CaM would have a similar effect on induction of IKCa in animals with reduced SHAB.
To downregulate nuclear signaling specifically in motor neurons, we expressed a nuclear-targeted “molecular sponge” containing multiple CaM-binding motifs (Weislogel et al. 2013) to reduce the concentration of free nuclear CaM, as well as a dominant negative form of CREB (dnCREB; Yin et al. 1994) to block CREB-dependent transcription. Expressed by themselves, both transcriptional disruptors reduced IKd (compare current amplitudes in Fig. 5 with those in Fig. 4A). For the CaM Sponge, the decrease in IKd was not statistically significant compared with that for GAL4 control neurons (17.9 ± 2.2 vs. 25.8 ± 1.1 pA/pF; P = 0.062), whereas for dnCREB, the reduction was larger and reached significance (16.2 ± 2.0; P < 0.03). Neither transgene reduced IKCa (P > 0.2) compared with the GAL4 control strain. These data suggest that activity-dependent transcription factors likely contribute to the “resting” levels of some, but not all, channels.
Fig. 5.

Inhibition of activity-regulated transcriptional processes reduces potassium currents and partially blocks homeostasis. IKd and IKCa current densities for control and experimental genotypes at 20-mV holding potential. Both dnCREB and CaM Sponge reduce outward currents and partially block the increase in IKCa normally produced by Shab RNAi (compare with Fig. 4). Data are means ± SE; n = 4 for OK371/+;UAS-CaMSponge/+, n = 5 for OK371/UAS-ShabRNAi;UAS-CaMSponge, n = 4 for OK371/+;UAS-dnCREB/+, and n = 3 for OK371/UAS-ShabRNAi;UAS-dnCREB/+. *P < 0.05, Shab RNAi + CaM Sponge vs. CaM Sponge alone; Student’s t-test.
Coexpression of CaM Sponge with Shab RNAi decreased IKd (P < 0.05). Interestingly, it did not completely block the ability of the SHAB knockdown to increase IKCa (P < 0.05) but did blunt the amplitude of the change (compare Fig. 5 with Fig. 4A). Coexpression of Shab RNAi with dnCREB elicited a similar increase in IKCa, but the difference was not statistically significant, likely due to low number of cells. These data suggest that activity-stimulated transcriptional pathways may contribute to the increase in IKCa stimulated by loss of SHAB but that there are other factors involved since loss of IKCa upregulation was not complete.
Loss of SHAB and SLO generate distinct transcriptional responses.
To determine if the increase in IKCa seen in Shab null mutants was due to direct transcriptional upregulation of slo mRNA, we performed RNA sequencing (RNAseq) on samples prepared from brains of Canton S wild type, Shab3, and slo1 third instar larvae. Statistically significant differences from pairwise comparisons of the expression profiles (P < 0.05) and differences corrected for false discovery rate (Q < 0.05) are listed in Supplemental Table S1. Comparison of wild-type and slo mutant expression profiles identified 129 genes, whose expression was altered (Q < 0.05); in Shab mutant brains, there were 240 genes that had Q < 0.05 compared with wild type. Interestingly, the overlap between the two sets included only 38 genes (29% of the genes changed in slo, and 16% of the genes changed in Shab). Only one of the shared genes was an ion channel, CG42260. This gene encodes a member of the cyclic nucleotide-gated cation channel family, and its expression is increased almost threefold in both mutants relative to wild type.
Loss of SHAB and SLO alter expression of distinct sets of ion channels.
Although there was little other commonality in the sets of genes changed, both mutants did show alterations in the expression of ion channels (Table 3). The first notable result is that Shab mutants have significantly higher levels of slo mRNA than either wild-type or slo animals (P < 0.05, up 1.68- and 1.96-fold, respectively). This is consistent with the increase in IKCa seen in Shab mutants or in neurons where Shab has been knocked down with RNAi in MN-1b. The other ion channels whose expression is increased in Shab mutants are all cation channels. One of them, cacophony, is a synaptic voltage-gated Ca2+ channel; it provides the main source of Ca2+ for activation of SLO (Ford and Davis 2014; Peng and Wu 2007b).
In contrast, being mutant for slo does not alter Shab expression (Q > 0.05 compared with either wild type or Shab mutants). This is consistent with the finding from the electrophysiological experiments that there is no reciprocal relationship in the homeostatic compensations triggered by loss of these two conductances. The other ion channels that change in slo1 are distinct from those that change in Shab3 and include two potassium channels (Table 3). slo1 animals have increases in CG42732, which encodes a Ca2+-activated potassium channel (1.61-fold), and CG8713, a two-pore potassium channel (13.94-fold), which has been implicated in sleep homeostasis (Pimentel et al. 2016).
Channel mutations alter membrane transport and signaling genes.
Gene ontology (GO) analysis of the data sets suggests that, compared with wild type, both mutants have changes in non-channel genes associated with ion flux. Nine percent of the genes altered in Shab3 have transport among their GO terms, and functional annotation with DAVID (https://david.ncifcrf.gov; Huang da et al. 2009a; Huang da et al. 2009b) shows that the enrichment for this term is significant (P = 0.011). In the slo1 data set, only 5% of the genes were classified as transporters, and this group was not significantly enriched. Signaling genes were well represented in both data sets (16% of Shab3 and 19% of slo1 lists were signaling molecules). Both mutant lists have a statistically significant enrichment for genes involved in bioamine metabolism (P = 0.018 for the Shab3 list; P = 0.028 for the slo1 list). These data suggest that there is a widespread network of gene changes that affect nervous system communication. The presence of changes in intercellular signaling and neurotransmitter metabolism pathways indicates that homeostatic changes are not limited to cell autonomous processes but also involve changes in brain circuit function.
DISCUSSION
Homeostatic compensation of Shab loss by induction of slowpoke.
Homeostatic compensation by alterations in ion channel function after both acute and chronic changes in neuronal excitability have been well documented (Davis 2006; Marder and Goaillard 2006; Turrigiano 2011). The basic idea that mutations or drugs that cause hyperexcitability will trigger processes that dampen activity, and vice versa, provides a mechanism for stabilizing neuronal activity. Although we do not actually know the nature of the target(s) of the homeostat, the direction of the compensatory changes seen with this type of manipulation is consistent across neurons and systems.
In this study we have shown that loss of SHAB, a noninactivating voltage-dependent potassium channel, causes an upregulation of a slowly activating, sustained Ca2+-dependent outward current. The upregulation of IKCa with Shab perturbations was a robust effect, with both null mutants and RNAi knockdowns displaying large increases. The fact that RNAseq from whole brain shows a significant increase in slo suggests that this mode of compensation is not limited to MN-1b cells. Furthermore, by examining the double knockdown of both genes, we were able to show that nearly all of the IKCa increase induced in motor neurons by Shab knockdown comprised SLO. This was in line with observations in other neurons where a chronically missing conductance was replaced with one that had similar properties (Bergquist et al. 2010; Kimm et al. 2015). The identification of slo as a homeostatic agent was also not unprecedented. Increases in slo transcription have been shown to underlie rapid tolerance to the sedative effects of benzyl alcohols (Ghezzi et al. 2004), and SLO currents are important for maintaining action potential width after loss of the Shaker IA current (Ford and Davis 2014).
Nonreciprocal homeostasis: IKd does not compensate for loss of slowpoke.
After demonstrating that SHAB loss is compensated by an IKCa current, we wondered if knockdown of slo would stimulate compensation by an IKd-like current. Reciprocal compensation has been observed previously between Shaker and Shal, the two genes encoding IA conductances in Drosophila (Bergquist et al. 2010). This simple, reciprocal model assumes that all ion channel genes are equally poised to stabilize activity, and given that there was an apparent role for both CaM-dependent transcription and dCREB in setting IKd levels (compare IKd current densities in Fig. 4A and Fig. 5), this was a reasonable hypothesis. Contrary to this simple expectation, however, knockdown of slo did not increase IKd, and in fact, total outward current was reduced in the slo RNAi motor neurons, suggesting that homeostatic compensations involved other classes of effectors in this cell type. Although it remains possible that reciprocal compensation can occur in neurons other than MN-1b, the fact that Shab is not upregulated in slo1 mutants argues against it being a widespread mode of response.
Nonreciprocal compensations have also been observed in other systems. In crustacean stomatogastric ganglion neurons, artificially increasing the Shal IA channel stimulates an increase in Ih, a hyperpolarization-gated nonspecific cation channel, counterbalancing the increased outward current and normalizing firing (MacLean et al. 2003). In contrast, overexpression of Ih does not increase IA (or any other outward current) and leaves neurons with altered firing properties (Zhang et al. 2003). This is supportive of the idea that compensations are tied to the identity of the dysfunctional component even if they do not result in perfect one-to-one replacement of the lost current.
Role of gene expression in homeostasis of excitability.
On long timescales, plasticity processes, including homeostatic ones, generally rely on transcriptional and translational changes to stabilize neuronal activity (Davis et al. 1996; Ibata et al. 2008; Parrish et al. 2014; Schulz et al. 2006; Wang et al. 2009). To compare the gene expression changes triggered by loss of SHAB and SLO, we performed RNAseq experiments on larval brains from wild-type animals and from Shab3 and slo1 mutants. Interestingly, although both Shab and slo encode slowly activating, slowly deactivating channels, the gene expression profiles of the two mutants were more different from each other than they were from wild type (for Q < 0.5, there were 344 genes differentially expressed between Shab3 and slo1; see Supplemental Table 1). The list of transcripts that changed in both mutants compared with wild type was quite small, consisting of 38 genes with only one ion channel. This suggests that each mutant responds to the loss of its cognate channel uniquely and that there is not a “generic” compensation program for loss of a potassium channel. This is in line with both theoretical models that show that a particular activity state can be built with many different combinations of conductances (Prinz et al. 2004) and with experimental studies at the larval neuromuscular junction, which show unique transcriptional responses to different channel mutations (Parrish et al. 2014).
The changes in gene expression for the individual mutants compared with wild type were more in line with expectations for homeostatic compensation. For both Shab3 and slo1, there were significant changes in genes encoding ion channels (Table 3) and membrane transporters, classes of proteins that could be expected to jointly modulate ion balance and excitability (Neverisky and Abbott 2015). For slo1, there were increases in two potassium channels, one of which, CG8713, has been previously implicated in homeostasis (Pimentel et al. 2016) and a decrease in an amiloride-sensitive Na+ channel previously shown to be important for mechanosensation (Tsubouchi et al. 2012). All of these changes would be expected to dampen excitability. For Shab3, the only obviously excitability-dampening channel change was an increase in slo.
All other channels that were significantly changed were ones that might be expected to be excitatory: increased levels of several cation channels and decreased levels of a chloride channel. Although this might seem counterintuitive, for Shab3 mutants, which have elevated SLO, increased Ca2+ might actually help suppress excitability (Ford and Davis 2014; Peng and Wu 2007b). For both Shab3 and slo1, it is also important to note that the RNAs were collected from whole brains, not just from neurons expressing the lost potassium channel. This means that the gene expression changes we see reflect the entire circuit and that there may be cells for which an increase in excitability is adaptive for the overall function of the mutant animal. RNAseq of isolated motor neurons after RNAi would be a first step in distinguishing cell-autonomous from circuit-level compensations.
The involvement of circuit-level compensations becomes even more obvious when classes of genes other than ion channels and transporters are considered. For both mutants, there was an enrichment for signaling molecules, and in particular for molecules involved in bioamine metabolism. Whether these changes reflect primary compensations or are a response to alterations in the activity of specific circuits is unknown, because homeostatic processes can operate at the level of the individual cell involved or at the level of brain circuits. For most global perturbations such as channel mutations, it is likely that both levels are engaged and that they interact (Bergquist et al. 2010; Grashow et al. 2010), but there are theoretical grounds on which to believe that the cell autonomous processes can drive the circuit-level changes (O’Leary et al. 2014).
Homeostatic programs are highly specific.
The idea that changes in excitability provoke compensations that move toward stabilization of neuronal function is well established. Most models have focused on global mechanisms and posit that overall activity (usually with Ca2+ as the effector) is the driving force for homeostasis (LeMasson et al. 1993; Marder and Prinz 2002; Yu and Goda 2009). This would suggest that perturbations that produce the similar global changes in activity would be compensated in similar ways. This assumption is challenged by the data presented in this report and in a number of other studies (e.g., Lee et al. 2014; MacLean et al. 2003; Nerbonne et al. 2008; Parrish et al. 2014; Ueda and Wu 2006; Zhang et al. 2003). It is clear that manipulations that in theory produce similar activity changes can evoke very different molecular compensations and can have varying levels of success in compensation. The timescale of the perturbation can also dictate the nature of the compensation; acute loss of a conductance may be dealt with quite differently than a long-term and developmental alteration such as a genetic deletion of the same channel (Nerbonne et al. 2008; Swensen and Bean 2005; Yuan et al. 2005). Even more interestingly, for clearly defined compensations where loss of one channel induces a particular second channel, loss of the second channel does not necessarily induce the first (this study and Zhang et al. 2003). The lack of reciprocity and the nonuniformity of homeostatic compensations suggest that the regulation of excitability is complex and not simply proportional to alterations in firing rate.
How can this level of specificity be achieved and still involve activity? One possibility is that the normal distribution and role of a channel provides a unique signature that can be read by the homeostatic machinery. Even if the biophysical properties of two channels are similar, their role in neuronal function can be quite different if they localize to different cellular compartments e.g., vertebrate IA encoding Kv1 and Kv4 channels (Rivera et al. 2003). Differences in localization could underlie differences in homeostatic response, because signaling mechanisms, including ones that ultimately regulate transcription, can be specific to subcellular compartments (Yu and Goda 2009) or even to activation of different types of Ca2+ channels (Deisseroth et al. 1998). There are of course many other possibilities, but what is clear is that there is currently no understanding of the genesis of homeostatic diversity. Decoding these basic mechanisms underlying channel-specific homeostatic compensation will be important for understanding the dysregulation of neuronal function in many disease states.
GRANTS
This work was supported by National Institute of Neurological Disorders and Stroke Grant P01 NS079419 (project 2; to L. C. Griffith). E. Z. Kim was supported by National Science Foundation-Integrative Graduate Education and Research Traineeship DGE-0549390. J. Vienne was supported by the Swiss National Science Foundation and a grant from the Ellison Foundation (to M. Rosbash).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
E.Z.K. and L.C.G. conceived and designed research; E.Z.K. and J.V. performed experiments; E.Z.K., J.V., and L.C.G. analyzed data; E.Z.K. and L.C.G. interpreted results of experiments; E.Z.K. and L.C.G. prepared figures; E.Z.K. drafted manuscript; E.Z.K., J.V., M.R., and L.C.G. edited and revised manuscript; E.Z.K., J.V., M.R., and L.C.G. approved final version of manuscript.
Supplementary Material
ACKNOWLEDGMENTS
We thank Eve Marder and Sacha Nelson for helpful advice on data analysis.
REFERENCES
- Abou Tayoun AN, Pikielny C, Dolph PJ. Roles of the Drosophila SK channel (dSK) in courtship memory. PLoS One 7: e34665, 2012. doi: 10.1371/journal.pone.0034665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson NS, Robertson GA, Ganetzky B. A component of calcium-activated potassium channels encoded by the Drosophila slo locus. Science 253: 551–555, 1991. doi: 10.1126/science.1857984. [DOI] [PubMed] [Google Scholar]
- Bellen HJ, Tong C, Tsuda H. 100 years of Drosophila research and its impact on vertebrate neuroscience: a history lesson for the future. Nat Rev Neurosci 11: 514–522, 2010. doi: 10.1038/nrn2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belvin MP, Zhou H, Yin JC. The Drosophila dCREB2 gene affects the circadian clock. Neuron 22: 777–787, 1999. doi: 10.1016/S0896-6273(00)80736-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergquist S, Dickman DK, Davis GW. A hierarchy of cell intrinsic and target-derived homeostatic signaling. Neuron 66: 220–234, 2010. doi: 10.1016/j.neuron.2010.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand AH, Dormand EL. The GAL4 system as a tool for unravelling the mysteries of the Drosophila nervous system. Curr Opin Neurobiol 5: 572–578, 1995. doi: 10.1016/0959-4388(95)80061-1. [DOI] [PubMed] [Google Scholar]
- Choi JC, Park D, Griffith LC. Electrophysiological and morphological characterization of identified motor neurons in the Drosophila third instar larva central nervous system. J Neurophysiol 91: 2353–2365, 2004. doi: 10.1152/jn.01115.2003. [DOI] [PubMed] [Google Scholar]
- Chopra M, Gu GG, Singh S. Mutations affecting the delayed rectifier potassium current in Drosophila. J Neurogenet 14: 107–123, 2000. doi: 10.3109/01677060009083478. [DOI] [PubMed] [Google Scholar]
- Davis GW. Homeostatic control of neural activity: from phenomenology to molecular design. Annu Rev Neurosci 29: 307–323, 2006. doi: 10.1146/annurev.neuro.28.061604.135751. [DOI] [PubMed] [Google Scholar]
- Davis GW, Müller M. Homeostatic control of presynaptic neurotransmitter release. Annu Rev Physiol 77: 251–270, 2015. doi: 10.1146/annurev-physiol-021014-071740. [DOI] [PubMed] [Google Scholar]
- Davis GW, Schuster CM, Goodman CS. Genetic dissection of structural and functional components of synaptic plasticity. III. CREB is necessary for presynaptic functional plasticity. Neuron 17: 669–679, 1996. doi: 10.1016/S0896-6273(00)80199-3. [DOI] [PubMed] [Google Scholar]
- Deisseroth K, Heist EK, Tsien RW. Translocation of calmodulin to the nucleus supports CREB phosphorylation in hippocampal neurons. Nature 392: 198–202, 1998. doi: 10.1038/32448. [DOI] [PubMed] [Google Scholar]
- Elkins T, Ganetzky B, Wu CF. A Drosophila mutation that eliminates a calcium-dependent potassium current. Proc Natl Acad Sci USA 83: 8415–8419, 1986. doi: 10.1073/pnas.83.21.8415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford KJ, Davis GW. Archaerhodopsin voltage imaging: synaptic calcium and BK channels stabilize action potential repolarization at the Drosophila neuromuscular junction. J Neurosci 34: 14517–14525, 2014. doi: 10.1523/JNEUROSCI.2203-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French LB, Lanning CC, Matly M, Harris-Warrick RM. Cellular localization of Shab and Shaw potassium channels in the lobster stomatogastric ganglion. Neuroscience 123: 919–930, 2004. doi: 10.1016/j.neuroscience.2003.08.036. [DOI] [PubMed] [Google Scholar]
- Ghezzi A, Al-Hasan YM, Larios LE, Bohm RA, Atkinson NS. slo K+ channel gene regulation mediates rapid drug tolerance. Proc Natl Acad Sci USA 101: 17276–17281, 2004. doi: 10.1073/pnas.0405584101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giachello CN, Baines RA. Regulation of motoneuron excitability and the setting of homeostatic limits. Curr Opin Neurobiol 43: 1–6, 2017. doi: 10.1016/j.conb.2016.09.014. [DOI] [PubMed] [Google Scholar]
- Grashow R, Brookings T, Marder E. Compensation for variable intrinsic neuronal excitability by circuit-synaptic interactions. J Neurosci 30: 9145–9156, 2010. doi: 10.1523/JNEUROSCI.0980-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Arnold FJ, Bading H. Nuclear calcium signaling controls CREB-mediated gene expression triggered by synaptic activity. Nat Neurosci 4: 261–267, 2001. doi: 10.1038/85109. [DOI] [PubMed] [Google Scholar]
- Hegde P, Gu GG, Chen D, Free SJ, Singh S. Mutational analysis of the Shab-encoded delayed rectifier K+ channels in Drosophila. J Biol Chem 274: 22109–22113, 1999. doi: 10.1074/jbc.274.31.22109. [DOI] [PubMed] [Google Scholar]
- Hodge JJ, Choi JC, O’Kane CJ, Griffith LC. Shaw potassium channel genes in Drosophila. J Neurobiol 63: 235–254, 2005. doi: 10.1002/neu.20126. [DOI] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37: 1–13, 2009a. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57, 2009b. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Ibata K, Sun Q, Turrigiano GG. Rapid synaptic scaling induced by changes in postsynaptic firing. Neuron 57: 819–826, 2008. doi: 10.1016/j.neuron.2008.02.031. [DOI] [PubMed] [Google Scholar]
- Jan LY, Jan YN. Properties of the larval neuromuscular junction in Drosophila melanogaster. J Physiol 262: 189–214, 1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36, 2013. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimm T, Khaliq ZM, Bean BP. Differential regulation of action potential shape and burst-frequency firing by BK and Kv2 channels in substantia nigra dopaminergic neurons. J Neurosci 35: 16404–16417, 2015. doi: 10.1523/JNEUROSCI.5291-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Ueda A, Wu CF. Pre- and post-synaptic mechanisms of synaptic strength homeostasis revealed by slowpoke and shaker K+ channel mutations in Drosophila. Neuroscience 154: 1283–1296, 2008. doi: 10.1016/j.neuroscience.2008.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Ueda A, Wu CF. Distinct roles of Drosophila cacophony and Dmca1D Ca2+ channels in synaptic homeostasis: genetic interactions with slowpoke Ca2+-activated BK channels in presynaptic excitability and postsynaptic response. Dev Neurobiol 74: 1–15, 2014. doi: 10.1002/dneu.22120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeMasson G, Marder E, Abbott LF. Activity-dependent regulation of conductances in model neurons. Science 259: 1915–1917, 1993. doi: 10.1126/science.8456317. [DOI] [PubMed] [Google Scholar]
- Ma M, Koester J. The role of K+ currents in frequency-dependent spike broadening in Aplysia R20 neurons: a dynamic-clamp analysis. J Neurosci 16: 4089–4101, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean JN, Zhang Y, Johnson BR, Harris-Warrick RM. Activity-independent homeostasis in rhythmically active neurons. Neuron 37: 109–120, 2003. doi: 10.1016/S0896-6273(02)01104-2. [DOI] [PubMed] [Google Scholar]
- Mahr A, Aberle H. The expression pattern of the Drosophila vesicular glutamate transporter: a marker protein for motoneurons and glutamatergic centers in the brain. Gene Expr Patterns 6: 299–309, 2006. doi: 10.1016/j.modgep.2005.07.006. [DOI] [PubMed] [Google Scholar]
- Marder E, Bucher D. Understanding circuit dynamics using the stomatogastric nervous system of lobsters and crabs. Annu Rev Physiol 69: 291–316, 2007. doi: 10.1146/annurev.physiol.69.031905.161516. [DOI] [PubMed] [Google Scholar]
- Marder E, Goaillard JM. Variability, compensation and homeostasis in neuron and network function. Nat Rev Neurosci 7: 563–574, 2006. doi: 10.1038/nrn1949. [DOI] [PubMed] [Google Scholar]
- Marder E, Prinz AA. Modeling stability in neuron and network function: the role of activity in homeostasis. BioEssays 24: 1145–1154, 2002. doi: 10.1002/bies.10185. [DOI] [PubMed] [Google Scholar]
- Mi H, Poudel S, Muruganujan A, Casagrande JT, Thomas PD. PANTHER version 10: expanded protein families and functions, and analysis tools. Nucleic Acids Res 44, D1: D336–D342, 2016. doi: 10.1093/nar/gkv1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nataraj K, Le Roux N, Nahmani M, Lefort S, Turrigiano G. Visual deprivation suppresses L5 pyramidal neuron excitability by preventing the induction of intrinsic plasticity. Neuron 68: 750–762, 2010. doi: 10.1016/j.neuron.2010.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nerbonne JM, Gerber BR, Norris A, Burkhalter A. Electrical remodelling maintains firing properties in cortical pyramidal neurons lacking KCND2-encoded A-type K+ currents. J Physiol 586: 1565–1579, 2008. doi: 10.1113/jphysiol.2007.146597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neverisky DL, Abbott GW. Ion channel-transporter interactions. Crit Rev Biochem Mol Biol 51: 257–267, 2016. doi: 10.3109/10409238.2016.1172553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Leary T, Williams AH, Franci A, Marder E. Cell types, network homeostasis, and pathological compensation from a biologically plausible ion channel expression model. Neuron 82: 809–821, 2014. [Erratum in Neuron 88: 1308, 2015]. doi: 10.1016/j.neuron.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish JZ, Kim CC, Tang L, Bergquist S, Wang T, Derisi JL, Jan LY, Jan YN, Davis GW. Krüppel mediates the selective rebalancing of ion channel expression. Neuron 82: 537–544, 2014. doi: 10.1016/j.neuron.2014.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng IF, Wu CF. Differential contributions of Shaker and Shab K+ currents to neuronal firing patterns in Drosophila. J Neurophysiol 97: 780–794, 2007a. doi: 10.1152/jn.01012.2006. [DOI] [PubMed] [Google Scholar]
- Peng IF, Wu CF. Drosophila cacophony channels: a major mediator of neuronal Ca2+ currents and a trigger for K+ channel homeostatic regulation. J Neurosci 27: 1072–1081, 2007b. doi: 10.1523/JNEUROSCI.4746-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perazzona B, Isabel G, Preat T, Davis RL. The role of cAMP response element-binding protein in Drosophila long-term memory. J Neurosci 24: 8823–8828, 2004. doi: 10.1523/JNEUROSCI.4542-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pimentel D, Donlea JM, Talbot CB, Song SM, Thurston AJ, Miesenböck G. Operation of a homeostatic sleep switch. Nature 536: 333–337, 2016. doi: 10.1038/nature19055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ping Y, Tsunoda S. Homeostatic plasticity in Drosophila central neurons, and implications in human diseases. Fly (Austin) 6: 153–157, 2012. doi: 10.4161/fly.20775. [DOI] [PubMed] [Google Scholar]
- Prinz AA, Bucher D, Marder E. Similar network activity from disparate circuit parameters. Nat Neurosci 7: 1345–1352, 2004. doi: 10.1038/nn1352. [DOI] [PubMed] [Google Scholar]
- Rivera JF, Ahmad S, Quick MW, Liman ER, Arnold DB. An evolutionarily conserved dileucine motif in Shal K+ channels mediates dendritic targeting. Nat Neurosci 6: 243–250, 2003. [Erratum in Nat Neurosci 6: 899, 2003]. doi: 10.1038/nn1020. [DOI] [PubMed] [Google Scholar]
- Salkoff LB, Tanouye MA. Genetics of ion channels. Physiol Rev 66: 301–329, 1986. [DOI] [PubMed] [Google Scholar]
- Schulz DJ, Goaillard JM, Marder E. Variable channel expression in identified single and electrically coupled neurons in different animals. Nat Neurosci 9: 356–362, 2006. doi: 10.1038/nn1639. [DOI] [PubMed] [Google Scholar]
- Singh A, Singh S. Unmasking of a novel potassium current in Drosophila by a mutation and drugs. J Neurosci 19: 6838–6843, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S, Wu C-F. Complete separation of four potassium currents in Drosophila. Neuron 2: 1325–1329, 1989. doi: 10.1016/0896-6273(89)90070-6. [DOI] [PubMed] [Google Scholar]
- Srinivasan S, Lance K, Levine RB. Contribution of EAG to excitability and potassium currents in Drosophila larval motoneurons. J Neurophysiol 107: 2660–2671, 2012. doi: 10.1152/jn.00201.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swensen AM, Bean BP. Robustness of burst firing in dissociated Purkinje neurons with acute or long-term reductions in sodium conductance. J Neurosci 25: 3509–3520, 2005. doi: 10.1523/JNEUROSCI.3929-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7: 562–578, 2012. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsubouchi A, Caldwell JC, Tracey WD. Dendritic filopodia, Ripped Pocket, NOMPC, and NMDARs contribute to the sense of touch in Drosophila larvae. Curr Biol 22: 2124–2134, 2012. doi: 10.1016/j.cub.2012.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunoda S, Salkoff L. The major delayed rectifier in both Drosophila neurons and muscle is encoded by Shab. J Neurosci 15: 5209–5221, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tubon TC Jr, Zhang J, Friedman EL, Jin H, Gonzales ED, Zhou H, Drier D, Gerstner JR, Paulson EA, Fropf R, Yin JC. dCREB2-mediated enhancement of memory formation. J Neurosci 33: 7475–7487, 2013. doi: 10.1523/JNEUROSCI.4387-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano G. Too many cooks? Intrinsic and synaptic homeostatic mechanisms in cortical circuit refinement. Annu Rev Neurosci 34: 89–103, 2011. doi: 10.1146/annurev-neuro-060909-153238. [DOI] [PubMed] [Google Scholar]
- Turrigiano G. Homeostatic synaptic plasticity: local and global mechanisms for stabilizing neuronal function. Cold Spring Harb Perspect Biol 4: a005736, 2012. doi: 10.1101/cshperspect.a005736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano G, Abbott LF, Marder E. Activity-dependent changes in the intrinsic properties of cultured neurons. Science 264: 974–977, 1994. doi: 10.1126/science.8178157. [DOI] [PubMed] [Google Scholar]
- Ueda A, Wu CF. Distinct frequency-dependent regulation of nerve terminal excitability and synaptic transmission by IA and IK potassium channels revealed by Drosophila Shaker and Shab mutations. J Neurosci 26: 6238–6248, 2006. doi: 10.1523/JNEUROSCI.0862-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Ghezzi A, Yin JC, Atkinson NS. CREB regulation of BK channel gene expression underlies rapid drug tolerance. Genes Brain Behav 8: 369–376, 2009. doi: 10.1111/j.1601-183X.2009.00479.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei A, Covarrubias M, Butler A, Baker K, Pak M, Salkoff L. K+ current diversity is produced by an extended gene family conserved in Drosophila and mouse. Science 248: 599–603, 1990. doi: 10.1126/science.2333511. [DOI] [PubMed] [Google Scholar]
- Weislogel JM, Bengtson CP, Müller MK, Hörtzsch JN, Bujard M, Schuster CM, Bading H. Requirement for nuclear calcium signaling in Drosophila long-term memory. Sci Signal 6: ra33, 2013. doi: 10.1126/scisignal.2003598. [DOI] [PubMed] [Google Scholar]
- Worrell JW, Levine RB. Characterization of voltage-dependent Ca2+ currents in identified Drosophila motoneurons in situ. J Neurophysiol 100: 868–878, 2008. doi: 10.1152/jn.90464.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin JCP, Wallach JS, Del Vecchio M, Wilder EL, Zhou H, Quinn WG, Tully T. Induction of a dominant negative CREB transgene specifically blocks long-term memory in Drosophila. Cell 79: 49–58, 1994. doi: 10.1016/0092-8674(94)90399-9. [DOI] [PubMed] [Google Scholar]
- Yu LM, Goda Y. Dendritic signalling and homeostatic adaptation. Curr Opin Neurobiol 19: 327–335, 2009. doi: 10.1016/j.conb.2009.07.002. [DOI] [PubMed] [Google Scholar]
- Yuan W, Burkhalter A, Nerbonne JM. Functional role of the fast transient outward K+ current IA in pyramidal neurons in (rat) primary visual cortex. J Neurosci 25: 9185–9194, 2005. doi: 10.1523/JNEUROSCI.2858-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Oliva R, Gisselmann G, Hatt H, Guckenheimer J, Harris-Warrick RM. Overexpression of a hyperpolarization-activated cation current (Ih) channel gene modifies the firing activity of identified motor neurons in a small neural network. J Neurosci 23: 9059–9067, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Y, Wu C-F. Alteration of four identified K+ currents in Drosophila muscle by mutations in eag. Science 252: 1562–1564, 1991. doi: 10.1126/science.2047864. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.