

Abstract
Here we present a technique called single-molecule high-resolution colocalization (SHREC) of fluorescent dyes that allows the measurement of interfluorophore distances in macromolecules and macromolecular complexes with better than 10-nm resolution. By using two chromatically differing fluorescent molecules as probes, we are able to circumvent the Rayleigh criterion and measure distances much smaller than 250 nm. The probes are imaged separately and localized individually with high precision. The registration between the two imaging channels is measured by using fiduciary markers, and the centers of the two probes are mapped onto the same space. Multiple measurements can be made before the fluorophores photobleach, allowing intramolecular and intermolecular distances to be tracked through time. This technique's lower resolution limit lies at the upper resolution limit of single molecule FRET (smFRET) microscopy. The instrumentation and fluorophores used for SHREC can also be used for smFRET, allowing the two types of measurements to be made interchangeably, covering a wide range of interfluorophore distances. A dual-labeled duplex DNA molecule (30 bp) was used as a 10-nm molecular ruler to confirm the validity of the method. We also used SHREC to study the motion of myosin V. We directly observed myosin V's alternating heads while it walked hand-over-hand along an actin filament.
Keywords: centroid tracking, dynamic conformational changes, fluorescence, molecular motors, total internal reflection microscopy
Single molecule fluorescence techniques are bridging the areas of structural biology and biochemistry (1). They yield structural constraints for an enzyme during its biochemical cycle. To probe close to this interface between structure and function, distances must be resolved on the order of the size of the enzyme. However, far field fluorescence microscopy is limited in its resolution by the Rayleigh criterion at ≈250 nm. On the other end of the size spectrum, single molecule FRET (smFRET) provides a way to estimate intramolecular distances <10 nm and has yielded insights on a range of biological molecules from the Tetrahymena ribozyme to the ribosome (2-4).
Recent methods, single-molecule high-resolution imaging with photobleaching (SHRImP) and nanometer-localized multiple single-molecule (NALMS), have been developed in an attempt to bridge this “gap” in distance resolution. These methods are capable of measuring an intramolecular distance to high precision, but only once, because of the intrinsic destruction of one of the probes (5, 6). A time series of the intramolecular distance is not possible with these methods, removing one of the greatest potential benefits of single molecule imaging.
Addressing the gap in imaging techniques suitable for studying macromolecules, we introduce a technique, single-molecule high-resolution colocalization (SHREC) of fluorescent probes, that can precisely measure intramolecular and intermolecular distances through time. By separately imaging two chromatically different fluorophores (Cy3 and Cy5) conjugated to a single molecule, we are able to localize both probes simultaneously. A transformation mapping for the two imaging channels is applied to the localization results, and the relative positions of the two probes in the plane of the microscope stage can then be accurately determined to better than 10 nm, as was confirmed by measuring the end-to-end distance of dual-labeled duplex DNA.
SHREC uses small probes (fluorescent molecules) that allow a direct intramolecular distance measurement because they can be located specifically on the domains being studied. The dramatic decrease in photons emitted by single fluorescent dyes compared with large fluorescent beads and nanocrystals used in previous multicolor colocalization studies (7) presents challenges to achieving high resolution. The development of fluorescence imaging with one nanometer accuracy (FIONA) has addressed the challenges involved in precisely localizing a single fluorescent dye (8-11). FIONA measures the location of a single molecular domain through time with high precision but cannot measure how the distance between two points within a molecule are changing with time.
The power of SHREC is that it can probe distances on the scale of biological macromolecules with relevant time resolution. Distance measurements in the range of 10-200 nm, difficult to probe by other techniques, are now accessible. This is precisely the range of distances that applies to the wide variety of macromolecules and macromolecular complexes that are the players in establishing the processes fundamental to all of cell and developmental biology. These processes are dynamic, and, therefore, the changes in the distance measurements need to be determined with good time resolution. To illustrate SHREC's capability in measuring intramolecular distances through time, we use it here to analyze the myosin V walking mechanism. Myosin V is a member of the myosin family of molecular motors and is responsible for cargo transport along actin filaments in a wide variety of cell types. Myosin V is an ideal enzyme to study with the SHREC technique because it undergoes multiple catalytic cycles per encounter with actin, each of which is coupled to a large conformational change in the protein. The myosin V heavy chain consists of three major domains: the catalytic head domain; the light chain binding domain, which binds six light chains and serves as a lever arm; and the C-terminal tail domain that forms a dimer with another myosin V heavy chain by means of coiled-coil interactions. Recent studies on the myosin V walking mechanism strongly implied that myosin V uses its lever arms to walk hand-over-hand along actin (9, 12-15, 17-19). The hand-over-hand walking mechanism predicts an alternation of the catalytic heads while the protein walks along the actin filament. Here we directly show the alternation of the heads by differentially labeling the two lever arms and imaging them by using the SHREC technique.
Materials and Methods
Duplex DNA Preparation. To make the 30-bp duplex DNA molecules, two oligonucleotides with the following sequence and modifications were hybridized (Integrated DNA Technologies, Coralville, IA). The oligonucleotide 5′-GGGTATGGAGATTTTTAGCGGAGTGACAGC-3′ was labeled with Cy5 at its 5′ end and with Cy3 at its 3′ end. The complementary oligonucleotide was labeled with biotin at both ends.
Protein Expression and Purification. The yellow fluorescent protein (YFP) gene in the p2Bac/pFastBac-YFP-M5-CaM plasmid (a gift from H. Lee Sweeney, University of Pennsylvania, Philadelphia) was deleted by PCR. The resulting p2Bac/pFastBac-M5-CaM plasmid codes for chicken myosin V that is truncated at Glu-1099. A leucine zipper followed the native coiled coil to ensure dimerization. To facilitate purification, the myosin V protein was N-terminally tagged with a FLAG-tag (DYKDDDDK). Two recombinant baculoviruses were generated for protein expression in Sf9 cells. One encoded the truncated myosin V and the Drosophila melanogaster calmodulin derived from the p2Bac/pFastBac-M5-CaM plasmid. The second virus encoded the human essential light chain derived from the p2Bac/pFastBac-ELC plasmid. Both viruses were used for coinfection of Sf9 cells. The protein was expressed and purified as described by Sweeney et al. (20).
Calmodulin Labeling and Exchange. Calmodulin was expressed and labeled with Cy3 or Cy5 as follows: A single cysteine was introduced in sea urchin calmodulin by means of the mutation Q143C. This calmodulin was expressed in Escherichia coli and purified as described in ref. 21. The calmodulin [800 μM in 50 mM Tris·HCl, pH 7.4/1 mM Tris-(2-carboxyethyl)phosphine] was labeled with either Cy3-maleimide or Cy5-maleimide (Amersham Biosciences) stock solutions (in DMSO) at a 1.4-fold molar ratio for 20 min. Excess dye was removed by gel filtration into exchange buffer (below), followed by overnight hydrophobic absorption onto BioBeads (Bio-Rad). Label incorporation was 102% for Cy3-calmodulin and 75% for Cy5-calmodulin. Aliquots were stored at -80°C.
The exchange of labeled calmodulin onto myosin V was performed as described but with some modifications (22). Myosin V (150 nM) was incubated with 0.7 nM Cy3-labeled calmodulin and 0.8 nM Cy5-labeled calmodulin in exchange buffer (25 mM KCl/20 mM imidazole HCl, pH 7.4/2 mM MgCl2/1 mM EGTA) at 22°C for 2 min. To exchange calmodulins, 0.9 mM CaCl2 was added and the mixture was incubated at 22°C for 5 min. The reaction was quenched with 7 mM EGTA. The reaction mixture was applied onto a 100K MWCO Nanocep ultrafiltration device (Pall) and washed three times with equal volumes of AB (below) to purify myosin V from excess calmodulin.
Flow Cell Preparation. The flow cell was constructed with a glass microscope slide and highly refracting coverslips made of NLAF21 (VA Optical Labs, San Anselmo, CA) held together by pieces of double-sided Scotch tape.
For the myosin V experiments, 15 μl of biotin-BSA (1 mg·ml-1) was incubated in the flow cell for 2 min, after which 30 μl of AB buffer (25 mM KCl/25 mM imidazole HCl, pH 7.4/1 mM EGTA/4 mM MgCl2/10 mM DTT/1 mg·ml-1 BSA) was passed through the flow cell to wash it out. Fifteen microliters of 0.5 mg/ml NeutrAvidin (Molecular Probes) was added to the cell and was allowed to incubate for 2 min, after which another wash was done with AB buffer. The flow cell was incubated with 15 μl of biotinylated phalloidin actin filaments (250 nM) for 5 min, followed by a 100-μl wash with AB buffer. Finally, the cell was loaded with 20 μl of imaging buffer [10 mM Tris, pH 8.0/50 mM NaCl/10 mM MgCl2/an oxygen scavenger system consisting of 17 units of glucose oxidase (Sigma) and 260 units of catalase (Roche) from a stock that had been filtered with a 0.2-μm syringe filter and centrifuged for 5 min at 13,000 × g/0.4% (vol/vol) glucose/1% (vol/vol) β-mercaptoethanol] including calmodulin-exchanged myosin V, 5 μM calmodulin, 300 nM ATP, an ATP regeneration system (0.1 mg·ml-1 creatine phosphokinase/1 mM creatine phosphate), and 0.5% (vol/vol) Triton-X 100 to reduce nonspecific binding. The cell was sealed with vacuum grease and imaged immediately. The final motor concentration resulted in sparsely decorated actin and thus allowed an analysis of single myosin V molecules.
For the DNA experiments, the biotin-BSA and NeutrAvidin were loaded in the same manner as for the myosin V experiments, but the washes were done with T50 buffer (10 mM Tris, pH 8.0/50 mM NaCl). Once the flow cell had a NeutrAvidin coating, 15 μlof dsDNA (30 nM) in T50 buffer with 1 mg·ml-1 BSA were flowed in and incubated for 2 min, after which 100 μl of imaging buffer was flowed in. The flow cell was then sealed with vacuum grease and promptly imaged. The resulting decoration of DNA molecules on the surface was sufficiently sparse that fluorescent spots from different molecules rarely overlapped.
Microscope Setup. The total internal reflection fluorescence (TIRF) microscope was set up as described in ref. 8, with some modifications (Fig. 5, which is published as supporting information on the PNAS web site). Excitation source beams at 532 nm (Coherent, Santa Clara, CA) and 633 nm (JDS Uniphase, San Jose, CA) were combined by a dichroic mirror and expanded to a 7-mm diameter. These sources were focused (focal length = 500 mm) on the back focal plane of an Olympus 1.65 NA ×100 TIRF objective by means of a laser line dichroic on a linear translation stage that allows microscope operation in either epifluorescence or TIRF modes. The objective was positioned under a closed-loop, two-axis, piezo nanotranslation stage equipped with capacitive sensors for position measurement (Physik Instrumente, Auburn, MA). The reflected light exiting the back aperture of the objective was directed on a quadrant photodiode to provide a signal for a focus feedback loop that clamped the distance between the objective and sample with an electrostrictive actuator (Newport, Fountain Valley, CA). Fluorescence emission was collected by the objective, passed through two StopLine thin film notch filters (Semrock, Rochester, NY), one for each excitation source, and transmitted through a dual-view apparatus that allowed simultaneous imaging of the Cy3 and Cy5 channels on a single iXon DV 887 EMCCD camera (Andor Technology, Belfast, Ireland). All data were imaged with a 0.5-sec integration time. Crosstalk between the two channels (10%) presumably biases the distance measurements toward smaller values. Our experimental error, however, renders it undetectable, as shown by Monte Carlo simulations in which 100 pairs of modeled images of single fluorophores separated by 10 nm were created and analyzed identically as with the DNA data (described below). Simulated images of fluorescent point spread functions were calculated by generating an integrated 2D Gaussian intensity distribution with a constant background to which shot noise was added. The simulated peaks had the same dimensions and signal-to-noise ratio as the real peaks. Simulated data with 10% crosstalk and simulated data without crosstalk are indistinguishable by a Kolmogorov-Smirnov test (N1 = 100, N2 = 100, p > 0.05).
Data Analysis. A registration mapping of the Cy3 and Cy5 channels was performed with fiducials consisting of 100-nm TransFluoSphere beads (Molecular Probes). They were excited at 532 nm, and they emit with a broad emission spectrum. The bead was detectable in both of our channels. We located a single bead associated to the coverslip, and with our piezo stage we stepped it with nanometer precision in a grid pattern (with a 0.5-μm spacing), taking an image at every stop. The end result was a stack of 312 images that shows the bead in both channels at different positions (Fig. 1a).
Fig. 1.

Precise alignment of the two imaging channels to within 3.3 nm. Fiducial grids with 0.5-μm spacing were made by translating a fluorescent bead detectable in both channels by a piezo stage. At each point in the grid, an image of the bead was taken. (a) The resulting stack of images was projected through z to observe the positions that the calibration series visited. (b) The center locations of the beads were found by means of a fit to a 2D Gaussian function. (c) The subsequent pairs of locations were used to calculate a transformation mapping of the Cy5 channel onto the Cy3 channel.
The centers of the beads were found by a least squares fit to a 2D elliptical Gaussian function with a background,
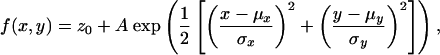 |
[1] |
to determine its location in its respective space (Fig. 1b). In practice, σx and σy were nearly identical. These locations allowed us to calculate a local weighted mean mapping (23) from the Cy5 channel to the Cy3 channel (Fig. 1c). This mapping is a weighted sum of second-order polynomials determined locally around each fiducial (within a six fiducial radius). It corrects registration errors that arise locally without allowing their influence to extend to the rest of the space. The target registration error (TRE) that accompanies this mapping is calculated in the following way. Each fiducial is set aside one at a time, and a mapping is calculated by using the others. The left-out fiducial's position in the Cy5 channel is then mapped into the Cy3 channel's space, and the deviation of the mapped location to the fiducial's location in the Cy3 channel is recorded. The mean value of the deviations' magnitudes calculated for each fiducial is the TRE. This mapping can then be applied to any dye localized in the Cy5 channel to determine where it resides relative to positions found in the Cy3 channel. The dyes' locations are found in units of pixels and must be converted by means of the pixel size. Because we know the differences in the locations of the bead in real space as we move it around with the piezo stage, we are also able to get an accurate measurement of the pixel size (110 nm) with this same calibration. We found that, in general, this calibration yields a transformation mapping that is valid over a few weeks. All data reported here, however, were collected on the same day that a calibration was performed.
The DNA data were analyzed by homemade software using matlab (Mathworks, Natick, MA). The routine automatically locates peaks in the image by determining the pixels of high intensity and ensures that the surrounding pixels have intensities as expected for a single fluorophore. Before fitting the peaks to a 2D Gaussian function to get its center locations, we search for pairs of peaks. The pixels found in the Cy5 channel are mapped onto the Cy3 channel, and a search for peaks in the surrounding Cy3 pixels is performed. If a peak is found, then the Cy5 peak and the Cy3 peak are considered a pair for further analysis. Each peak is fit to a 2D Gaussian function in its respective space as described for the beads above (Eq. 1). The error on the fit mean location is calculated with the number of photons (Nγ) collected,
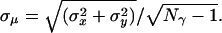 |
[2] |
The Cy5's location is mapped to the Cy3 channel, and the distance between them is calculated. After the computational analysis of the DNA data, the identified fluorophore pairs were screened manually to ensure that the pairs were not close to any other fluorophores that would have interfered in the pairs' analysis.
The myosin V data were analyzed by first identifying a moving motor by eye. Homemade software written in matlab then fit the starting pair of fluorescent peaks to 2D Gaussian functions as described above and continued to track the peaks for as long as the user specified. Motors whose conjugated dyes each photobleached in a single step were analyzed, as was the case for most of the motors observed, which ensured that we indeed were studying motors with only one Cy3 label and one Cy5 label. The resulting trajectories were registered by mapping the Cy5 trajectory onto the Cy3 channel. A least squares linear fit to the points from both trajectories gave the orientation of the actin filament to which the trajectories were projected. Distances along the linear fit (the actin filament) were plotted against time to see the heads' relative distances (Fig. 4).
Fig. 4.

Time trace of a differentially labeled myosin V molecule walking along an actin filament. The labels (Cy3 and Cy5) are covalently attached to calmodulins that were exchanged onto the myosin V molecule. In this trace, both of the fluorescent probe's locations are taking 72-nm steps, indicating that the calmodulins were exchanged close to the motor domain. The alternating positions of the probes provide a direct observation of myosin V's hand-over-hand walking mechanism.
Results
Image Registration Achieved to Within 3.3 nm. Here we describe imaging and colocalization of two different dyes with distinct emission spectra, Cy3 and Cy5, with high resolution. In our dual view imaging setup, the images of the spectrally separated fluorophores are transmitted through two separate light paths containing separate sets of optics that distort the images in different ways (Fig. 5). We mapped this distortion by using fluorescent beads that have a broad emission spectrum (see Materials and Methods). These beads can be imaged in both of our detection channels and thus are considered fiducial markers. By finding the center locations of the beads at multiple positions in the two channels, we were able to calculate a mapping from the Cy5 channel to the Cy3 channel with a target registration error of 3.3 nm (Fig. 1).
End-to-End Length Measurements of dsDNA 30-mers. We created a molecular ruler consisting of 30 bp of dsDNA with Cy5 at one end and Cy3 at the other. We then imaged the Cy5 and the Cy3 dyes on each end of the DNA molecule simultaneously and on separate halves of the CCD camera. The locations of each dye were determined by fitting to a 2D Gaussian function. The Cy5's location was mapped onto the Cy3 channel, and then the distance between the dyes was calculated by using the center locations found from the registered 2D Gaussian fits, (xCy3, yCy3) and (xCy5, yCy5):
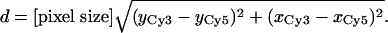 |
[3] |
We solved the expected distance distribution numerically by means of Monte Carlo simulations. We modeled a Gaussian distribution of points around two positions separated by a fixed distance. Pairs of those points were picked at random, and the distance between them was calculated and graphed as histograms (Fig. 2a). The reason for the skewed shape of the simulated histogram is evident from geometric considerations. For a dsDNA molecule of length s lying along the x axis with one end at the origin and the other at the position x = s (Fig. 2b), the experimental error gives a probability of a position measurement of these ends that is different from their true values. This probability decays from the true location in a Gaussian manner (see gray cloud in Fig. 2b). If we measure the first end of the molecule to be at the origin, to measure the correct end-to-end separation s, the second point will need to lie on the circumference of the semicircle originating at (0, 0) and cutting through the Gaussian gray cloud at a radius s. The points in the Gaussian gray cloud lying inside this semicircle will give distances <s, and the points lying outside will give distances >s. More of the Gaussian gray cloud lies outside the circle, so it is more likely to measure a distance >s; hence, the distribution of possible distance measurements from the origin will have a skew toward large values.
Fig. 2.

Determining the accurate end-to-end distance from the skewed distribution of distance measurements. (a) The distance probability distribution was calculated by means of Monte Carlo simulations. (b) The skew in the histograms toward large values can be understood from a geometric argument. If one end of the DNA molecule is measured to reside at the origin, then only points lying on the circumference of a circle with radius s and origin (0, 0) will yield the true end-to-end distance. It is more likely that a point will lie outside of the dashed-line semicircle than inside it, which gives the distance distribution a long tail. (c) Despite the non-Gaussian nature of the distance distribution, the end-to-end separation can be calculated using the geometric mean, variance, and localization errors.
A useful formula can be derived relating the end-to-end distance (or separation s) to measured quantities in SHREC. By mathematically defining the mean distance squared between a point from a 2D Gaussian probability distribution with a variance σ21 and a point from a second 2D Gaussian probability distribution with a variance σ22 a distance s away as
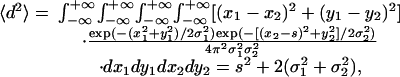 |
one can derive an expression for the end-to-end separation, s, based on the mean of the distribution and the variance of the distribution along with the localization errors:
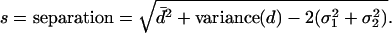 |
[4] |
The above formula was tested with Monte Carlo simulations of various absolute separations, and it correctly yields the true separation (Fig. 2c).
We measured the end-to-end lengths of 482 DNA molecules (Fig. 3). The data are skewed toward large values, which is expected from the above description. Applying Eq. 4 to our data, we measure an end-to-end distance of 10 ± 1 nm (95% confidence limits were determined from 1,000 bootstrap distributions) using the following mean errors on our localizations:
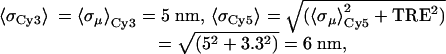 |
where σμ is the error on the fit mean location and TRE is the target registration error. With a persistence length of 50 nm (16), the 30-bp duplex DNA molecule can be considered a rigid rod with a contour length of 10 nm, assuming a 0.34-nm rise per base. All data were imaged with a 0.5-sec integration time, which is long enough such that all thermal motion of the DNA molecules, their conjugated probes, and linkers are averaged out. The expected measurement of the end-to-end length of the DNA molecules is thus the contour length, 10 nm, which is in excellent agreement with our result.
Fig. 3.

End-to-end distances of 482 duplex DNA molecules (30 bp) as measured by SHREC.
Watching both Heads of Myosin V Walk Hand-over-Hand. Myosin V is an excellent model enzyme with which to test SHREC's ability to track distances through time, because its hand-over-hand walking is confirmed and well understood (9, 12-15, 17-19). It takes 36-nm steps, which is well above our experimental error. By exchanging the native calmodulins with a mixture of Cy3- and Cy5-labeled calmodulins, we can observe two different legs of the same myosin molecule. The Cy3 and the Cy5 data are obtained by analyzing each fluorescent channel's “movie” separately, mapping the results on top of one another, and then projecting each of the trajectories onto the inferred position of the actin filament. A time trace is made by plotting the location of each probe along the actin filament against time. The sample trace shows the trajectory of a single myosin V molecule as an alternating staircase, which is expected from the hand-over-hand model of processivity (Fig. 4). Each leg's step size is ≈72 nm, as expected for calmodulins exchanged close to the motor domain. Additionally, the motor domains are separated from each other by ≈36 nm along the axis of the actin filament, as expected.
Discussion
The distance that SHREC measures is a 2D projection of the true intramolecular distance. In our studies, we have arranged the geometries such that the intramolecular distances lie in the x-y plane, a common geometry for molecular motor studies and other single molecule measurements. In other cases, an absolute distance might not be measurable due to limitations in the possible experimental geometries. Nevertheless, a change in the 2D projection of the intramolecular distance would also yield valuable information.
The limited number of emitted photons per fluorescent dye determines the possible temporal resolution and the total observation time. We show here that temporal resolutions of interest in biomolecular processes are achievable. Thus, it is possible to track the colocalization of two single fluorophores through time with high resolution.
Knowledge of an enzyme's structural constraints while it progresses through its biological function is an excellent complement to a crystal structure, which gives only a snapshot of the structure in one of its conformations. SHREC measurements can be performed on the same microscopes used in smFRET studies and with the same fluorophores, providing a smooth transition from smFRET analysis to SHREC analysis. When smFRET begins to lose effectiveness (≈10 nm), SHREC can resolve the fluorophores' locations. Combining smFRET and SHREC, structural information can be tracked for long periods of time and over broad distance scales.
Supplementary Material
Acknowledgments
We thank Lee Sweeney for the pFastBac-YFP-M5-CaM plasmid construct. We are grateful to Michael Levene (Yale University, New Haven, CT) for his thoughtful comments on the manuscript. We thank Zev Bryant, David Altman, and other members of J.A.S.'s laboratory for their critical comments on the work. This work was supported by Boehringer Ingelheim Fonds (Z.Ö.), a Burroughs Wellcome Career Award at the Scientific Interface (to R.S.R.), the Natural Sciences and Engineering Research Council of Canada (J.F.D.), and National Institutes of Health Grant GM33289 (to J.A.S.).
Abbreviations: smFRET, single molecule FRET; SHREC, single molecule high-resolution colocalization.
References
- 1.Weiss, S. (1999) Science 283, 1676-1683. [DOI] [PubMed] [Google Scholar]
- 2.Blanchard, S. C., Kim, H. D., Gonzalez, R. L., Jr., Puglisi, J. D. & Chu, S. (2004) Proc. Natl. Acad. Sci. USA 101, 12893-12898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blanchard, S. C., Gonzalez, R. L., Kim, H. D., Chu, S. & Puglisi, J. D. (2004) Nat. Struct. Mol. Biol. 11, 1008-1014. [DOI] [PubMed] [Google Scholar]
- 4.Zhuang, X., Bartley, L. E., Babcock, H. P., Russell, R., Ha, T., Hershlag, D., Chu, S. (2000) Science 288, 2048-2051. [DOI] [PubMed] [Google Scholar]
- 5.Qu, X., Wu, D., Mets, L. & Scherer, N. F. (2004) Proc. Natl. Acad. Sci. USA 101, 11298-11303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gordon, M. P., Ha, T. & Selvin, P. R. (2004) Proc. Natl. Acad. Sci. USA 101, 6462-6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lacoste, T. D., Michalet, X., Pinaud, F., Chemla, D. S., Alivisatos, A. P. & Weiss, S. (2000) Proc. Natl. Acad. Sci. USA 97, 9461-9466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Okten, Z., Churchman, L. S., Rock, R. S. & Spudich, J. A. (2004) Nat. Struct. Mol. Biol. 11, 884-887. [DOI] [PubMed] [Google Scholar]
- 9.Yildiz, A., Forkey, J. N., McKinney, S. A., Ha, T., Goldman, Y. E. & Selvin, P. R. (2003) Science 300, 2061-2065. [DOI] [PubMed] [Google Scholar]
- 10.Yildiz, A., Tomishige, M., Vale, R. D. & Selvin, P. R. (2004) Science 303, 676-678. [DOI] [PubMed] [Google Scholar]
- 11.Yildiz, A., Park, H., Safer, D., Yang, Z., Chen, L. Q., Selvin, P. R. & Sweeney, H. L. (2004) J. Biol. Chem. 279, 37223-37226. [DOI] [PubMed] [Google Scholar]
- 12.De La Cruz, E. M., Wells, A. L., Rosenfeld, S. S., Ostap, E. M. & Sweeney, H. L. (1999) Proc. Natl. Acad. Sci. USA 96, 13726-13731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mehta, A. D., Rock, R. S., Rief, M., Spudich, J. A., Mooseker, M. S. & Cheney, R. E. (1999) Nature 400, 590-593. [DOI] [PubMed] [Google Scholar]
- 14.Rief, M., Rock, R. S., Mehta, A. D., Mooseker, M. S., Cheney, R. E. & Spudich, J. A. (2000) Proc. Natl. Acad. Sci. USA 97, 9482-9486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moore, J. R., Krementsova, E. B., Trybus, K. M. & Warshaw, D. M. (2001) J. Cell Biol. 155, 625-635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hagerman, P. J. (1988) Annu. Rev. Biophys. Biophys. Chem. 17, 265-286. [DOI] [PubMed] [Google Scholar]
- 17.Purcell, T. J., Morris, C., Spudich, J. A. & Sweeney, H. L. (2002) Proc. Natl. Acad. Sci. USA 99, 14159-14164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Veigel, C., Wang, F., Bartoo, M. L., Sellers, J. R. & Molloy, J. E. (2002) Nat. Cell Biol. 4, 59-65. [DOI] [PubMed] [Google Scholar]
- 19.Forkey, J. N., Quinlan, M. E., Alexander Shaw, M., Corrie, J. E. & Goldman, Y. E. (2003) Nature 422, 399-404. [DOI] [PubMed] [Google Scholar]
- 20.Sweeney, H. L., Rosenfeld, S. S., Brown, F., Faust, L., Smith, J., Xing, J., Stein, L. A. & Sellers, J. R. (1998) J. Biol. Chem. 273, 6262-6270. [DOI] [PubMed] [Google Scholar]
- 21.Gopalakrishna, R. & Anderson, W. B. (1982) Biochem. Biophys. Res. Commun. 104, 830-836. [DOI] [PubMed] [Google Scholar]
- 22.Sakamoto, T., Amitani, I., Yokota, E. & Ando, T. (2000) Biochem. Biophys. Res. Commun. 272, 586-590. [DOI] [PubMed] [Google Scholar]
- 23.Goshtasby, A. (1988) Image and Vision Computing 6, 255-261. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}