

Abstract
Background:
Placental multidrug resistance-associated protein 2 (MRP2), encoded by ABCC2 gene in human, plays a significant role in regulating drugs’ transplacental transfer rates. Studies on placental MRP2 regulation could provide more therapeutic targets for individualized and safe pharmacotherapy during pregnancy. Currently, the roles of epigenetic mechanisms in regulating placental drug transporters are still unclear. This study aimed to investigate the effect of histone deacetylases (HDACs) inhibition on MRP2 expression in the placental trophoblast cell line and to explore whether HDAC1/2/3 are preliminarily involved in this process.
Methods:
The human choriocarcinoma-derived trophoblast cell line (Bewo cells) was treated with the HDAC inhibitors-trichostatin A (TSA) at different concentration gradients of 0.5, 1.0, 3.0, and 5.0 μmol/L. Cells were harvested after 24 and 48 h treatment. Small interfering RNA (siRNA) specific for HDAC1/HDAC2/HDAC3 or control siRNA was transfected into cells. Total HDAC activity was detected by colorimetric assay kits. HDAC1/2/3/ABCC2 messenger RNA (mRNA) and protein expressions were determined by real-time quantitative polymerase chain reaction and Western-blot analysis, respectively. Immunofluorescence for MRP2 protein expression was visualized and assessed using an immunofluorescence microscopy and ImageJ software, respectively.
Results:
TSA could inhibit total HDAC activity and HDAC1/2/3 expression in company with increase of MRP2 expression in Bewo cells. Reduction of HDAC1 protein level was noted after 24 h of TSA incubation at 1.0, 3.0, and 5.0 μmol/L (vs. vehicle group, all P < 0.001), accompanied with dose-dependent induction of MRP2 expression (P = 0.045 for 1.0 μmol/L, P = 0.001 for 3.0 μmol/L, and P < 0.001 for 5.0 μmol/L), whereas no significant differences in MRP2 expression were noted after HDAC2/3 silencing. Fluorescent micrograph images of MRP2 protein were expressed on the cell membrane. The fluorescent intensities of MRP2 in the control, HDAC2, and HDAC3 siRNA-transfected cells were week, and no significant differences were noticed among these three groups (all P > 0.05). However, MRP2 expression was remarkably elevated in HDAC1 siRNA-transfected cells, which displayed an almost 3.19-fold changes in comparison with the control siRNA-transfected cells (P < 0.001).
Conclusions:
HDACs inhibition could up-regulate placental MRP2 expression in vitro, and HDAC1 was probably to be involved in this process.
Keywords: Epigenetic Regulation, Histone Deacetylases, Multidrug Resistance-associated Protein 2, Placenta
Introduction
Drug consumption during pregnancy is increasingly common and often unavoidable for the treatment of various maternal and fetal diseases. Epidemiological studies showed that 60–70% of pregnant women are prescribed on one or more drugs except for minerals and vitamins during pregnancy, and 5–10% are taking medications categorized D or X by the Food and Drug Administration (FDA) which are suspected to be teratogenic to the fetus.[1,2,3] Individualized pharmacotherapy during pregnancy has become a major clinical issue in the field of perinatology. Depending on the intended action of the drugs, their transfer across the placenta may be termed as either desired or undesirable. Individualized pharmacotherapy requires proper transplacental rates for balancing the drug's efficacy and its side-effects when deciding treatment regimen in pregnant women. Therefore, proper understanding of the transplacental passage of drugs and its influence factors is vital and helpful in guiding clinicians to more accurate and safer pharmacotherapy during pregnancy.
Accumulating evidence have confirmed that the passage of drugs across the placenta cannot simply be predicted on the basis of their physical-chemical properties. Several drug transporters have been discovered in the placenta and are widely proved to play a significant role in controlling the drugs’ transplacental rates. Functional expression of those placental drug transporters must be considered to optimize pharmacotherapy during pregnancy.[4] Analogous to P-glycoprotein and breast cancer resistance protein, the multidrug resistance-associated protein 2 (MRP2), encoded by ABCC2 gene in human, has also been found to be highly expressed in placenta and of great importance in controlling drugs’ transplacental rates recently.[5] It is localized to the maternal-facing apical membrane of placental syncytiotrophoblast and possesses the capacity to actively extrude a wide range of drugs back to the maternal circulation.[5] More studies on the regulation of placental MRP2 are of great significance to the individualized and safe pharmacotherapy during pregnancy.
Recent studies have highlighted the potential importance of epigenetic effects on the regulation of placental gene expression, particularly in the contexts of fetoplacental development, trophoblast differentiation, fetal programming, and placental pathophysiology.[6,7,8] However, the roles of epigenetic mechanisms in regulating placental drug transporters are still unclear.
As an important chromatin-modifying enzyme, histone deacetylases (HDACs) could remove acetyl groups from histone lysine tails, stabilize nucleosome structure, and compact chromatin, thereby blocking access of transcriptional activators to the DNA template and repressing gene transcription.[9] Totally, there are four classes of HDACs according to phylogenetic analysis and sequence homologies. Until now, only HDAC1/2/3, which are core members of Class I HDACs, have been proved to be extremely abundant in trophoblast cells and involved in placental development by regulating trophoblastic fusion and embryogenesis.[10,11,12] Emerging studies have revealed that HDAC inhibitors, such as suberoylanilide hydroxamic acid and trichostatin A (TSA), could alter MRP2 expression in tumor cells.[13,14] These findings imply that HDACs, particularly the HDAC1/2/3, might play a significant role in placental MRP2 regulation. However, as the HDACs might exhibit cell type-specific manners in gene regulation, whether these processes are also involved in the regulation of placental MRP2 still need to be further investigated. Therefore, the aim of this study was to investigate the effect of HDAC inhibition on the expression of MRP2 in placental trophoblast cell line and to explore whether HDAC1/2/3 are preliminarily involved in this process or not, which might illuminate the pathway of MRP2 regulation by epigenetics in placenta.
Methods
Cell line and culture conditions
The human choriocarcinoma-derived trophoblast cell line (Bewo cells) obtained from the Cell Bank of Chinese Academy of Science were cultured in 10% fetal bovine serum-DMEM/F-12 (Thermo Fisher Scientific, USA) supplemented with 100 units/ml penicillin and 100 μg/ml streptomycin (Gibco, USA) at 37°C in a humidified atmosphere of 95% air and 5% CO2.
Histone deacetylase inhibitors-trichostatin A treatment
HDAC inhibitors-TSA was widely used as a HDAC inhibitor and has been validated in many studies. TSA (WXBC0707V, Vetec, USA) was first dissolved in dimethylsulfoxide (DMSO) at the concentration of 1 mmol/L and stored at −70°C for use. To determine the sensitivity of cells to TSA, we used a tetrazolium reagent, 2-(4-indophenyl)-3-(4-nitrophenyl)-5-(2,4-disulphophenyl)-2H-tetrazolium monosodium salt (WST-1, Cell Counting Kit, Beyotime, Beijing, China). In brief, the Bewo cells were seeded in triplicate at a density of 5000 cells per well into 96-well plates and grown overnight. The cells were treated with or without different concentrations of TSA (0.5, 1.0, 3.0, and 5.0 μmol/L) for 24, 48, or 72 h. At the end of experiments, 10 μl WST-1 reagents were added to each well and cells were incubated at 37°C for an additional 4 h. The absorbance of each sample was measured by a microplate reader (Varioskan Flash, Thermo Scientific, USA) under a wavelength of 450 nm. The percent cell viability was expressed using the following formula: percent cell viability = ([absorbance of the experimental well] – [absorbance of the blank])/([absorbance of the vehicle well] − [absorbance of the blank]) × 100%. The experiments were performed in triplicate. From these studies, it was noted that Bewo cells were sensitive to TSA treatment, and 72 h of incubation with TSA reduced its cell viability to about 10% [Supplementary Figure 1 (189.9KB, tif) ]. Thus, only the 24 h and 48 h time points were selected.
Effect of TSA on Bewo cell viability at different time points of exposure. The cells were treated with or without different concentrations of TSA (0.5, 1.0, 3.0, and 5.0 μmol/L) for 24, 48, or 72 h. Cell viability was evaluated using the WST-1 assay. n = 3 for each group. Data were expressed as mean ± SE. *P < 0.001, versus vehicle. TSA: Trichostatin A; SE: Standard error.
In further studies, cells were grown in six-well plates until 70–80% confluence was reached. Then, the medium was changed and 2 ml fresh medium was added to each well. The 1, 2, 6, and 10 μl of TSA were diluted into cell culture medium at the desired concentrations of 0.5, 1.0, 3.0, and 5.0 μmol/L, respectively. Subsequently, 9, 8, 4, and 0 μl of DMSO were individually added to respective well to guarantee an equivalent concentration of DMSO (0.5%) in different groups while culture medium with DMSO (0.5%) was used as control. The medium was changed daily with new medium carrying new test compounds, and cellular status was observed as well. The cells were washed with ice-cold PBS and harvested at 24 and 48 h for next step tests. All experiments were repeated three times.
Transfection of histone deacetylase 1/2/3 small interfering RNA
An amount of 1 × 105 or 4 × 105 cells were seeded initially in 24-well or 6-well plates for real-time polymerase chain reaction (PCR) or Western-blot analysis, respectively. After cells were grown to 30–50% confluence, 80 nmol/L of small interfering RNA (siRNA) specific for HDAC1 (stB0001570A, GuangZhou RiboBio. Co., China), HDAC2 (stB0001571A, GuangZhou RiboBio. Co.), HDAC3 (stB0001590A, GuangZhou RiboBio. Co.), or control siRNA (siN05815122147, GuangZhou RiboBio. Co.) was transfected into Bewo cells using LipofectamineR RNAiMAX Reagent (13778-150, Invitrogen, Life technologies, Carlsbad, CA, USA) according to the manufacturer's instruction. Cells were harvested after 48 h of transfection for next step tests. All experiments were repeated three times. The siRNA sequences used were as follows: siRNA HDAC1 sense: 5’-GCGACUGUUUGAGAACCUU dTdT-3’, anti-sense: 3’-dTdTCGCUGACAAACUCUUGGAA-5’; siRNA HDAC2 sense: 5’-CCGUAAUGUUGCUCGAUGU dTdT-3’, anti-sense: 3’-dTdT GGCAUUACAACG AGCUACA-5’; siRNA HDAC3 sense: 5’-GAGCAA CCCAGCUGAACAA dTdT-3’, anti-sense: 3’-dTdT CUC GUUGGGUCGACUUGUU-5’.
Real-time quantitative real-time polymerase chain reaction
Total RNA was isolated and purified using the Trizol reagent (Invitrogen, Life technologies, Carlsbad, CA, USA). The concentration of purified RNA samples was assessed spectrophotometrically using the Nanodrop_2000 instrument (Thermo Scientific). RNA (1 μg) was reverse transcribed using PrimeScript™ RT Reagent Kit with gDNA eraser (RR0047A, Takara, Japan) according to the manufacturer's instructions.
Amplification of complementary DNA (cDNA) was performed with SsoFast EvaGreen Supermixture (Bio-Rad Laboratories, Hercules, CA, USA) using 5 μl reaction mixture, 0.5 μl forward primer, 0.5 μl reverse primer, 3 μl nuclease-free H2O, and 1 μl cDNA in a total volume of 10 μl. PCR conditions were 39 cycles of 30 s at 95°C, 10 s at 55°C for HDAC1, HDAC2, HDAC3, ABCC2, and GAPDH, preceded by an initial denaturation of 3 min at 95°C, and followed by a continuous melt curve from 65°C to 95°C. A validation experiment had been undertaken in which equal quantities of cDNA were used. Similar amplification efficiencies and cycle threshold (Ct) values of GAPDH were obtained between the control group and the treatment group. The stability of GAPDH expression between the control group and the treatment group guaranteed its use as an appropriate endogenous control for normalization. In addition, we have ascertained the efficiencies of amplifications for all genes involved in our study (HDAC1, HDAC2, HDAC3, ABCC2, and GAPDH), which were consistent across a range of template concentrations. All the slope of the amplification efficiency curves were more than 95% and efficiencies for the target genes (HDAC1, HDAC2, HDAC3, and ABCC2) and the endogenous control (GAPDH) were approximately equal (0.954–0.982). All samples were amplified in triplicates. Gene expression was represented for the Ct value by the mean of triple tests. Relative expressions of the target genes in each sample were normalized to expression of GAPDH using 2−ΔΔCt. The primer sequences specific for target genes were as follows: HDAC1: 5’- CACCCATTCTTCCCGTTCT-3’ (forward), 5’-GCACTTGGCATTTCAGGAGT-3’ (reverse); HDAC2: 5’-GTTCTGGCATCCTCCCTGT-3’ (forward), 5’-TTCCATCTCCTCCATCCACT-3’ (reverse); HDAC3: 5’-GAGGGATGAACGGGTAGACA-3’ (forward), 5’-CAGGTGTTAGGGAGCCAGAG-3’ (reverse); ABCC2: 5’-AGCACCGACTATCCAGCATC-3’ (forward), 5’-GAAACCAAAGGCACTCCAGA-3’ (reverse); GAPDH: 5’-GAAGGTGAAGGTCGGAGTC-3’ (forward), 5’-GAAGATGGTGATGGGATTTC-3’(reverse).
Western-blot analysis
Cells were lysed by RIPA (P0013B, Beyotime, China) with complete protease inhibitor cocktail (P8340, Sigma-Aldrich, USA) for 20 min at 4°C, centrifuged at 12,000 ×g for 5 min at 4°C. Supernants were collected and the protein concentration was measured using enhanced BCA protein assay kit (P0010S, Beyotime, China) according to the manufacturer's protocol. Cell lysates were boiled in 4× sample buffer for 5 min, and 50 μg protein/lane was subjected to 8% SDS-polyacrylamide gel. Proteins were transferred to polyvinylidene difluoride membranes (Millipore, Bedford, MA, USA). The membranes were blocked for 60 min in Tris-base buffer containing 0.1% Tween 20 (TBST) and 5% nonfat milk and incubated overnight at 4°C with primary antibodies against HDAC1 (10197-1-AP, Proteintech, 1:1000), HDAC2 (12922-3-AP, Proteintech, 1:1000), HDAC3 (10255-1-AP, Proteintech, 1:500), MRP2 (sc-20766, Santa Cruz, 1:100) and GAPDH (CW0100A, CWBIO, 1:500). Following extensive washing with TBST, the membranes were immunoblotted with horseradish peroxidase-conjugated goat anti-rabbit/goat anti-mouse immunoglobulin G secondary antibodies (diluted 1:2500) for 2 h at room temperature. Washed several times in TBST, membranes were exposed to enhanced chemiluminescene detection system. The protein expression levels were quantified by software Gelpro32 and normalized against the GAPDH as an endogenous control. We have confirmed the specificity of antibodies (HDAC1, HDAC2, HDAC3, and MRP2) through preincubation with the peptide epitope and prevention of binding at 60,000, 55,000, 49,000, and 200,000, respectively.
Histone deacetylase activity assay
The total HDAC activity was determined by HDAC activity colorimetric assay kits (K331-100, BioVision, USA) according to the manufacturer's protocol. The assay was based on a two-step colorimetric reaction. The first step of reaction was deacetylation of the acetylated lysine side chain of HDAC colorimetric substrate by a sample containing HDAC activity. Deacetylation of the substrate sensitized the substrate, so that, in the second step, treatment with the lysine developer produced a chromophore, analyzed by the microplate reader, which was proportional to the deacetylation activity of the sample. In brief, the cell was lysed and protein was extracted using the protocol mentioned above. Following measurement of protein concentration, 100 μg of cell lysate was incubated with HDAC colorimetric substrate for 1 h at 37°C. The lysine developer was added to above reaction mixture and incubated for another 30 min at 37°C. Total HDAC activity was determined by assess the absorbance at 405 nm using a microplate reader (Varioskan Flash, Thermo Scientific), expressed as the relative optical density value per μg protein. Then, based on the prepared standard curve using the known amount of deacetylated standard in the kit, the absolute amount of deacetylated lysine generated in the sample could be determined (μmol·L-1·μg-1 protein).
Immunofluorescence staining of multidrug resistance-associated protein 2 and quantification by ImageJ
Following 48 h transfection as described above, cells were washed three times with ice-cold PBS, fixed in 4% formaldehyde, and blocked for 30 min with 3% of BSA and 2% of fetal bovine serum in 0.2% Triton X-100/PBS. The cells were incubated overnight at 4°C with a 1:50 dilution of primary antibodies against MRP2 (sc-20766, Santa Cruz, USA) in the blocking buffer. Negative controls were obtained by omitting primary antibody. The cells were washed with PBS and incubated with 1:500 diluted Fluor 594 goat anti-rabbit secondary antibody (A11012, Invitrogen) for 40 min at room temperature. Nuclei were stained with 4’,6-diamidino-2-phenylindole (DAPI) (Sigma) at 1:500 dilution for 5 min. The slides were washed twice with PBS and fluorescence images for MRP2 were captured using an immunofluorescence microscopy (Nikon. Eclipse. 80i, Nikon, Japan).
Images were processed by Adobe Photoshop CS6, and MRP2 membrane immunofluorescence for each sample was quantified by ImageJ version 1.44 software (National Institutes of Health, Bethesda, USA). In brief, a library of JPEG images was imported into imageJ software. The original images were converted to 8-bit images before being auto-thresholded to binary photos by the “Make Binary” function in ImageJ. Using the “Add to Manager” function, regions of interests (ROIs) were selected around each DAPI-stained nuclei. These selections only enclose the cellular membranes where the MRP2 protein was found. Thereafter, the areas of ROIs in each of the binary images were calculated by “Analyze Particle” function, and the sum of integral optical density (IOD SUM) of MRP2 in the cells was measured after background fluorescence was dislodged by “Subtract Background” function. Finally, the mean IOD was calculated as a ratio of IOD SUM relative to area.
Statistical analysis
All data were shown as mean ± standard error (SE) and all analyses were conducted with SPSS version 17.0 (SPSS Inc., Chicago, IL, USA). Shapiro-Wilk test and homogeneity test of variance were used to confirm that quantitative data from different groups come from a normal distribution and meet the homogeneity of variance. The differences between two groups were determined by the independent sample t-test. The differences among different groups were determined by one-way analysis of variance followed by Tukey's honestly significant difference multiple range test. A two-tailed P < 0.05 was considered statistically significant.
Results
After 24 h treatment of TSA, in comparison with the vehicle group (incubated with DMSO), HDAC1 messenger RNA (mRNA) level was significantly decreased at concentrations 3.0 and 5.0 μmol/L (q = 5.702, P = 0.016 for 3.0 μmol/L; q = 6.613, P = 0.001 for 5.0 μmol/L), accompanied with prominent increase of ABCC2 mRNA expression at 1.0, 3.0, and 5.0 μmol/L (q = 5.714, P = 0.011 for 1.0 μmol/L; q = 6.619, P = 0.001 for 3.0 μmol/L; q = 11.521, P < 0.001 for 5.0 μmol/L), whereas mRNA expressions of HDAC2 and HDAC3 did not alter at all concentration gradients (P > 0.05; Figure 1a). The effect of TSA became more pronounced at 48 h, exhibiting inhibition of HDAC1/2/3 mRNA and induction of ABCC2 mRNA at all concentration gradients (HDAC1: q = 15.912, 17.905, 19.093, and 19.790 for 0.5, 1.0, 3.0, and 5.0 μmol/L, respectively, all P < 0.001; HDAC2: q = 6.152, P = 0.002 for 0.5 μmol/L; q = 8.663, P < 0.001 for 1.0 μmol/L; q = 8.105, P < 0.001 for 3.0 μmol/L; and q = 10.406, P < 0.001 for 5.0 μmol/L, respectively; HDAC3: q = 6.093, P = 0.004for 0.5 μmol/L; q = 6.570, P = 0.002 for 1.0 μmol/L; q = 6.617, P = 0.001 for 3.0 μmol/L; and q = 6.629, P = 0.001 for 5.0 μmol/L, respectively; ABCC2: q = 5.544, P = 0.033 for 0.5 μmol/L; q = 6.568, P = 0.002 for 1.0 μmol/L; q = 9.348, P < 0.001 for 3.0 μmol/L; and q = 12.877, P < 0.001 for 5.0 μmol/L; Figure 1b).
Figure 1.

TSA regulation of HDAC1/HDAC2/HDAC3/ABCC2 messenger RNA expressions after 24 (a) and 48 h (b) of incubation in Bewo cells. n = 3 for each group. Data were expressed as mean ± standard error. *P < 0.05, †P < 0.01, ‡P < 0.001, versus vehicle group. HDAC: Histone deacetylase; GAPDH: Glyceraldehyde phosphate dehydrogenase; SE: Standard error.
As shown in Figure 2, TSA repressed HDAC1/2/3 proteins and induced MRP2 expression. Compared with the vehicle group, reduction of HDAC1 protein level was noted after 24 h of TSA incubation at 1.0, 3.0, and 5.0 μmol/L (q = 11.202, 16.257, 16.900 for 1.0, 3.0, 5.0 μmol/L, respectively, all P < 0.001), accompanied with dose-dependent induction of MRP2 expression (q = 4.870, P = 0.045 for 1.0 μmol/L, q = 6.616, P = 0.001 for 3.0 μmol/L, q = 11.296, P < 0.001 for 5.0 μmol/L). However, HDAC2 and HDAC3 proteins were not altered (all P > 0.05; Figure 2a). In comparison with the vehicle group, treatment with TSA for up to 48 h dramatically reduced HDAC1/2/3 proteins at all concentration gradients (HDAC1: q = 6.566, P = 0.002 for 0.5 μmol/L; q = 12.449, 13.134, 12.276 for 1.0, 3.0, 5.0 μmol/L, respectively, all P < 0.001; HDAC2: q = 28.256, 32.713, 30.468, 34.919 for 0.5, 1.0, 3.0, 5.0 μmol/L, respectively, all P < 0.001; HDAC3: q = 25.035, 27.571, 24.650, 30.011 for 0.5, 1.0, 3.0, 5.0 μmol/L, respectively, all P < 0.001) and increased MRP2 expression at 1.0, 3.0 and 5.0 μmol/L (q = 18.111, 30.375, 38.298 for 1.0, 3.0, 5.0 μmol/L, respectively, all P < 0.001).
Figure 2.

Western-blot analysis of HDAC1/HDAC2/HDAC3/MRP2 in Bewo cells treated with TSA (0.5, 1.0, 3.0 and 5.0 μmol/L) for 24 (a) and 48 h (b) in comparison with vehicle group. n = 3 for each group. Data were expressed as mean ± SE. *P < 0.001, †P < 0.05, ‡P < 0.01, versus HDAC1. HDAC: Histone deacetylase; MRP2: Multidrug resistance-associated protein 2; GAPDH: Glyceraldehyde phosphate dehydrogenase; TSA: Trichostatin A; SE: Standard error.
As shown in Figure 3, TSA at all concentration gradients robustly ablated HDAC activity after 24 and 48 h of exposure with a concentration- and time-dependent manners as compared with the vehicle group (at 24 h: q = 6.041, P = 0.007 for 0.5 μmol/L; q = 6.419, P = 0.003 for 1.0 μmol/L; q = 6.618, P = 0.001 for 3.0 μmol/L; q = 7.963, P < 0.001 for 5.0 μmol/L; at 48 h: q = 9.340, 9.984, 10.413, 10.795 for 0.5, 1.0, 3.0, 5.0 μmol/L, respectively, all P < 0.001).
Figure 3.
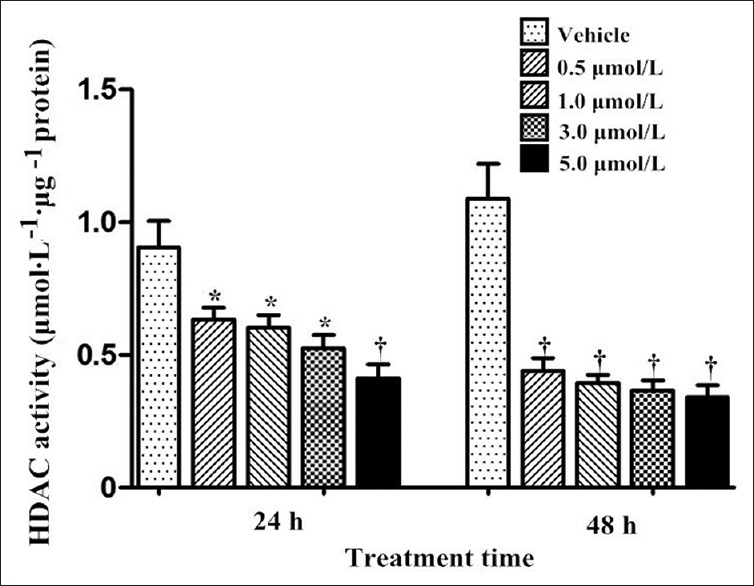
Total HDAC activity of Bewo cells treated with trichostatin A (0.5, 1.0, 3.0 and 5.0 μmol/L) for 24 and 48 h. n = 3 for each group. Data were expressed as mean ± SE. *P < 0.01, †P < 0.001, versus vehicle. HDAC: Histone deacetylase; SE: Standard error.
After transfection of HDAC1/2/3 siRNAs separately, endogenous expressions of HDAC1/2/3 were successfully inhibited, respectively, evidenced by quantitative reverse transcription polymerase chain reaction (mRNA: t = 12.435, P < 0.001 for HDAC1; t = 11.106, P < 0.001 for HDAC2; t = 13.001, P < 0.001 for HDAC3; Figure 4a) and Western-blot analysis (protein: t = 7.029, P = 0.002 for HDAC1; t = 8.157, P = 0.001 for HDAC2; t = 12.444, P < 0.001 for HDAC3; Figure 4b). Compared with the control, the inhibition of HDAC1 resulted in a noticeable elevation in MRP2 mRNA (q = 6.615, P = 0.001) and protein expressions (q = 17.845, P < 0.001), whereas no significant differences in MRP2 expression were noted after HDAC2 and HDAC3 silencing (mRNA: q = 0.269, P > 0.05 for HDAC2; q = 0.297, P > 0.05 for HDAC3; protein: q = 1.662, P > 0.05 for HDAC2; q = 0.633, P > 0.05 for HDAC3).
Figure 4.

Bewo cells were transfected with control or specific siRNA for HDAC1, HDAC2 or HDAC3. After 48 h of transfection, the levels of HDAC1/HDAC2/HDAC3/MRP2 messenger RNA (a) and protein levels (b) were analyzed by reverse transcription polymerase chain reaction and Western-blot analysis, respectively. n = 3 for each group. Data were expressed as mean ± SE. *P < 0.001, †P < 0.01, versus control siRNA. HDAC: Histone deacetylase; MRP2: Multidrug resistance-associated protein 2; GAPDH: Glyceraldehyde phosphate dehydrogenase; siRNA: Small interfering RNA; SE: Standard error.
Fluorescent micrograph images of MRP2 expression after transfection of the control, HDAC1, HDAC2, and HDAC3 siRNAs into Bewo cells are shown in Figure 5. Negative controls obtained by omitting primary antibody showed negligible background fluorescence [Figure 5e]. Fluorescence of MRP2 was mainly expressed on the cell membrane. The fluorescent intensities of MRP2 in the control, HDAC2, and HDAC3 siRNA-transfected cells were week, and no significant differences were noticed among these three groups (HDAC2 siRNA vs. control: q = 1.910, P > 0.05, HDAC3 siRNA vs. control siRNA: q = 2.472, P > 0.05; Figure 5a, 5c, and 5d). However, MRP2 expression was remarkably elevated in HDAC1 siRNA-transfected cells, which displayed an almost 3.19-fold changes in comparison with the control siRNA-transfected cells (q = 14.638, P < 0.001; Figure 5b). The quantitative analysis of MRP2 expression among different groups is shown in Figure 5f in detail.
Figure 5.

Effect of HDAC1/HDAC2/HDAC3 silencing on MRP2 expression in Bewo cells. Bewo cells were transfected with control siRNA (a) or HDAC1 (b)/HDAC2 (c)/HDAC3 (d) siRNA, respectively. Negative controls were obtained by omitting primary antibody (e). MRP2 was mainly on the cell membranes (in red). Nuclei were stained with DAPI (in blue). Quantitative folds changes in the MRP2 expression were shown in (f). n = 3 for each group. Data were expressed as mean ± SE. Scale bars: 25 μm. *P < 0.001 versus control siRNA. MRP2: Multidrug resistance-associated protein 2; siRNA: Small interfering RNA; SE: Standard error; HDAC: Histone deacetylase.
Discussion
To date, most studies on placental MRP2 have merely confined to its localization and mRNA/protein expressions affected by different physiological and pathological conditions, with comparatively little attention being paid to its possible regulation mechanisms, particularly the roles of epigenetics. To the best of our knowledge, our study first explored whether HDACs are involved in the MRP2 regulation in placental trophoblast cell line. The results revealed that the HDAC inhibition could induce placental MRP2 expression in company with inhibition of HDAC1/2/3 expression and total HDAC activity. In addition, the specific inhibition of HDAC1 could result in a noticeable elevation in MRP2 expression, whereas no significant differences in MRP2 expression were noted after HDAC2 and HDAC3 silencing. These findings strongly indicated that HDAC1 might be involved in the negative regulation of placental MRP2 expression.
In the past decade, in vitro and in vivo studies have widely proved that many dietary bioactive compounds (e.g., sulforaphane, butyrate, epigallocatechin, etc.), which could be administered during pregnancy, could inhibit HDAC1 expression and activity.[15] Given the findings in our study and wide range of MRP2 substrates, those dietary bioactive compounds might attract considerable clinical attention, particularly for the individualized and safe pharmacotherapy when MRP2 substrates are used during pregnancy. For instance, MRP2 substrates’ adverse effect on fetus might be minimized when those drugs and above dietary bioactive compounds are co-administrated for the treatment of maternal diseases during pregnancy because their transplacental transfer rates are most likely to be reduced. On the other hand, to achieve ideal drug concentrations in fetal compartments when treating fetal diseases, higher quantities of MRP2 substrates need to be administered theoretically in such conditions. In addition, many drugs or toxicants, which could induce fetal developmental malformations, have also been proved to be MRP2 substrates.[16,17,18,19] Up-regulation of placental MRP2 might reduce the risk of those compounds-induced fetal anomalies. Therefore, the findings in this study suggested that HDAC1 might be a promising target for the prevention of congenital malformations if these findings could be validated in animal studies.
Our results showed that TSA repressed HDAC1/2/3 mRNA and protein levels in Bewo cells in time and dose-dependent manners. Nevertheless, the mRNA and protein changes were not invariably in a synchronous manner. For instance, down-regulation of protein occurred, whereas its mRNA still remained unchanged (e.g., HDAC1 mRNA level at 1.0 μmol/L in Figure 1a compared with its protein level after exposure to the same drug concentration shown in Figure 2a), yet protein remained the same whereas their mRNA increased (e.g., ABCC2 mRNA level at 0.5 μmol/L shown in Figure 1b compared with its corresponding protein expression in Figure 2b). These results might suggest that TSA affected the rate of protein synthesis or degradation of targeted genes, such as proteasomal degradation after ubiquitination.[20,21] Furthermore, the mRNA and protein levels of HDAC1 and HDAC2/3 were ablated with different timing-effectiveness of TSA on HDAC1 being more rapid relative to that on HDAC2/3. Hence, these findings revealed that changes in HDAC expression patterns were influenced by HDAC types, as well as exposure length and concentration, indicating there were subtle differences of action mechanism of TSA on each individualized gene expression, which need to be further clarified.
HDACs’ biological functions are mainly dependent on their enzymatic activity.[22] The study has proved that TSA administration resulted in inhibition of total HDAC activity with a time and dose accumulation [Figure 3], which was consistent with that on HDAC1/2/3 repression. It implied that theses HDACs might play an important role in epigenetic regulation functionally on account of their high expressions in Bewo cells. To further clarify the correlation between HDAC1/2/3 and placental MRP2, specific siRNA for HDAC1, HDAC2 or HDAC3 had been transfected into Bewo cells, respectively. The results manifested that HDAC1 might acted as a repressor in placental MRP2 regulation, seeming to be consistent with that found in cancer cell lines.[23] As core epigenetic regulators, a well-known alternative target of HDACs is “histone protein,” HDACs are capable of removing acetyl groups from histone lysine tails, stabilizing nucleosome structure and compacting chromatin, thereby blocking access of transcriptional activators to the DNA template and repressing gene transcription. Moreover, nonhistone proteins are also susceptible to be modified by HDACs, binding with specific co-regulators, forming co-repressor complexes, and subsequently affecting transcriptional activity.[22] Furthermore, HDACs may have primarily nontranscriptional roles as a result of certain specific environmental stimuli.[22] Hence, on account of cell-specific regulation and individual roles in respective gene regulation of HDACs, the regulatory pathway of HDAC1 on MRP2 in placental cells are still needed to be further clarified and clearly elucidated, which could provide more novel therapeutic targets for controlling drug delivery across the placenta.
However, some limitations of this study must be considered. First, since total HDAC activity cannot accurately reflect the enzymatic activity of specific HDAC subtypes, HDAC1/2/3 activities should be investigated individually to provide more clues to establish the relationship between HDACs and placental MRP2. Moreover, as the discordance between gene expression and its function, it is unknown whether the elevated placental MRP2 expression is also accompanied with increased efflux function, which will offer more practical meanings. In addition, we only looked into the regulation of HDAC1/2/3 on placental MRP2. However, TSA could inhibit other HDACs except for HDAC1/2/3, whether other HDACs are also involved in the regulation of placental MRP2 or not still remain to be further clarified. Finally, on the grounds that Bewo cell line possesses some different properties in comparison with the primary trophoblast cells (i.e., origin, differentiation, relative expression of MRP2) and most appropriately model later gestation due to cell differentiation extent, exploration of those in vitro data to the pregnant woman through the whole gestational stage is possibly difficult. Taken together, the findings in this study were merely the first step toward placental MRP2 regulation from the perspective of epigenetic, which could offer a clue for further exploration. The results need to be further validated in primary placental cells, animal models, and human studies in different gestational stage. Meanwhile, more studies need to be carried out to explore different physiological and pathological factors on placental MRP2 and its possible mechanisms, which may provide brand-new therapeutic targets for individualized pharmacotherapy during pregnancy.
In conclusion, our study made a preliminary exploration of placental MRP2 regulation from the perspective of epigenetics, demonstrating that HDACs inhibition could up-regulate placental MRP2 expression in the trophoblast cells, and illustrated that HDAC1 was mainly involved in this process. These findings might provide some references for efficient and safe pharmacotherapy during pregnancy.
Supplementary information is linked to the online version of the paper on the Chinese Medical Journal website.
Financial support and sponsorship
This study was supported by grants from National Natural Science Foundation of China (No. 81602817, No. 81571515, and No. 81401233), Science-technology Support Plan Projects in Sichuan province (No. 2016FZ0088), and Research Projects of Health and Family Planing Commission of Sichuan Province (No. 140045).
Conflicts of interest
There are no conflicts of interest.
Footnotes
Edited by: Xin Chen
References
- 1.Bakker MK, Jentink J, Vroom F, Van Den Berg PB, De Walle HE, De Jong-Van Den Berg LT. Drug prescription patterns before, during and after pregnancy for chronic, occasional and pregnancy-related drugs in the Netherlands. BJOG. 2006;113:559–68. doi: 10.1111/j.1471-0528.2006.00927.x. doi: 10.1111/j.1471-0528.2006.00927.x. [DOI] [PubMed] [Google Scholar]
- 2.Irvine L, Flynn RW, Libby G, Crombie IK, Evans JM. Drugs dispensed in primary care during pregnancy: A record-linkage analysis in Tayside, Scotland. Drug Saf. 2010;33:593–604. doi: 10.2165/11532330-000000000-00000. doi: 10.2165/11532330-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 3.Lee E, Maneno MK, Smith L, Weiss SR, Zuckerman IH, Wutoh AK, et al. National patterns of medication use during pregnancy. Pharmacoepidemiol Drug Saf. 2006;15:537–45. doi: 10.1002/pds.1241. doi: 10.1002/pds.1241. [DOI] [PubMed] [Google Scholar]
- 4.Staud F, Cerveny L, Ceckova M. Pharmacotherapy in pregnancy; effect of ABC and SLC transporters on drug transport across the placenta and fetal drug exposure. J Drug Target. 2012;20:736–63. doi: 10.3109/1061186X.2012.716847. doi: 10.3109/1061186X.2012.716847. [DOI] [PubMed] [Google Scholar]
- 5.Vähäkangas K, Myllynen P. Drug transporters in the human blood-placental barrier. Br J Pharmacol. 2009;158:665–78. doi: 10.1111/j.1476-5381.2009.00336.x. doi: 10.1111/j.1476-5381.2009.00336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Waring RH, Harris RM, Mitchell SC. In utero exposure to carcinogens: Epigenetics, developmental disruption and consequences in later life. Maturitas. 2016;86:59–63. doi: 10.1016/j.maturitas.2016.01.008. doi: 10.1016/j.maturitas.2016.01.008. [DOI] [PubMed] [Google Scholar]
- 7.Huhta J, Linask KK. Environmental origins of congenital heart disease: The heart-placenta connection. Semin Fetal Neonatal Med. 2013;18:245–50. doi: 10.1016/j.siny.2013.05.003. doi: 10.1016/j.siny.2013.05.003. [DOI] [PubMed] [Google Scholar]
- 8.Staud F, Ceckova M. Regulation of drug transporter expression and function in the placenta. Expert Opin Drug Metab Toxicol. 2015;11:533–55. doi: 10.1517/17425255.2015.1005073. doi: 10.1517/17425255.2015.1005073. [DOI] [PubMed] [Google Scholar]
- 9.Eom GH, Kook H. Posttranslational modifications of histone deacetylases: Implications for cardiovascular diseases. Pharmacol Ther. 2014;143:168–80. doi: 10.1016/j.pharmthera.2014.02.012. doi: 10.1016/j.pharmthera.2014.02.012. [DOI] [PubMed] [Google Scholar]
- 10.Jamaladdina S, Kellya RD, O’Regana L, Doveyb OM, Hodsona GE, Millarda CJ, et al. The physiological roles of histone deacetylase (HDAC) 1 and 2: Complex co-stars with multiple leading parts. Biochem Soc Trans. 2013;41:741–9. doi: 10.1042/BST20130010. doi: 10.1042/BST20130010. [DOI] [PubMed] [Google Scholar]
- 11.Lagger G, O’Carroll D, Rembold M, Khier H, Tischler J, Weitzer G, et al. Essential function of histone deacetylase 1 in proliferation control and CDK inhibitor repression. EMBO J. 2002;21:2672–81. doi: 10.1093/emboj/21.11.2672. doi: 10.1093/emboj/21.11.2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chuang HC, Chang CW, Chang GD, Yao TP, Chen H. Histone deacetylase 3 binds to and regulates the GCMa transcription factor. Nucleic Acids Res. 2006;34:1459–69. doi: 10.1093/nar/gkl048. doi: 10.1093/nar/gkl048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ni X, Li L, Pan G. HDAC inhibitor-induced drug resistance involving ATP-binding cassette transporters (Review) Oncol Lett. 2015;9:515–21. doi: 10.3892/ol.2014.2714. doi: 10.3892/ol.2014.2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim H, Kim SN, Park YS, Kim NH, Han JW, Lee HY, et al. HDAC inhibitors downregulate MRP2 expression in multidrug resistant cancer cells: Implication for chemosensitization. Int J Oncol. 2011;38:807–12. doi: 10.3892/ijo.2010.879. doi: 10.3892/ijo.2010.879. [DOI] [PubMed] [Google Scholar]
- 15.Vahid F, Zand H, Nosrat-Mirshekarlou E, Najafi R, Hekmatdoost A. The role dietary of bioactive compounds on the regulation of histone acetylases and deacetylases: A review. Gene. 2015;562:8–15. doi: 10.1016/j.gene.2015.02.045. doi: 10.1016/j.gene.2015.02.045. [DOI] [PubMed] [Google Scholar]
- 16.May K, Minarikova V, Linnemann K, Zygmunt M, Kroemer HK, Fusch C, et al. Role of the multidrug transporter proteins ABCB1 and ABCC2 in the diaplacental transport of talinolol in the term human placenta. Drug Metab Dispos. 2008;36:740–4. doi: 10.1124/dmd.107.019448. doi: 10.1124/dmd.107.019448. [DOI] [PubMed] [Google Scholar]
- 17.Bridges CC, Joshee L, Zalups RK. Placental and fetal disposition of mercuric ions in rats exposed to methylmercury: Role of Mrp2. Reprod Toxicol. 2012;34:628–34. doi: 10.1016/j.reprotox.2012.10.001. doi: 10.1016/j.reprotox.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neumanova Z, Cerveny L, Ceckova M, Staud F. Interactions of tenofovir and tenofovir disoproxil fumarate with drug efflux transporters ABCB1, ABCG2, and ABCC2;role in transport across the placenta. AIDS. 2014;28:9–17. doi: 10.1097/QAD.0000000000000112. doi: 10.1097/QAD.0000000000000112. [DOI] [PubMed] [Google Scholar]
- 19.Vinot C, Gavard L, Tréluyer JM, Manceau S, Courbon E, Scherrmann JM, et al. Placental transfer of maraviroc in an ex vivo human cotyledon perfusion model and influence of ABC transporter expression. Antimicrob Agents Chemother. 2013;57:1415–20. doi: 10.1128/AAC.01821-12. doi: 10.1128/AAC.01821-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krämer OH, Zhu P, Ostendorff HP, Golebiewski M, Tiefenbach J, Peters MA, et al. The histone deacetylase inhibitor valproic acid selectively induces proteasomal degradation of HDAC2. EMBO J. 2003;22:3411–20. doi: 10.1093/emboj/cdg315. doi: 10.1093/emboj/cdg315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wakabayashi-Nakao K, Tamura A, Furukawa T, Nakagawa H, Ishikawa T. Quality control of human ABCG2 protein in the endoplasmic reticulum: Ubiquitination and proteasomal degradation. Adv Drug Deliv Rev. 2009;61:66–72. doi: 10.1016/j.addr.2008.08.008. doi: 10.1016/j.addr.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 22.Gallinari P, Di Marco S, Jones P, Pallaoro M, Steinkühler C. HDACs, histone deacetylation and gene transcription: From molecular biology to cancer therapeutics. Cell Res. 2007;17:195–211. doi: 10.1038/sj.cr.7310149. doi: 10.1038/sj.cr.7310149. [DOI] [PubMed] [Google Scholar]
- 23.Xu Y, Jiang Z, Yin P, Li Q, Liu J. Role for class I histone deacetylases in multidrug resistance. Exp Cell Res. 2012;318:177–86. doi: 10.1016/j.yexcr.2011.11.010. doi: 10.1016/j.yexcr.2011.11.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Effect of TSA on Bewo cell viability at different time points of exposure. The cells were treated with or without different concentrations of TSA (0.5, 1.0, 3.0, and 5.0 μmol/L) for 24, 48, or 72 h. Cell viability was evaluated using the WST-1 assay. n = 3 for each group. Data were expressed as mean ± SE. *P < 0.001, versus vehicle. TSA: Trichostatin A; SE: Standard error.