

Abstract
The brain and muscle aryl hydrocarbon receptor nuclear translocator–like protein (BMAL)-1 constitutes a major transcriptional regulator of the circadian clock. Here, we explored the impact of conditional deletion of Bmal1 in endothelium and hematopoietic cells in murine models of microvascular and macrovascular injury. We used two models of Bmal1fx/fx;Tek-Cre mice, a retinal ischemia/reperfusion model and a neointimal hyperplasia model of the femoral artery. Eyes were enumerated for acellular capillaries and were stained for oxidative damage markers using nitrotyrosine immunohistochemistry. LSK (lineage-negative, stem cell antigen-1–positive, c-Kit–positive) cells were quantified and proliferation assessed. Hematopoiesis is influenced by innervation to the bone marrow, which we assessed using IHC analysis. The number of acellular capillaries increased threefold, and nitrotyrosine staining increased 1.5-fold, in the retinas of Bmal1fx/fx;Tek-Cre mice. The number of LSK cells from the Bmal1fx/fx;Tek-Cre mice decreased by 1.5-fold and was accompanied by a profound decrease in proliferative potential. Bmal1fx/fx;Tek-Cre mice also exhibited evidence of bone marrow denervation, demonstrating a loss of neurofilament-200 staining. Injured femoral arteries showed a 20% increase in neointimal hyperplasia compared with similarly injured wild-type controls. Our study highlights the importance of the circadian clock in maintaining vascular homeostasis and demonstrates that specific deletion of BMAL1 in endothelial and hematopoietic cells results in phenotypic features similar to those of diabetes.
Most organisms possess an endogenous circadian system that drives the daily oscillations of a variety of physiologic and behavioral processes with a 24-hour period. A variety of cardiovascular and hemodynamic parameters, such as heart rate, blood pressure, endothelial function, and fibrinolytic activity, exhibit such diurnal patterns.1 The frequency of occurrence of acute cardiovascular events, such as acute myocardial infarction, myocardial ischemia, cardiac arrest, ventricular tachycardia after myocardial infarction, and sudden death, varies according to the time of day.2, 3
Although the suprachiasmatic nucleus orchestrates the neuroendocrine physiology of the circadian rhythm, at the molecular level the circadian rhythm is orchestrated by autoregulatory transcriptional/translational loops of a set of clock genes. About 10% of the transcriptome in all tissues is under the control of these clock genes.4 Genetic studies on clock gene–mutant mice recapitulate phenotypic changes observed, similar to those in cardiovascular5 and metabolic disorders.6 Mice deficient in brain and muscle aryl hydrocarbon receptor nuclear translocator-like protein (Bmal)-1 possess a range of disorders, including reduced lifespan, premature aging, monocytosis, and cataracts.
Diabetes is a multifactorial disease characterized by metabolic abnormalities, including hyperglycemia, insulin resistance, and an increase in fatty acids, with each abnormality provoking a molecular arsenal that contributes to vascular diseases.7, 8 Most importantly, there is an interplay between the circadian system and metabolic disorders.9, 10 A disrupted circadian rhythm in shift workers and frequent flyers leads to an increased susceptibility to metabolic diseases,11 and metabolic diseases such as diabetes lead to a disrupted circadian rhythm.12
Endothelial dysfunction is central to the underlying pathology of diabetic vascular diseases, along with reduced vascular repair, which is mediated in large part by bone marrow–derived progenitor cells (BMPCs). We12 and others13, 14 have reported that diabetes is a disease of circadian dysfunction. Our studies using a rat model of type 2 diabetes have demonstrated that core circadian regulatory genes are crucial for the vascular health of the retina12 and that dysfunctional release and intrinsic dysfunction of BMPCs contribute to the development of acellular capillaries, the pathologic hallmark of diabetic retinopathy. Using a clock gene (Per2)-mutant mouse model, we demonstrated that a lack of functional period circadian protein homolog-2 recapitulates a vascular phenotype similar to that of diabetic retinopathy.15, 16 For this study, we asked whether a BMAL1 deficiency in endothelial cells is linked to accelerated microvascular and macrovascular injury. Because the findings from a previous study demonstrated that angiopoietin-1 receptor (TIE-2; alias TEK) is also expressed in a fraction of BMPCs [LSK (lineage-negative, stem cell antigen-1–positive, c-Kit–positive) cells],17 we hypothesized that the function of the BMPC population in Bmal1fx/fx;Tek-Cre mice would also be adversely affected.
Materials and Methods
Animals
The animal studies were performed in accordance with the Use of Animals in Ophthalmic and Vision Research statement from the Association for Research in Vision and Ophthalmology. The study was approved by the institutional animal care and use committees at the University of Florida and Michigan State University. The Bmalfx/fx and Tek-Cre mice were purchased from The Jackson Laboratory (Bar Harbor, ME). The Bmal1fx/fx mice were crossed with Tek-Cre transgenic mice to generate either Bmal1fx/fx;Tek-Cre or Bmal1fx/fx controls [wild type (WT)]. All animals were maintained under 12-hour light/dark conditions. All of the studies were performed when the animals were aged between 8 and 10 months.
Ischemia and Reperfusion Injury of the Retina
The Bmal1fx/fx;Tek-Cre and WT mice were kept under inhalational anesthesia. The anterior chamber of the eye was cannulated with a 30-gauge needle attached to an infusion line of sterile saline, and the eye was subjected to 2 hours of hydrostatic pressure in the anterior chamber. After 2 hours, the needle was withdrawn and the intraocular pressure was normalized, resulting in reperfusion injury. Seven days after injury, at which time retinal capillary damage is detectable, the retinas were harvested and processed for trypsin digestion.
Trypsin Digestion of Retinas
The retinas were processed using a trypsin digestion protocol as described previously.18 The retinal digests were mounted on presalinized slides and stained with periodic acid-Schiff base, followed by counterstaining with hematoxylin (Sigma-Aldrich, St. Louis, MO). Acellular capillaries, defined as naked basement membrane tubes lacking both endothelial cells and pericytes, were counted at ×20 magnification by investigators blinded to the respective groups, and the number per square milliliter was quantified.
Immunofluorescence Staining
The ischemia/reperfusion (I/R)-injured retinas were paraffin embedded and sectioned. After a citrate buffer epitope retrieval, the retinal sections were stained for either endothelial NO synthase (eNOS), Akt, or phospho-Akt antibodies (Cell Signaling, Danvers, MA); Bmal1 (Novus Biologicals LLC, Littleton, CO); CD31 (Dianova, Hamburg, Germany); and CD45 (Sigma-Aldrich). The immunofluorescence images were quantified for the degree of respective staining using image analysis software (Zen lite version 2; Carl Zeiss Microscopy LLC, Thornwood, NY).
Neointimal Hyperplasia
The Bmal1fx/fx;Tek-Cre and WT mice were kept under inhalational anesthesia. The endoluminal macrovascular injury was performed by introducing a 0.015 mm–diameter angioplasty guidewire into the femoral artery. The animals were euthanized after 4 weeks for the harvesting of the femoral artery. The degree of neointimal hyperplasia was evaluated on transverse sections of the femoral artery by evaluation of the thickness of the luminal area of the injured artery.
Quantification of BMPCs
Femurs were isolated from Bmal1fx/fx;Tek-Cre and WT mice. The bone marrow was flushed in phosphate-buffered saline containing 2% fetal bovine serum. After serum blocking for 10 minutes, mononuclear cells were stained with biotin mouse lineage panel (BD Pharmingen, San Diego, CA), followed by staining with Streptavidin APC (eBioscience, San Diego, CA). Stem cell antigen-1 and c-Kit staining were performed using PE-rat anti-mouse Ly-6A/E and PE-Cy7 rat anti-mouse CD117 antibodies (BD Pharmingen). The stained cells were analyzed using an LSR II flow cytometer (BD Biosciences), and data were analyzed using FlowJo software version 9.9.4 (FlowJo, Ashland, OR).
Neurofilament-200 and Tyrosine Hydroxylase Staining of Bone Marrow
Femurs from Bmal1fx/fx;Tek-Cre and WT mice were isolated and embedded in paraffin. Sections were obtained on a microtome at 4 μm and stained for tyrosine hydroxylase using polyclonal rabbit anti–tyrosine hydroxylase antibody (EMD Millipore, Billerica, MA), and for neurofilament (NF)-200 using polyclonal rabbit anti–NF-200 antibody (Sigma-Aldrich). The images of stained sections were obtained using an Olympus IX-70 inverted microscope (Olympus, Center Valley, PA).
Protein Estimation in Bone Marrow Supernatants
The bone marrow was flushed with phosphate-buffered saline; the cellular fraction was pelleted by centrifugation. The supernatant was concentrated using ultracentrifugation filter units (EMD Millipore). The supernatants were analyzed using a Luminex bead-based cytokine array (AssayGate, Ijamsville, MD). The data are shown in picograms of analyte per milligram of total protein.
Statistical Analysis
The data are expressed as means ± SEM. Statistical analysis was performed using Prism software version 6 (GraphPad Software, La Jolla, CA), and data were analyzed using one-way analysis of variance followed by the Tukey-Kramer post hoc test or t-test.
Results
Decrease in Bmal1 Staining in Endothelial Cells of Bmal1fx/fx;Tek-Cre Mice
To confirm endothelial deletion of Bmal1, we stained retinal sections with Bmal1 and CD31 antibodies (platelet endothelial adhesion molecule-1, an endothelial marker). Bmal1 staining was observed in the ganglion cell layer, inner nuclear layer, and outer plexiform layer. However, the vessels of Bmal1fx/fx;Tek-Cre mice exhibited a marked decrease in staining for Bmal1 compared with that in WT controls (Supplemental Figure S1).
Bmal1fx/fx;Tek-Cre Mice Demonstrate an Increase in Acellular Capillaries
Increased numbers of acellular capillaries represent the hallmark feature of ischemic and diabetic retinopathies. In the acute model of I/R injury used in this study, capillary degeneration mimics that observed in diabetic retinopathy.19 Specifically, the I/R model recapitulates the small vessel disease seen in diabetes, but without the long time course required for diabetic eye changes and without the confounding metabolic effects associated with diabetes.19 WT eyes that underwent retinal I/R injury exhibited an increase (1.86-fold; P < 0.01) in acellular capillaries compared with those in control uninjured eyes. The uninjured Bmal1fx/fx;Tek-Cre eyes demonstrated retinal pathology under baseline conditions, as they had 2.07-fold more (P < 0.001) acellular capillaries compared with those in WT controls. However, after I/R injury, the Bmal1fx/fx;Tek-Cre mice showed a dramatic increase in retinal damage compared with that in the injured WT mice. The rate of the fold increase in acellular capillaries was accelerated, showing 2.28-fold in the Bmal1fx/fx;Tek-Cre mice compared with 1.31-fold in WT mice P < 0.001 (Figure 1).
Figure 1.

Enhanced development of acellular capillaries in Bmal1fx/fx;Tek-Cre retinas after ischemia/reperfusion (I/R) injury. Bmalfx/fx;Tek-Cre and WT mice underwent an I/R injury, and 7 days after the injury, the retinas were harvested and trypsin digested. A: Representative images of trypsin digests of untreated or treated eyes from respective groups. Acellular capillaries are indicated by arrowheads. B: Quantification of acellular capillaries. Data are expressed as means ± SEM. n = 6 Bmalfx/fx;Tek-Cre; n = 10 WT.∗∗P < 0.01, ∗∗∗P < 0.001. Scale bars = 100 μm.
Increase in CD45+ Leukocytes in Bmal-1fx/fx;Tek-Cre Mice
To explore any leukocyte-mediated involvement in the I/R injury, we stained retinal cross sections with CD45+ antibodies (a pan-leukocyte marker) and CD31 antibodies (platelet endothelial adhesion molecule-1, an endothelial marker). With I/R injury in WT controls, there was a 2.9-fold (P < 0.001) increase in leukocytes compared with those in uninjured WT-controls. In Bmal1fx/fx;Tek-Cre mice, the increase was also 2.9-fold (P < 0.01) compared with that in the uninjured Bmal1fx/fx;Tek-Cre mice. There was no significant difference in leukocyte numbers between WT and Bmal1fx/fx;Tek-Cre mice (Figure 2).
Figure 2.

Increased leukocytes in ischemia/reperfusion (I/R)–injured retinas. The retinas of I/R-injured Bmalfx/fx;Tek-Cre and WT mice were stained with CD45-Alexa 488 and CD31-Alexa 555 antibodies. A: Representative images of retinal sections showing an increase in CD45 stain (arrows) in injured retinas. B: Quantification of CD45 clusters. Data are expressed as means ± SD. n = 3 per group. ∗∗P < 0.01, ∗∗∗P < 0.001. Scale bars = 50 μm. GCL, ganglion cell layer; INL, inner nuclear layer; IPL, inner plexiform layer; ONL, outer nuclear layer; OPL, outer plexiform layer; PR, photoreceptor.
Bmal1fx/fx;Tek-Cre Mice Exhibit an Increase in Neointimal Hyperplasia
To explore whether Bmal1 deficiency adversely affects the macrovasculature, we used an animal model of neointimal hyperplasia. Four weeks after the injury, the femoral artery was assessed for degree of neointimal hyperplasia. The aortas of Bmal1fx/fx;Tek-Cre mice exhibited thicker walls compared with those in injured WT mice (P < 0.05) (Figure 3).
Figure 3.

Increase in neointimal hyperplasia in Bmal1fx/fx;Tek-Cre mice. Endoluminal macrovascular injury was performed into the femoral artery, and the degree of neointimal hyperplasia was evaluated 4 weeks after injury. A: Transverse sections of WT and Bmal-1fx/fx;Tek-Cre femoral arteries, showing a degree of intimal hyperplasia in untreated and treated femoral arteries. B: Percentage of the neointimal lesion in each respective group. Bmal1fx/fx;Tek-Cre mice show accelerated injury in arteries compared with that in WT controls. Data are expressed as means ± SEM. n = 3 Bmal1fx/fx;Tek-Cre; n = 4 WT. ∗P < 0.05 versus WT. Scale bars = 20 μm.
Decrease in Bmal1 Staining in Bone Marrow
We next characterized the expression of Bmal1 protein in bone marrow by staining the femurs of Bmal1fx/fx;Tek-Cre mice with Bmal1 antibodies. We observed a marked decrease in Bmal1 staining in bone marrow sections of Bmal1fx/fx;Tek-Cre mice (Supplemental Figure S2).
Bmal1fx/fx;Tek-Cre Mice Demonstrate Decreased Levels of LSK Cells
BMPCs play an important role in vascular repair by expression of growth factors and cytokines needed by the injured endothelium. TEK is expressed in a fraction of LSK cells, which have the ability to generate long-term, reconstituting, colony-forming cells.17 Thus, we reasoned that functional deletion of BMAL1 in LSK cell fraction will adversely affect the LSK cell numbers, leading to compromised vascular repair. LSK numbers were quantitated using flow cytometry, and after omission of the lineage, negative fraction, stem cell antigen-1+, and c-Kit+, they were enumerated as dual-positive cells (Figure 4A). The Bmal1fx/fx;Tek-Cre mice demonstrated a significant decrease (1.46-fold; P < 0.05) in the percentage of LSK cell numbers compared with that in WT control mice (Figure 4B).
Figure 4.

Decrease in LSK cells in Bmal1fx/fx;Tek-Cre mice. The bone marrow from WT and Bmal1fx/fx;Tek-Cre mice were harvested, and the percentages of LSK cells were determined using flow cytometry. A: A representative flow cytometry dot plot showing selecting gating strategy used for separate the LSK population. B: Decreased LSK cells in Bmal1fx/fx;Tek-Cre mice. Data are expressed as means ± SEM. n = 5 per group. ∗P < 0.05 versus WT. LK, lineage and c-Kit; LS, lineage and sca-1; LSK, lineage-negative, stem cell antigen-1–positive, c-Kit–positive.
Decrease in Stem Cell Mobilization Factors in the Bone Marrow of Bmal1fx/fx;Tek-Cre Mice
To identify a possible mechanism for the decrease in LSK cell numbers, we analyzed the bone marrow supernatants using a multiplex array panel. We observed that, with a lack of Bmal1 in bone marrow (Bmal1fx/fx;Tek-Cre mice), there was a 1.8-fold (P < 0.05) decrease in granulocyte-monocyte colony stimulating factor, a slight decrease in stem cell factor, and a marginal elevation in stromal derived factor-1 or chemokine (C-X-C motif) ligand-12 (Supplemental Figure S3).
Notably, we observed decreases in a variety of inflammatory markers, such as tumor necrosis factor-α, IL-6, IL-1α, IL-1β, and a macrophage activator, interferon-γ. There was a nonsignificant increase in macrophage inflammatory protein-2 (1.2-fold) or chemokine (C-X-C motif) ligand-2 (Supplemental Figure S4).
Bmal-1fx/fx;Tek-Cre Mice Show Decreased Proliferation of BMPCs
To further characterize the bone marrow cell defect, the proliferative capacity of the mononuclear fraction was evaluated using methylcellulose colony forming units (CFUs). The potential for proliferation of BMPCs from the Bmal1fx/fx;Tek-Cre mice was markedly reduced, as evidenced by reductions in CFUs, specifically a threefold decrease (P < 0.05) in CFU–granulocyte-monocytes (Figure 5A); a fourfold (P < 0.05) decrease in CFU–granulocyte-erythrocyte-monocyte/macrophage-megakaryocytes (Figure 5C); and a sixfold decrease (P < 0.05) in CFU-granulocytes (Figure 5D), but a nonsignificant increase in CFU-monocytes (Figure 5B).
Figure 5.

Decreased proliferation in Bmal1fx/fx;Tek-Cre mice. Bone marrow mononuclear cells were isolated from WT and Bmal-1fx/fx;Tek-Cre mice and were propagated in methylcellulose semisolid cultures; the number of colonies was enumerated after one week. Representative images and bar charts showing decreases in CFU-GM (granulocyte monocyte) (A), CFU-GEMM (granulocyte, erythrocyte, monocyte/macrophage, megakaryocyte) (C), and CFU-G (granulocyte) (D), and an increase in CFU-monocytes (M) in Bmal-1fx/fx (B). Data are expressed as means ± SEM. n = 5 WT; n = 6 Bmal1fx/fx;Tek-Cre. ∗P < 0.05 versus WT. Scale bars = 200 μm.
Retinas of Bmal1fx/fx;Tek-Cre Mice Show an Increase in Nitrotyrosine Staining
To identify potential mechanism(s) for the increased microvascular injury observed in the Bmal1fx/fx;Tek-Cre mice, the levels of oxidative injury in the injured retinas were examined. In the presence of the reactive oxidant species, such as superoxides, nitric oxide (NO) rapidly forms peroxynitrite. The major product of the reaction of proteins with peroxynitrite is a nitro group added to tyrosine to form nitrotyrosine. We reasoned that the combination of a lack of Bmal1 and I/R injury would result in an increase in the levels of nitrotyrosine. The retinas of uninjured WT mice exhibited minimal nitrotyrosine staining, whereas nitrotyrosine was markedly increased in I/R-injured WT retinas (Figure 6A). The increase in fluorescence intensity was observed in the outer plexiform layer, consisting of synapses of horizontal cells, bipolar cells, and photoreceptor inner segments, and in the inner plexiform layer, consisting of synapses of amacrine cells and ganglion cells. Uninjured retinas from Bmal1fx/fx;Tek-Cre mice demonstrated increased nitrotyrosine compared with that in uninjured WT retinas, and nitrotyrosine staining was markedly intensified in the I/R-injured Bmal1fx/fx;Tek-Cre mice (Figure 6A). Quantification of fluorescence intensity demonstrated a 1.5-fold increase (P < 0.05) in injured Bmal1fx/fx;Tek-Cre retinas compared with that in WT injured retinas (Figure 6B).
Figure 6.

Bmal1fx/fx;Tek-Cre mice show an increase in nitrative stress. The retinas of WT and Bmal1fx/fx;Tek-Cre mice after an ischemia/reperfusion injury were paraffin-embedded, sectioned, and stained for nitrotyrosine. A: Representative images of retinal mounts showing a profound increase in nitrotyrosine staining in injured Bmal1fx/fx;Tek-Cre mice (arrows). B: Quantification fluorescence intensity for nitrotyrosine. Data are expressed as means ± SEM. n = 5 WT; n = 6 Bmal1fx/fx;Tek-Cre. ∗P < 0.05. Scale bars = 20 μm. GCL, ganglion cell layer; INL, inner nuclear layer; IPL, inner plexiform layer; ONL, outer nuclear layer; OPL, outer plexiform layer; PR, photoreceptor layer.
Bmal1fx/fx;Tek-Cre Mice Exhibit Bone Marrow Pathology
We previously reported that bone marrow neuropathy adversely affected BMPC function and release into the circulation in a model of type 2 diabetes.12 The bone marrow neuropathy was mediated by neuronal damage due to a dysfunctional vasa nervorum. We reasoned that a lack of BMAL1 in the vascular endothelium of the vasa nervorum would induce a neuropathic phenotype resulting in compromised BMPC functioning. Femurs were stained and quantified for NF-200 and tyrosine hydroxylase. The NF-200 staining of the bone marrow was decreased by 1.5-fold (P < 0.01) (Figure 7A); however, the tyrosine hydroxylase staining remained unchanged (Figure 7B).
Figure 7.

Bone marrow neuropathy in Bmal1fx/fx;Tek-Cre mice. Decalcified bones were sectioned and stained for tyrosine hydroxylase (TH) and neurofilament (NF)-200. Representative images showing a decrease in NF-200 (A, arrows) and TH (B, arrows). Insets show TH-positive areas at higher magnification. Bar chart showing quantification of TH and NF-200 staining. Data are expressed as means ± SEM. n = 5 per group. ∗P < 0.05 versus WT. Scale bars = 50 μm.
Bmalfx/fx;Tek-Cre Mice Exhibit Defects in Akt and eNOS Signaling in the Retina
Activation of the serine-threonine kinase Akt represents a crucial prosurvival signal for endothelial cells by increasing NO generation via phosphorylation of eNOS. We asked whether the lack of BMAL1 could lead to a reduction in expression of AKT, phospho-AKT, or eNOS immunoreactivity. Retinal sections were stained for eNOS, Akt, and phospho-Akt, and the intensity of staining around blood vessels was evaluated using image morphometry. Profound decreases in the expressions of total Akt (Figure 8A) and phospho-Akt (Figure 8B) (P < 0.05), but not of eNOS (Figure 8C), were observed in the retinal vessels from Bmalfx/fx;Tek-Cre mice.
Figure 8.

Decrease in Akt–endothelial NO synthase (eNOS) signaling in retinal vasculature of Bmal1fx/fx;Tek-Cre mice. The retinal sections of WT and Bmal1fx/fx;Tek-Cre stained with Akt (A), phospho-Akt (B), and eNOS (C) antibodies. Representative photomicrographs show a staining pattern of respective antibodies. The degree of staining in the area around the blood vessel representing vascular endothelium (highlighted in red with brightly fluorescent red blood cells) was quantified using image analysis software (Zen 2 lite). Bar charts show quantification of intensity for Akt, phospho-Akt, and endothelial NO synthase (eNOS). Data are expressed as means ± SEM. n = 3 per group. ∗P < 0.05. Scale bars = 50 μm.
Discussion
It is widely appreciated that the biological clock interacts with environmental cues to influence a variety of cardiovascular and hemodynamic parameters. The findings from our study demonstrate that a loss of endothelial BMAL1 orchestrates the progression of microvascular and macrovascular damage. In agreement with findings from a previous study by Anea et al,20 which reported endothelial dysfunction in whole-body Bmal1-knockout mice, the findings from our study support the concept that endothelial Bmal1 is necessary for vascular health. In our study, the conditional deletion of Bmal1 in vascular endothelium accelerated both retinal microvascular and femoral arterial macrovascular injury. The lack of Bmal1 in BMPCs further aggravated endothelial injury due to the inability of these cells to adequately participate in vascular repair.
In this study, we generated a conditional knockout mouse using the Tie2-Cre (Tek-Cre) transgene. In this animal model, the mouse endothelial-specific receptor, tyrosine kinase (Tie-2 or Tek) promoter directed the expression of Cre recombinase. The TIE-2 gene (TEK) encodes an angiopoietin receptor, a member of the receptor tyrosine kinase family.21 Murine Tie-2 expression is detected as the first endothelial cells arise, remains positive in endothelial cells throughout development, and is detectable in virtually all endothelial cells in adult tissues.22, 23, 24 However, there are reports that TIE-2 is also expressed in subpopulations of BMPCs.25 Thus, we considered that the crossing of these animals with Bmalfx/fx mice generated a conditional knockout mouse specifically lacking Bmal1 in endothelial cells and BMPCs. The loss of BMAL1 in LSK cells and BMPCs, as well as in endothelial cells, affects their functioning, and endothelial dysfunction would lead to worsening neointimal hyperplasia. The neointimal process is affected by endothelial repair, and monocytes infiltrate (early on), but by 4 weeks the majority of the cells within the lesion are smooth muscle cells. Although the degree of intimal hyperplasia was only a 20% increase, this was in addition to a robust 135% neointimal lesion. The findings from our study suggest that retinal deterioration was increased at baseline due to the endothelial loss of the Bmal1. As the studies were performed in animals 8 to 10 months of age, we cannot rule out that these changes were not partially developmental. Additional studies using younger animals could elucidate this further.
The eyes of the Bmal1fx/fx;Tek-Cre mice that underwent I/R injury exhibited an accelerated increase in retinal acellular capillaries 7 days after injury compared with that in the WT controls. Findings from a previous study suggest that an increase in acellular capillaries in I/R-injured retinas is due to enhanced expression of inducible NOS.26 In our study, the I/R-injured retinas from Bmal1fx/fx;Tek-Cre mice were stained intensely for nitrotyrosine, suggesting that NO signaling plays a crucial role in mediating the observed injury response in these mice. NO generated by glutamate accumulates in diabetes27 and plays an important role in the degeneration of retinal capillaries. In the presence of superoxide, NO rapidly forms a strong oxidant peroxynitrite that rapidly interacts with proteins, resulting in the formation of nitrotyrosine.27 We speculate that NO and BMAL1 form a tight regulatory loop, wherein both components are closely dependent on each other. Anea et al28 previously reported that the whole-body Bmal1 knockout mice exhibit aberrant vascular remodeling and enhanced susceptibility to thrombosis in the carotid arteries of older animals. They showed that a lack of BMAL1 results in increased generation of superoxide radical.28 We believe our data support their work, as we observed an increase in retinal nitrotyrosine expression in Bmal1fx/fx;Tek-Cre mice both at baseline and after I/R injury. An increase in nitrotyrosine staining in retinas from Bmal1fx/fx;Tek-Cre mice was not only limited to retinal vasculature but also prominent in retinal neuropile.
The vessel remodeling in the whole-body Bmal1 knockout mice was linked to a decrease in Akt and eNOS activity. Although not tested in our macrovascular injury model, we believe that the increase in neointimal hyperplasia observed in the Bmal1fx/fx;Tek-Cre mice may be similarly related to a decrease in eNOS and Akt activity.29 Immunofluorescence studies on retinal sections further support the involvement of eNOS-AKT signaling by the exaggerated injury response observed in Bmal1fx/fx;Tek-Cre mice. Findings from studies suggest that AKT is a putative target of the circadian clock,30 and that the expression of Akt1 is substantially reduced in Bmal1 knockout mice.20 It remains unclear how BMAL1 promotes the AKT activity; one plausible explanation is that the AKT signaling may be directly under the clock regulation. Another possibility is that BMAL1 directly affects the protein abundance of RICTOR (rapamycin-insensitive companion of mammalian target of rapamycin), a key component of mTORC2 (mammalian target of rapamycin complex 2), known to phosphorylate AKT2 at phospho-AKT (SER-473) in the liver. However, further studies are warranted to explain the involvement of BMAL1 in AKT signaling within the retina.
The findings from our study also suggest that the BMPC dysfunction due to the conditional deletion of BMAL1 is detrimental to the retinal and femoral artery vasculature and highlights the crucial role of BMAL1 in maintaining BMPC health. The Bmal1fx/fx;Tek-Cre mice showed a profound decrease in BMPC numbers and proliferation. Findings from a previous study suggest that, in the whole-body Bmal1 knockout animals, there exists a loss of diurnal rhythm in circulating progenitor cells and a decrease in levels of the cytokine chemokine (C-X-C motif) ligand-12, necessary for BMPC release from the bone marrow.31 However, we did not observe a significant change in chemokine (C-X-C motif) ligand-12 in our conditional knockout animals. Instead, we observed a profound decrease in granulocyte-monocyte colony stimulating factor, which is secreted by a variety of cell types, such as macrophages, T cells, mast cells, NK cells, endothelial cells, and fibroblasts and is known for its pleiotropic effects. We speculate that the decrease in granulocyte-monocyte colony stimulating factor may be attributed to the decrease in LSK cell numbers. In the Bmal1fx/fx;Tek-Cre mice, we observed significant decreases in interferon-γ, tumor necrosis factor-α, IL-6, IL-1α, and IL-1β compared with those in controls. We propose that these changes are due to a loss of mesenchymal stem cells, cells involved in the generation of these cytokines. Furthermore, there is a loss of innervation in the mesenchymal stem cells, reducing the generation and release of cytokines and growth factors from these cells.32, 33, 34 The loss of innervation is supported by the reduced levels of NF-200 immunostaining (Figure 7A). Thus, the reduction of these inflammatory factors supports longstanding bone marrow dysfunction in these mice and warrants further investigation.
Our study also showed decreases in CFU-granulocyte-monocytes, CFU-granulocyte-erythrocyte-monocyte/macrophage-megakaryocytes, and CFU-granulocytes, and an increase in CFU-monocytes, due to the conditional deletion of Bmal1. We believe that the decrease in BMPC numbers is, in part, due to bone marrow denervation, as indicated by an overall decrease in NF-200 staining. Bone marrow neuropathy results in specific denervation of bone marrow stromal cells, which in turn results in a change in the expression of the key cytokines and growth factors needed for hematopoietic stem cell–progenitor cell differentiation and function. However, in our studies, there was a down-regulation of several cytokines that are known to be increased with inflammation. One possible explanation for an increase in CFU-monocytes is that TIE-2 expression in BMPCs is required for maintaining quiescence and maintaining adherence in the osteoblastic niche of the bone marrow.25 The findings from our study support the concept that a lack of BMAL1 in TIE-2–expressing BMPCs alters the quiescent stage of hematopoietic stem cells, leading to an increase in myelomonocytic (CFU-monocyte) cells in the bone marrow. Additional studies are necessary for further characterizing bone marrow defects in Bmal1fx/fx;Tek-Cre mice.
In conclusion, the findings from our study highlight the importance of the circadian clock system in maintaining vascular homeostasis, and that endothelial- and BMPC-specific deletion of BMAL1 is detrimental to both micro- and macrovasculature beds and results in denervation of the bone marrow, leading to BMPC dysfunction.
Acknowledgment
We thank the Flow Cytometry Resource Facility at Indiana University Simon Cancer Center (Indianapolis, IN).
E.B. helped with the flow cytometry analysis; D.Y. performed experiments involving the femoral artery–injury model; J.C., Q.L., and A.A. performed immunofluorescence staining of the retinal sections and analyzed the data; S.C., J.M.D., and T.E.S. helped with the animal studies; J.V.B. provided the Bmalfx/fx;Tek-Cre mice; M.S.S. supervised the studies involving the femoral artery–injury model; A.D.B. and M.B.G. were involved in the conception and design of the study, collection, analysis, and interpretation of the data, and the writing, financial support, and final approval of the manuscript.
Footnotes
Supported by the Ralph and Grace Showalter Trust Fund, International Retinal Research Foundation, an unrestricted award from the Research to Prevent Blindness Foundation to the Department of Ophthalmology, Indiana University (A.D.B.), and NIH grants R01EY007739, R01EY012601, R01NHLBI110170, and R01 DK090730 (all to M.B.G). The Flow Cytometry Resource Facility at Indiana University Simon Cancer Center is partially funded by National Cancer Institute grant P30 CA082709.
Disclosures: None declared.
Supplemental material for this article can be found at http://dx.doi.org/10.1016/j.ajpath.2017.02.014.
Contributor Information
Ashay D. Bhatwadekar, Email: abhatwad@iupui.edu.
Maria B. Grant, Email: mabgrant@iupui.edu.
Supplemental Data
Supplemental Figure S1.
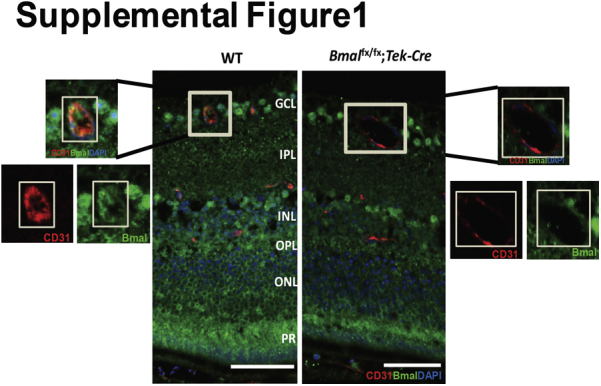
The decrease in brain and muscle aryl hydrocarbon receptor nuclear translocator–like protein (Bmal)-1 intensity in the blood vessels of Bmal1fx/fx;Tek-Cre mice. Retinal sections are stained with Bmal1 (green) and CD31 (red) in this representative photomicrograph showing a decrease in the staining intensity in vessels. n = 3 per group. Scale bars = 50 μm. GCL, ganglion cell layer; INL, inner nuclear layer; IPL, inner plexiform layer; ONL, outer nuclear layer; OPL, outer plexiform layer; PR, photoreceptor.
Supplemental Figure S2.
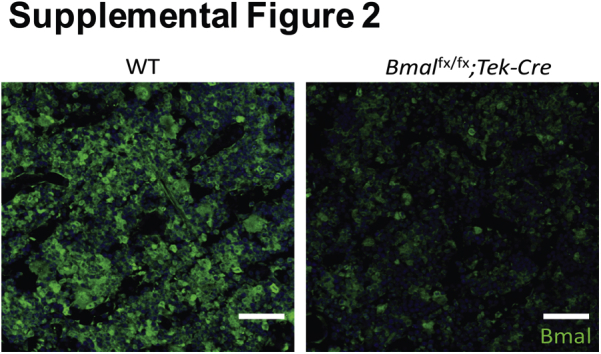
Brain and muscle aryl hydrocarbon receptor nuclear translocator–like protein (Bmal)-1 intensity in bone marrow. Representative images of Bmal1-stained femurs showing a decrease in Bmal1 expression in the bone marrow. n = 3 per group. Scale bars = 50 μm.
Supplemental Figure S3.

Decrease in stem cell mobilization factors in Bmal1fx/fx;Tek-Cre mice. Bar chart shows levels of mobilization factors in bone marrow supernatants. Data are expressed as means ± SEM. n = 5 WT; n = 6 Bmal1fx/fx;Tek-Cre. ∗P < 0.05 versus WT. GM-CSF, granulocyte-monocyte colony stimulating factor; ns, nonsignificant; SCF, stem cell factor; SDF, stromal derived factor.
Supplemental Figure S4.

Inflammatory markers in bone marrow supernatants of Bmal1fx/fx;Tek-Cre mice. Bar chart shows levels of a variety of inflammatory markers in bone marrow. Data are expressed as means ± SEM. n = 5 WT; n = 6 Bmal1fx/fx;Tek-Cre. ∗∗P < 0.01 versus WT. IFN, interferon; MIP, macrophage inflammatory protein; TNF, tumor necrosis factor.
References
- 1.Reilly D.F., Westgate E.J., FitzGerald G.A. Peripheral circadian clocks in the vasculature. Arterioscler Thromb Vasc Biol. 2007;27:1694–1705. doi: 10.1161/ATVBAHA.107.144923. [DOI] [PubMed] [Google Scholar]
- 2.Muller J.E., Stone P.H., Turi Z.G., Rutherford J.D., Czeisler C.A., Parker C., Poole W.K., Passamani E., Roberts R., Robertson T., Sobel B.E., Willerson J.T., Braunwald E., MILIS Study Group Circadian variation in the frequency of onset of acute myocardial infarction. N Engl J Med. 1985;313:1315–1322. doi: 10.1056/NEJM198511213132103. [DOI] [PubMed] [Google Scholar]
- 3.Willich S.N. Circadian variation and triggering of cardiovascular events. Vasc Med. 1999;4:41–49. doi: 10.1177/1358836X9900400108. [DOI] [PubMed] [Google Scholar]
- 4.Miller B.H., McDearmon E.L., Panda S., Hayes K.R., Zhang J., Andrews J.L., Antoch M.P., Walker J.R., Esser K.A., Hogenesch J.B., Takahashi J.S. Circadian and CLOCK-controlled regulation of the mouse transcriptome and cell proliferation. Proc Natl Acad Sci U S A. 2007;104:3342–3347. doi: 10.1073/pnas.0611724104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Young M.E., Razeghi P., Taegtmeyer H. Clock genes in the heart: characterization and attenuation with hypertrophy. Circ Res. 2001;88:1142–1150. doi: 10.1161/hh1101.091190. [DOI] [PubMed] [Google Scholar]
- 6.Turek F.W., Joshu C., Kohsaka A., Lin E., Ivanova G., McDearmon E., Laposky A., Losee-Olson S., Easton A., Jensen D.R., Eckel R.H., Takahashi J.S., Bass J. Obesity and metabolic syndrome in circadian Clock mutant mice. Science. 2005;308:1043–1045. doi: 10.1126/science.1108750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Creager M.A., Luscher T.F., Cosentino F., Beckman J.A. Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: part I. Circulation. 2003;108:1527–1532. doi: 10.1161/01.CIR.0000091257.27563.32. [DOI] [PubMed] [Google Scholar]
- 8.Luscher T.F., Creager M.A., Beckman J.A., Cosentino F. Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: part II. Circulation. 2003;108:1655–1661. doi: 10.1161/01.CIR.0000089189.70578.E2. [DOI] [PubMed] [Google Scholar]
- 9.Staels B. When the Clock stops ticking, metabolic syndrome explodes. Nat Med. 2006;12:54–55. doi: 10.1038/nm0106-54. discussion 5. [DOI] [PubMed] [Google Scholar]
- 10.Laposky A.D., Bradley M.A., Williams D.L., Bass J., Turek F.W. Sleep-wake regulation is altered in leptin-resistant (db/db) genetically obese and diabetic mice. Am J Physiol Regul Integr Comp Physiol. 2008;295:R2059–R2066. doi: 10.1152/ajpregu.00026.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zee P.C., Attarian H., Videnovic A. Circadian rhythm abnormalities. Continuum (Minneap Minn) 2013;19:132–147. doi: 10.1212/01.CON.0000427209.21177.aa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Busik J.V., Tikhonenko M., Bhatwadekar A., Opreanu M., Yakubova N., Caballero S., Player D., Nakagawa T., Afzal A., Kielczewski J., Sochacki A., Hasty S., Li Calzi S., Kim S., Duclas S.K., Segal M.S., Guberski D.L., Esselman W.J., Boulton M.E., Grant M.B. Diabetic retinopathy is associated with bone marrow neuropathy and a depressed peripheral clock. J Exp Med. 2009;206:2897–2906. doi: 10.1084/jem.20090889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kreier F., Kalsbeek A., Sauerwein H.P., Fliers E., Romijn J.A., Buijs R.M. “Diabetes of the elderly” and type 2 diabetes in younger patients: possible role of the biological clock. Exp Gerontol. 2007;42:22–27. doi: 10.1016/j.exger.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 14.Ando H., Fujimura A. Circadian clock disruption and diabetes mellitusNihon Rinsho. 2013;71:2114–2118. Japanese. [PubMed] [Google Scholar]
- 15.Jadhav V., Luo Q., M Dominguez J., II, Al-Sabah J., Chaqour B., Grant M.B., Bhatwadekar A.D. Per2-mediated vascular dysfunction is caused by the upregulation of the connective tissue growth factor (CTGF) PLoS One. 2016;11:e0163367. doi: 10.1371/journal.pone.0163367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bhatwadekar A.D., Yan Y., Qi X., Thinschmidt J.S., Neu M.B., Li Calzi S., Shaw L.C., Dominiguez J.M., Busik J.V., Lee C., Boulton M.E., Grant M.B. Per2 mutation recapitulates the vascular phenotype of diabetes in the retina and bone marrow. Diabetes. 2013;62:273–282. doi: 10.2337/db12-0172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yano M., Iwama A., Nishio H., Suda J., Takada G., Suda T. Expression and function of murine receptor tyrosine kinases, TIE and TEK, in hematopoietic stem cells. Blood. 1997;89:4317–4326. [PubMed] [Google Scholar]
- 18.Bhatwadekar A., Glenn J.V., Figarola J.L., Scott S., Gardiner T.A., Rahbar S., Stitt A.W. A new advanced glycation inhibitor, LR-90, prevents experimental diabetic retinopathy in rats. Br J Ophthalmol. 2008;92:545–547. doi: 10.1136/bjo.2007.127910. [DOI] [PubMed] [Google Scholar]
- 19.Zheng L., Gong B., Hatala D.A., Kern T.S. Retinal ischemia and reperfusion causes capillary degeneration: similarities to diabetes. Invest Ophthalmol Vis Sci. 2007;48:361–367. doi: 10.1167/iovs.06-0510. [DOI] [PubMed] [Google Scholar]
- 20.Anea C.B., Zhang M., Stepp D.W., Simkins G.B., Reed G., Fulton D.J., Rudic R.D. Vascular disease in mice with a dysfunctional circadian clock. Circulation. 2009;119:1510–1517. doi: 10.1161/CIRCULATIONAHA.108.827477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davis S., Aldrich T.H., Jones P.F., Acheson A., Compton D.L., Jain V., Ryan T.E., Bruno J., Radziejewski C., Maisonpierre P.C., Yancopoulos G.D. Isolation of angiopoietin-1, a ligand for the TIE2 receptor, by secretion-trap expression cloning. Cell. 1996;87:1161–1169. doi: 10.1016/s0092-8674(00)81812-7. [DOI] [PubMed] [Google Scholar]
- 22.Sato T.N., Qin Y., Kozak C.A., Audus K.L. Tie-1 and tie-2 define another class of putative receptor tyrosine kinase genes expressed in early embryonic vascular system. Proc Natl Acad Sci U S A. 1993;90:9355–9358. doi: 10.1073/pnas.90.20.9355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schnurch H., Risau W. Expression of tie-2, a member of a novel family of receptor tyrosine kinases, in the endothelial cell lineage. Development. 1993;119:957–968. doi: 10.1242/dev.119.3.957. [DOI] [PubMed] [Google Scholar]
- 24.Kisanuki Y.Y., Hammer R.E., Miyazaki J., Williams S.C., Richardson J.A., Yanagisawa M. Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev Biol. 2001;230:230–242. doi: 10.1006/dbio.2000.0106. [DOI] [PubMed] [Google Scholar]
- 25.Arai F., Hirao A., Ohmura M., Sato H., Matsuoka S., Takubo K., Ito K., Koh G.Y., Suda T. Tie2/angiopoietin-1 signaling regulates hematopoietic stem cell quiescence in the bone marrow niche. Cell. 2004;118:149–161. doi: 10.1016/j.cell.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 26.Hangai M., Yoshimura N., Hiroi K., Mandai M., Honda Y. Inducible nitric oxide synthase in retinal ischemia-reperfusion injury. Exp Eye Res. 1996;63:501–509. doi: 10.1006/exer.1996.0140. [DOI] [PubMed] [Google Scholar]
- 27.Du Y., Smith M.A., Miller C.M., Kern T.S. Diabetes-induced nitrative stress in the retina, and correction by aminoguanidine. J Neurochem. 2002;80:771–779. doi: 10.1046/j.0022-3042.2001.00737.x. [DOI] [PubMed] [Google Scholar]
- 28.Anea C.B., Cheng B., Sharma S., Kumar S., Caldwell R.W., Yao L., Ali M.I., Merloiu A.M., Stepp D.W., Black S.M., Fulton D.J., Rudic R.D. Increased superoxide and endothelial NO synthase uncoupling in blood vessels of Bmal1-knockout mice. Circ Res. 2012;111:1157–1165. doi: 10.1161/CIRCRESAHA.111.261750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Westgate E.J., Cheng Y., Reilly D.F., Price T.S., Walisser J.A., Bradfield C.A., FitzGerald G.A. Genetic components of the circadian clock regulate thrombogenesis in vivo. Circulation. 2008;117:2087–2095. doi: 10.1161/CIRCULATIONAHA.107.739227. [DOI] [PubMed] [Google Scholar]
- 30.Rudic R.D., McNamara P., Reilly D., Grosser T., Curtis A.M., Price T.S., Panda S., Hogenesch J.B., FitzGerald G.A. Bioinformatic analysis of circadian gene oscillation in mouse aorta. Circulation. 2005;112:2716–2724. doi: 10.1161/CIRCULATIONAHA.105.568626. [DOI] [PubMed] [Google Scholar]
- 31.Mendez-Ferrer S., Lucas D., Battista M., Frenette P.S. Haematopoietic stem cell release is regulated by circadian oscillations. Nature. 2008;452:442–447. doi: 10.1038/nature06685. [DOI] [PubMed] [Google Scholar]
- 32.Tay J., Levesque J.P., Winkler I.G. Cellular players of hematopoietic stem cell mobilization in the bone marrow niche. Int J Hematol. 2017;105:129–140. doi: 10.1007/s12185-016-2162-4. [DOI] [PubMed] [Google Scholar]
- 33.Prieto P., Fernandez-Velasco M., Fernandez-Santos M.E., Sanchez P.L., Terron V., Martin-Sanz P., Fernandez-Aviles F., Bosca L. Cell expansion-dependent inflammatory and metabolic profile of human bone marrow mesenchymal stem cells. Front Physiol. 2016;7:548. doi: 10.3389/fphys.2016.00548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roson-Burgo B., Sanchez-Guijo F., Del Canizo C., De Las Rivas J. Insights into the human mesenchymal stromal/stem cell identity through integrative transcriptomic profiling. BMC Genomics. 2016;17:944. doi: 10.1186/s12864-016-3230-0. [DOI] [PMC free article] [PubMed] [Google Scholar]