

Abstract
The cholesterol-dependent cytolysins (CDCs), a superfamily of pore-forming toxins, are characterized by a conserved undecapeptide motif that is believed to be critical for membrane recognition by means of cholesterol. Intermedilysin (ILY), an unusual member of the CDCs, exhibits specificity for human cells and contains nonconservative substitutions in the motif. We show that the cellular specificity of ILY is based on its ability to specifically bind to human cells and does not involve some other feature of the CDC mechanism. Furthermore, cellular recognition by ILY appears to be encoded in domain 4 alone but does not involve the variant undecapeptide of ILY. We show that the undecapeptide is involved in the prepore-to-pore conversion of ILY and so demonstrate a direct connection between the structure of the undecapeptide and the prepore-to-pore transition. We have determined the crystal structure of ILY, which, when compared to the known structure of a prototypical CDC, suggests that the basic aspects of its 3D structure are likely to be conserved in all CDCs.
Keywords: channels, crystallography, toxins
The cholesterol-dependent cytolysins (CDCs) are one of the most widely distributed toxins known, having been identified in five different genera of Gram-positive bacteria (1). The CDCs exhibit a number of unique features amongst pore-forming toxins, including an absolute dependence on the presence of cholesterol-rich membranes for their activity and the formation of oligomeric transmembrane pores >150 Å in diameter. More than 20 members of the CDC family have been identified so far, and there exists a high degree of sequence similarity (40-80%), suggesting that they all have similar activities and 3D structures. The crystal structure of one CDC, perfringolysin O (PFO) from Clostridium perfringens, has been determined (2). The structure revealed that CDCs are elongated, rod-shaped molecules rich in β-sheet and composed of four domains, but it provided few clues as to how the molecule formed pores. The high degree of similarity of the CDC sequences suggested they likely exhibit a closely similar 3D structure, although a structure of another CDC has not been elucidated to confirm this hypothesis.
A major advance in understanding the mechanism of CDC membrane insertion came with the discovery that domain 3 harbored regions that formed the transmembrane (TM) β-barrel (3-5). The studies of Shepard et al. (4) and Shatursky et al. (5) revealed that a series of small helices on either side of a central β-sheet could unfurl into β-hairpins, TM hairpin (TMH) 1 and TMH2, that were capable of spanning membrane bilayers. Such conformational changes were unprecedented amongst toxins and hence represented a new paradigm for how pore-forming proteins generated a membrane-spanning domain.
The CDCs are characterized by a conserved undecapeptide (ECTGLAWEWWR), termed the Trp-rich motif, which was found to correspond to a loop in domain 4 of the PFO 3D structure (2). Its interaction with membranes is a prerequisite for the insertion of the domain 3 TMHs that form the wall of the TM β-barrel (6). The conserved undecapeptide and three other short hydrophobic loops, all located at the tip of domain 4 of PFO, penetrate the surface of the membrane and anchor the molecule for the subsequent conformational changes during pore formation (6-8). Domains 3 and 4 are conformationally coupled, although they are not in direct physical contact; mutations in domain 3 can affect the rate at which domain 4 interacts with the membrane, even though the domain 4 undecapeptide interacts with the membrane before the insertion of the domain 3 TMHs. The cytolytic activity of CDCs is highly sensitive to changes in the Trp-rich motif, especially mutations of the conserved tryptophans or chemical modification of the thiol of conserved cysteine-produced derivatives that are incapable of membrane binding (7, 9-11). However, some undecapeptide mutations or modifications appear to affect the global structure of CDCs, thus suggesting that its conformation is important to the proper function of CDCs.
Streptococcus intermedius secretes the CDC intermedilysin (ILY) (12), and ILY appears to be a major virulence factor of S. intermedius, because it is expressed up to 10-fold more in abscesses (13). ILY exhibits three features that distinguish it from most other CDCs. First, it has a variant undecapeptide sequence (GATGLAWEPWR). The normally conserved cysteine of the CDC undecapeptide is replaced by an alanine residue, resulting in an oxidation-resistant toxin (14), and a proline residue is substituted for the second conserved tryptophan. Second, ILY is specific for human cells (12, 14). Nagamune et al. (14) showed that animal erythrocytes from nine species are not susceptible to ILY and that non-human primate erythrocytes are 100-fold less susceptible to ILY than are human erythrocytes. Third, free cholesterol is only weakly inhibitory. Although the presence of membrane cholesterol is still essential to the activity of ILY, it does not appear to function as a receptor as it does for PFO (15). Giddings et al. (15) showed that depletion of ≈90% of the membrane cholesterol did not affect binding of ILY or the related CDC, streptolysin O (SLO), to human erythrocytes but that it did completely abrogate the hemolytic activity of both toxins. It was determined by fluorescence spectroscopy that cholesterol depletion prevented the prepore-to-pore conversion of ILY and SLO. Therefore, cholesterol was necessary for ILY insertion but not its membrane binding.
The unique aspects of the ILY mechanism, its variant undecapeptide and human cell specificity, prompted us to investigate whether the two aspects were linked. We have pursued a detailed biological and structural investigation of ILY with the aim of understanding how these distinguishing features impact its interaction with cells. The biological data and the crystal structure show that the fundamental structural features of the CDCs are maintained in at least two members of this toxin family. Furthermore, these studies reveal that the ILY undecapeptide is not involved in membrane binding but facilitates the prepore-to-pore conversion.
Materials and Methods
Hemolytic Activity. The hemolytic activity of each toxin on human erythrocytes was determined as described in ref. 4.
Generation and Purification of ILY and Hybrids. The cysteineless gene for ILY was cloned from the Gram-positive bacteria S. intermedius by using the following pTrcHisA vector primers for ILY: BamHI-GGGCCGGATCCGAAACACCTACCAAACCAAAAGC and EcoRI-GGGCCCTTAAGTTAATCAGTGTTATCTTTCACAAC. Amino acid substitutions in the ILY gene were generated by using PCR quick-change mutagenesis (Stratagene). Expression and purification of all proteins used in this study were carried out as described in ref. 4 for PFO.
Modification of Proteins with Fluorescent Probes. Derivatization of Cys-substituted PFOA215C and ILYH242C and their Ala-substituted variants with thiol-specific fluorescent dyes Alexa Fluor 488 C5 maleimide (Molecular Probes) or iodoacetamido-NBD (N,N′-dimethyl-N-(iodoacetyl)-N′-(7-nitrobenz-2-oxa-1,3-diazolyl)ethylene-diamine) was carried out as described in ref. 15. All Cys-substituted toxin derivatives were labeled at a 1:1 molar ratio of dye to protein. The dye-labeled protein samples were made up in 10% (vol/vol) glycerol, quick-frozen in liquid nitrogen, and stored at -80°C.
Erythrocyte Ghost Membrane Preparation. Erythrocyte ghost membranes were prepared as described in ref. 15. Membrane protein concentration was determined by the method of Bradford (16) (Bio-Rad) by using BSA as a standard.
Fluorescence Spectroscopy. All fluorescence intensity measurements were performed by using an SLM-8100 photon-counting spectrofluorimeter as described in ref. 4. An excitation wavelength of 470 nm was used for NBD, and a wavelength of 488 nm was used for Alexa Fluor 488, with a bandpass of 2 nm. Emission scans of each sample were carried out at a resolution of 1 nm with an integration time of 1 s. Membrane insertion of TMH1 was determined as described in ref. 15. Binding of proteins to erythrocyte ghost membranes was determined by using FRET between the NBD-labeled toxin (donor) and membranes labeled with the membrane probe octadecyl rhodamine B (ORB) chloride (acceptor). NBD and rhodamine are used in FRET experiments as donor and acceptor dyes, respectively, because the emission wavelength of NBD overlaps the excitation wavelength of rhodamine. Erythrocyte ghost membranes were labeled with rhodamine by equilibrating the membranes (equivalent to 10 μg of protein) with 0.684 nmol of ORB for 120 min at room temperature. The ORB spontaneously inserts into the membranes and places the rhodamine B dye on the membrane surface (17). Unincorporated probe was removed by three rounds of centrifugation and resuspension of the membranes in PBS (10 mM Na2HPO4, pH 7.5/147 mM NaCl/3 mM KCl).
The FRET experiments were carried out as described for intensity measurements, except that the NBD-labeled toxin was also mixed with the equivalent amount of the ORB-labeled membranes. The net fluorescence intensity of the donor dye was calculated by subtracting the fluorescence emission of unlabeled toxin mixed with the ORB-labeled membranes. If the NBD-labeled toxin bound to the membranes, then the emission intensity of the NBD was quenched by FRET from the NBD to the membrane-associated ORB.
Flow Cytometry. The ability of PFOA215C or ILYH215C containing the various Ala substitutions within the undecapeptides to bind to human erythrocyte cells was determined by using flow cytometry as described in ref. 15. The percentage of binding was determined by fixing the geometric mean of the fluorescence (GMF) bound per cell for dye-labeled PFOA215C or ILYH242C (wild type) as 100% binding, and, therefore, relative binding of each mutant was equal to GMF mutant/GMF wild-type toxin.
Gel Electrophoresis and Immunoblot Analysis. Monomers and oligomers of PFO and ILY were separated by using denaturing SDS-agarose gel electrophoresis, and the protein bands were identified by immunoblot analysis as described in refs. 4 and 15.
Structure Determination and Refinement. ILY was overexpressed, purified, and crystallized as described in ref. 18. The structure was determined by single isomorphous replacement and multiple anomalous dispersion (see supporting information, which is published on the PNAS web site). The model has been refined to a crystallographic R-factor of 24.5% (Rfree of 29.3%) to 2.6-Å resolution. The final model includes residues 56-528 for each ILY molecule in the asymmetric unit, 139 waters, and 4 sulfate ions.
Results
Mechanistic Basis for ILY Cellular Specificity. Two possibilities currently exist to explain the lack of activity of ILY on animal cells (12, 14). The first option is that ILY does not bind to animal cells because they lack a receptor for it. Although cholesterol has been generally accepted to be the CDC receptor, Giddings et al. (15) have recently shown that cholesterol is not involved in membrane recognition by ILY and that cholesterol appears to be important for prepore-to-pore conversion for most, if not all, CDCs. Therefore, it is likely that ILY requires a specific receptor that is not present on animal cells. The second option is that ILY may bind to animal cells but is not able to assemble into a pore-forming complex. This scenario has been observed for the unrelated pore-forming toxin α-hemolysin, which has been shown to bind to resistant cells but did not insert its TM β-barrel into the membrane (19). Therefore, we examined the ILY and PFO mechanism to determine the basis of resistance of animal cells to the cytolytic mechanism of ILY.
As expected, PFO lysed both human and horse erythrocytes, whereas ILY lysed only human erythrocytes (Table 1). Each toxin was then examined for its ability to bind to and insert its TM β-hairpins into the membranes of human and horse erythrocytes. Initially, the insertion of the hairpins into the membrane was examined because, if insertion was observed, then binding also had to occur before insertion. To perform these analyses, PFO was labeled with the environmentally sensitive dye NBD at Ala 215, which had been substituted with Cys (PFOA215C). Residue 215 is located in one of the two TM β-hairpins of PFO and has been shown to enter the nonpolar core of the membrane when PFO forms a pore. Therefore, if PFOA215C is derivatized with NBD, the fluorescence intensity of the NBD increases significantly as this residue moves from its polar environment in the monomer to the nonpolar environment of the membrane (4). Similarly, we modified the equivalent residue His 242 (ILYH242C) in ILY with NBD (15). We measured both insertion of the TM β-barrels of PFO and ILY and membrane binding. The insertion of the β-barrels into the isolated ghost membranes was measured by following the change in the fluorescence intensity of NBD-derivatized PFOA215C or ILYH242C as they make the transition from soluble monomer to the membrane-embedded pore. We have previously shown that an increase in the fluorescence intensity of the NBD-labeled derivatives of these two toxins reflects membrane insertion (4, 5, 15). As can be seen in Fig. 1A, both ILY and PFO insert into the ghost membranes from human erythrocytes. PFO also inserted its TM domains into horse erythrocyte membranes, whereas ILY did not, consistent with the cytolytic activity measurements in Table 1.
Table 1. Hemolytic activity of PFO and ILY and their chimeras.
| HD50, fmol
|
|||
|---|---|---|---|
| Toxin | Human erythrocytes | Horse erythrocytes | Fold change, horse/human |
| PFO | 14 | 26.7 | 2 |
| PFOILYD4 | 21 | >180,000 | >8500 |
| ILY | 9 | >173,000 | >19,000 |
| ILYPFOD4 | 169 | 203.3 | 1 |
The HD50 (hemolytic dose) is defined as the concentration of toxin required to lyse 50% of the erythrocytes in 100 μl of PBS containing ≈3 × 107 human erythrocytes. PFOILYD4 is the chimera of PFO containing domains 1–3 and domain 4 of ILY; ILYPFOD4 is ILY that has PFO domain 4 substituted for its native sequence.
Fig. 1.

Membrane binding and insertion into human or horse erythrocyte (RBC) ghost membranes of PFO and ILY and their chimeras. (A) Membrane insertion of the TMβ-hairpins was assessed by the increase in the fluorescence intensity of the fluorescent probe (NBD) that was attached to a membrane-facing residue of TMH1 of PFO (4), ILY (15), and their derivatives, PFOILYD4 and ILYPFOD4. The emission intensity of NBD is shown for the membrane-inserted toxin on either human or horse erythrocyte membranes (dashed lines) or in the absence of membranes (solid lines). (B) Binding of the toxins to human or horse erythrocyte membranes was determined by FRET between NBD and the membrane probe ORB. The emission of the NBD-derivatized toxins is shown at 520 nm after equilibration with membranes with (gray bars) or without (black bars) ORB.
To determine whether ILY was in intimate contact with the horse membranes but unable to insert its β-hairpins, we performed the same experiments with membranes that had been labeled with the fluorescence lipid ORB. Hence, if the NBD-labeled toxin bound to the membrane, its fluorescent emission would be quenched by the presence of ORB in the ghost membranes by means of FRET. The Förster distance, R0, (the distance at which FRET efficiency is 50%) for this dye pair is ≈31-35 Å (20). Because FRET 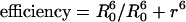 , where r is the distance between donor and acceptor dyes, then a separation of the dyes by >2r will exhibit an efficiency that effectively drops to 0. Therefore, the donor and acceptor dyes must be closely juxtaposed to exhibit FRET, as would be the case for donor-labeled toxin being bound to the acceptor-labeled membranes.
, where r is the distance between donor and acceptor dyes, then a separation of the dyes by >2r will exhibit an efficiency that effectively drops to 0. Therefore, the donor and acceptor dyes must be closely juxtaposed to exhibit FRET, as would be the case for donor-labeled toxin being bound to the acceptor-labeled membranes.
Consistent with the insertion analysis shown in Fig. 1A and the activity measurements presented in Table 1, the fluorescence intensity of the NBD-labeled PFO was nearly completely quenched when it was incubated with ORB-labeled human or horse ghost membranes, showing that it bound to the membranes (Fig. 1B). Also consistent with these measurements, ILY bound only to the human ghost membranes, because no change in the fluorescence intensity of the NBD was recorded when the dye-labeled ILY was incubated with horse membranes with ORB (Fig. 1B).
Domain 4 Encodes the Species Specificity of PFO and ILY Cytolytic Activity. Previous studies have shown that CDC membrane binding is mediated by means of domain 4 (21-23). Was the unique cellular specificity encoded in domain 4, or was it dependent on another functional domain? To determine whether domain 4 was responsible for the species specificity of ILY, chimeras of PFO and ILY were generated in which domain 4 was exchanged at a conserved tyrosine at the domain 2-4 junction. As shown in Table 1, the PFO chimera, PFOILYD4 (PFO with domain 4 of ILY), lysed only human cells, whereas the ILY chimera, ILYPFOD4 (ILY with domain 4 of PFO), lysed both human and horse erythrocytes. We further showed that the chimeras exhibited the characteristics of the domain 4 parent molecule with respect to membrane binding and insertion (Fig. 1). These results show that domain 4 is responsible for cellular specificity of each toxin.
A Functional Undecapeptide Is Not Involved in Cellular Specificity but Is Required for Conversion of Prepore to Pore. The highly conserved undecapeptide region of the CDCs is known to be of paramount importance to their mechanism, and mutations within this sequence usually result in the loss of cytolytic activity (10). The highly conserved undecapeptide (ECTGLAWEWWR) of the CDCs, located in domain 4, is modified in ILY to GATGLAWEPWR, suggesting that this difference may be responsible for the unique human cell specificity of ILY. The function of the two undecapeptides was examined by performing a complete alanine scan of the residues of both undecapeptides. The hemolytic activity and binding of each mutant to human erythrocytes was then compared to wild type. As can be seen in Fig. 2, the PFO undecapeptide was extremely sensitive to substitution by Ala with two exceptions: Substitution of Glu 458 and Cys 459 with Ala resulted in no significant loss of hemolytic and binding activity. In contrast, the ILY undecapeptide was relatively insensitive to mutation with alanine; only the mutation of Trp 491 and Trp 494 resulted in a major loss of hemolytic activity, but both appeared to bind to erythrocytes, similar to native ILY. Similarly, the PFO Glu 465 mutant appeared to retain most of its binding activity but lost ≈90% of its hemolytic activity.
Fig. 2.

Hemolytic and binding activity of PFO (Lower) and ILY (Upper) and their Ala-substituted undecapeptide derivatives on human erythrocytes. The HD50 (hemolytic dose or the toxin concentration required for 50% lysis) was determined for native toxin and assigned a value of 100%. The solid bars show the fraction of hemolytic activity based on the HD50 of each toxin compared with wild type. The hatched bars show the fraction of bound toxin compared to wild type (set as 100% binding) as determined by flow cytometry of the individually fluorescently labeled derivatives of native toxin and each mutant.
The fact that ILYW491A and PFOE465A both retained similar binding activity to wild type but lost ≥90% of their hemolytic activity suggested that these mutants would be useful in dissecting the function of the undecapeptide. Because both mutants appeared to be largely unaffected in membrane binding, we examined the next step in the CDC mechanism: formation of the prepore. The CDCs have been found to form a prepore intermediately before the formation of the pore complex (24, 25). Formation of the prepore involves membrane binding, the lateral diffusion of the bound monomers, and the polymerization of the monomers on the surface into the large CDC prepore oligomer. Once assembled, the monomers cooperate to insert their amphipathic β-hairpins to form the TM β-barrel (26). Because both ILYW491A and PFOE465A retained binding activity, we examined their capacity to form an oligomer on the membrane. The formation of oligomers by native ILY and PFO and their mutants ILYW491A and PFOE465A was examined by SDS-agarose gel electrophoresis to measure oligomer formation, as has been done previously for both wild-type toxins (15). Both mutants appear to form oligomers similar to wild type, showing that they form prepore structures but apparently are unable to convert to a pore-forming complex (data not shown). Hence, the mutation of the undecapeptide interferes with the transition of the prepore to pore in both toxins.
Crystal Structure of ILY. To gain a molecular explanation of the biological data, the crystal structure of ILY was determined. ILY is an elongated, boomerang-shaped molecule, rich in β-sheet and comprising four discontinuous domains (Fig. 3). The two molecules in the asymmetric unit superimpose with a rms deviation of 2.2 Å on Cα positions. This large deviation is due to a relative 10° rotation of domain 4 with respect to the rest of the molecule; the tips of domain 4 are nearly 16 Å apart when domain 1 of each molecule is superimposed. Superposition of domains 1-3 yields a rms deviation of 0.7 Å, and superposition of domain 4s yields a deviation of 0.6 Å. Hence, aside from the rotation of domain 4, the two molecules are very similar. Four sulfate ions have been identified in the asymmetric unit and are found to bind to the same two sites on each of the two protein molecules (Fig. 3). This observation suggests that the sulfate binding may be more than just an artifact of the crystallization process. One sulfate ion is bound to Arg 92 and Arg 477 and, hence, bridges domains 2 and 4. Another sulfate is bound in an unusual manner; it is coordinated by two consecutive residues, Lys 87 and Asn 88, both from domain 2 (through both side-chain and main-chain atoms), as well as Arg 477 from the opposing molecule. It is conceivable that these sites might represent binding pockets for the putative human protein receptor.
Fig. 3.

Crystal structure of ILY. Stereo ribbon representation indicates the location of domains by color. Domain 1 is blue, domain 2 is green, domain 3 is red, and domain 4 is orange. Bound sulfate ions and ligands are shown in ball-and-stick formation. The figure was drawn by using molscript (30) and raster3d (31).
Special Structural Features of ILY. ILY and PFO share an overall pairwise sequence identity of 40% (197 of 532 residues) that is distributed relatively evenly over the entire sequence of each molecule. As expected, ILY has an overall fold and topology similar to that of PFO (Fig. 4A). The most striking difference is the highly bent shape of ILY compared with PFO, with domain 4 rotated between 20° and 30° away from the main axis of the ILY molecule (Fig. 4A). (The variation depends on which ILY molecule in the asymmetric unit is used in the superposition.) Another prominent difference between ILY and PFO is the conformation of the Trp-rich loop in domain 4 that adopts an extended conformation in ILY but is curled up against the β-sheet in PFO (Fig. 4B). The extended conformation does not appear to be an artifact of the crystallization, because the loop of one of the two ILY molecules in the asymmetric unit is not involved in crystal contacts and the loops of both molecules superimpose almost exactly. There also is a movement of a hairpin loop by almost 10 Å at the other end of this domain because of differences in domain 2 (Fig. 4B and supporting information).
Fig. 4.

Stereo superpositions of ILY and PFO. (a) Cα traces of ILY (blue) and PFO (brown), based on a domain 1 superposition. (b)Cα trace of superimposed domain 4s of ILY (blue) and PFO (brown). The equivalent Trp residue at the tip of each undecapeptide loop is highlighted.
Location of a Putative Cholesterol-Binding Site in PFO That Is Absent in ILY. Previous work has implicated cholesterol as the receptor for the CDC family, whereby it is thought to act by concentrating the toxin in cholesterol-rich domains of the target cell membrane to promote oligomerization (27). We have previously hypothesized that cholesterol could bind to PFO by displacing the Trp-rich loop and binding to one face of the β-sheet in domain 4 (2). A striking feature of the ILY structure is the extended conformation of the Trp-rich loop (Fig. 5A), a conformation predicted for PFO when it binds cholesterol. We decided to computationally dock a cholesterol molecule into the ILY structure by using autodock (28) and found no preferred clustering of cholesterol molecules into the putative binding site. The conformation of the Trp-rich loop in ILY provides a basis for modeling the equivalent loop (Fig. 5B), in its sprung-out form, in PFO (Fig. 5 and supporting information). We then docked cholesterol into the modified PFO structure and found that 87 of 100 docks yielded a close clustering of cholesterol molecules (rms deviation of <1.5 Å of the second-lowest docked energy run) with the same orientation and position in the putative binding pocket. The cluster of putative cholesterol complex structures made chemical sense, with the cholesterol molecule stacking against the side chains of Trp 466 and 467 and the 3β-hydroxy moiety forming a hydrogen bond to Glu 407 of PFO. A comparison of the key residues involved in interacting with cholesterol in PFO (Fig. 5C) with the equivalent residues in ILY (Fig. 5A) shows that some are not conserved. Glu 407 (PFO) is replaced by Tyr 434 in ILY, and this residue does not appear capable of providing a hydrogen bond to the 3β-hydroxyl of cholesterol. Trp 466 (PFO) is substituted by Pro 493 in ILY; the latter residue is likely responsible for the extended conformation of the undecapeptide observed in ILY and also disrupts the van der Waals interaction between cholesterol and the tryptophans suggested in the PFO model.
Fig. 5.

The undecapeptide region and cholesterol binding. (a) Domain 4 of ILY. (b) Domain 4 of PFO. (c) Domain 4 of PFO, with the Trp-rich loop conformation modeled based on the conformation seen in ILY. A molecule of cholesterol has been docked onto the domain by using autodock (28). The figures were drawn by using bobscript (33) and raster3d (32).
Discussion
The CDCs are one of the most studied families of toxins because of their ability to not only transform from water-soluble proteins into large oligomeric pore-forming complexes but also to be, in many instances, a major virulence factor of the bacterium that produces them (1). Two aspects of the molecular mechanism of the CDCs have been particularly difficult to elucidate: the roles of cholesterol and the conserved undecapeptide in the cytolytic mechanism. Past studies have suggested that the undecapeptide participates in membrane binding by virtue of an interaction by means of cholesterol (9). Recently, Giddings et al. (15) have shown that the primary effect of cholesterol on CDC activity occurs downstream of membrane binding and that its presence in the membrane is necessary for prepore-to-pore conversion. The discovery of ILY has changed our perception of how CDCs recognize membranes and the roles of cholesterol and the conserved undecapeptide in the CDC cytolytic mechanism. The studies herein have revealed that the undecapeptide functions in the conversion of the prepore-to-pore complex in ILY.
ILY differs from other CDCs in that it is specific for human cells (12, 14). We have now shown that the specificity of ILY for human cells resides in its capacity to specifically recognize and bind to human cells by means of domain 4. Swapping domain 4 of ILY and PFO results in the exchange of their cellular specificity. Therefore, differences in the domain 4 structure alone are responsible for the cellular specificity of PFO and ILY. Comparison of the crystal structures of ILY and PFO shows that domain 4 is the most highly conserved structure of the two toxins, with the most significant difference within the domain 4 structure in the undecapeptide loop. However, when residues of the undecapeptide of ILY were substituted by alanine, the resulting mutants retained their human cell specificity. Hence, the ILY cellular specificity must result in differences of residues that are outside of the motif, although comparison of the domain 4 primary structures did not provide any obvious candidate regions that may be involved in ILY cellular specificity.
The role of the undecapeptide in the CDC mechanism appears complex and has been difficult to elucidate because mutations within the undecapeptide of PFO often result in a significant decrease in activity. Furthermore, interpretation of these results has been complicated by the fact that global structural changes in the structure of the CDC can result from mutations introduced into the undecapeptide (9) or chemical modification of the conserved cysteine residue (11). To more fully explore the function of the undecapeptide in PFO and ILY, we generated alanine substitutions for each residue and examined the activity of each mutant. The PFO undecapeptide was extremely sensitive to alanine substitution, and only residues Glu 458 and Cys 459 were unaffected by substitution. The remaining residues lost >85% of their binding capacity, with most substitutions resulting in a complete loss of hemolytic activity. In contrast, the ILY undecapeptide was relatively insensitive to substitution, with most mutants retaining a substantial level of hemolytic and binding activity. Of significance is the observation that none of the alanine substitutions within the ILY undecapeptide resulted in a significant loss of binding. Therefore, the ILY undecapeptide apparently was not involved in cell binding.
One mutant of PFO, PFOE465A, and two mutants of ILY, ILYW491A and ILYW494A, were of particular interest. All three mutants exhibited a significant loss of haemolytic activity but retained near wild-type binding. Further analyses of PFOE465A and ILYW491A showed that these defects were similar, because both were able to bind and oligomerize on the membrane but were not able to form a pore complex, as evidenced by the inability to lyse erythrocytes. Therefore, these undecapeptide mutants were locked in the prepore state. Sekino-Suzuki et al. (9) have also shown that substitution of each undecapeptide tryptophan of PFO with phenylalanine resulted in inactive mutants that could still bind cholesterol and form membrane oligomers.
The inability of PFOE465A and ILYW491A to convert the prepore to the pore complex suggests that a functional undecapeptide is important for this step of the CDC mechanism. These results also are consistent with our previous study that showed that conformational changes in the PFO undecapeptide were coupled to the insertion of the domain 3 TM β-hairpins (6). The results here show that specific mutations in the undecapeptide can prevent prepore-to-pore conversion, a process that requires the insertion of the domain 3 β-hairpins. Hence, we have shown that mutations in the undecapeptide directly affect the conversion of the prepore to the pore complex, thus establishing a function of the undecapeptide in prepore-to-pore conversion in ILY.
There are now two features of CDCs that have been shown to be critical for prepore-to-pore conversion: a functional undecapeptide (shown in this study) and the presence of cholesterol (15). Is there a connection between these two key features? We show here that PFO possesses a plausible binding site in domain 4 for a cholesterol molecule if it is assumed that the undecapeptide adopts an extended conformation, as observed in the crystal structure of ILY (Fig. 5). Surprisingly, ILY is not predicted to possess such a site. We hypothesize that ILY does not need a cholesterol-binding site because its undecapeptide is already “sprung out.” These suggestions are in keeping with earlier work that has shown that free cholesterol is only weakly inhibitory for ILY cytolytic activity in contrast to PFO (14). Nevertheless, cholesterol does play a role in the insertion process of ILY (15), indicating that cholesterol may play multiple roles in CDC activity.
Finally, the elucidation of the ILY structure demonstrates that the basic features of the CDC 3D structure appear to be conserved among the CDCs. A phylogenetic tree generated by the alignment of the known CDC primary structures (15 in all) by using the nearest-neighbor method of Saitou and Nei (29) shows that PFO and ILY are two of the most distantly related CDCs (data not shown), and yet they exhibit similar 3D structures. Therefore, it is likely that all CDCs will exhibit 3D structures similar to those of PFO and ILY.
In summary, we have shown that the cellular specificity of ILY is based on its ability to specifically bind to human cells and does not involve some other feature of the CDC mechanism. Furthermore, cellular recognition by ILY appears to be encoded in domain 4 alone but does not involve the variant undecapeptide of ILY. This finding led to the revelation that the undecapeptide is involved in the prepore-to-pore conversion of ILY, demonstrating a direct connection between the structure of the undecapeptide and the prepore-to-pore transition. Finally, the structural similarity of the ILY and PFO crystal structures suggests that the basic aspects of the CDC 3D structure are likely to be conserved in all CDCs.
Supplementary Material
Acknowledgments
We acknowledge the excellent technical assistance of Amy Marpoe. We thank Harry Tong, other BioCARS staff, and Michelle Dunstone for their help at the Advanced Photon Source. This work was supported by the Australian Synchrotron Research Program, which is funded by the Commonwealth of Australia under the Major National Research Facilities Program; a grant from the National Health and Medical Research Council of Australia (NHMRC) (to M.W.P); and National Institutes of Health Grant NIAID AI37657 (to R.K.T.). Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Basic Energy Sciences, Office of Energy Research. G.P. is supported by an NHMRC R. D. Wright Research Fellowship, and M.W.P. is an NHMRC Fellow.
Author contributions: R.K.T. and M.W.P. designed research; G.P. and K.S.G. performed research; G.P., K.S.G., R.K.T., and M.W.P. analyzed data; and G.P., R.K.T., and M.W.P. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: CDCs, cholesterol-dependent cytolysins; ILY, intermedilysin; PFO, perfringolysin O; NBD, 7-nitrobenz-2-oxa-1,3-diazolyl; TM, transmembrane; TMH, TM hairpin; ORB, octadecyl rhodamine B.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 1S3R).
References
- 1.Tweten, R. K., Parker, M. W. & Johnson, A. E. (2001) Curr. Top. Microbiol. Immunol. 257, 15-33. [DOI] [PubMed] [Google Scholar]
- 2.Rossjohn, J., Feil, S. C., McKinstry, W. J., Tweten, R. K. & Parker, M. W. (1997) Cell 89, 685-692. [DOI] [PubMed] [Google Scholar]
- 3.Palmer, M., Saweljew, P., Vulicevic, I., Valeva, A., Kehoe, M. & Bhakdi, S. (1996) J. Biol. Chem. 271, 26662-26667. [DOI] [PubMed] [Google Scholar]
- 4.Shepard, L. A., Heuck, A. P., Hamman, B. D., Rossjohn, J., Parker, M. W., Ryan, K. R., Johnson, A. E. & Tweten, R. K. (1998) Biochemistry 37, 14563-14574. [DOI] [PubMed] [Google Scholar]
- 5.Shatursky, O., Heuck, A. P., Shepard, L. A., Rossjohn, J., Parker, M. W., Johnson, A. E. & Tweten, R. K. (1999) Cell 99, 293-299. [DOI] [PubMed] [Google Scholar]
- 6.Heuck, A. P., Hotze, E. M., Tweten, R. K. & Johnson, A. E. (2000) Mol. Cell, 6, 1233-1242. [DOI] [PubMed] [Google Scholar]
- 7.Nakamura, M., Sekino, N., Iwamoto, M. & Ohno-Iwashita, Y. (1995) Biochemistry 34, 6513-6520. [DOI] [PubMed] [Google Scholar]
- 8.Ramachandran, R., Heuck, A. P., Tweten, R. K. & Johnson, A. E. (2002) Nat. Struct. Biol. 11, 823-827. [DOI] [PubMed] [Google Scholar]
- 9.Sekino-Suzuki, N., Nakamura, M., Mitsui, K. I. & Ohno-Iwashita, Y. (1996) Eur. J. Biochem. 241, 941-947. [DOI] [PubMed] [Google Scholar]
- 10.Alouf, J. E. (2000) Int. J. Med. Microbiol. 290, 351-356. [DOI] [PubMed] [Google Scholar]
- 11.Iwamoto, M., Ohno-Iwashita, Y. & Ando, S. (1987) Eur. J. Biochem. 167, 425-430. [DOI] [PubMed] [Google Scholar]
- 12.Macey, M. G., Whiley, R. A., Miller, L. & Nagamune, H. (2001) Infect. Immun. 69, 6102-6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nagamune, H., Whiley, R. A., Goto, T., Inai, Y., Maeda, T., Hardie, J. M. & Kourai, H. (2000) J. Clin. Microbiol. 38, 220-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nagamune, H., Ohnishi, C., Katsuura, A., Fushitani, K., Whiley, R. A., Tsuji, A. & Matsuda, Y. (1996) Infect. Immun. 64, 3093-3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Giddings, K. S., Johnson, A. E. & Tweten, R. K. (2003) Proc. Natl. Acad. Sci. USA 100, 11315-11320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bradford, M. M. (1976) Anal. Biochem. 72, 248-254. [DOI] [PubMed] [Google Scholar]
- 17.Hoekstra, D., de Boer, T., Klappe, K. & Wilschut, J. (1984) Biochemistry 23, 5675-5681. [DOI] [PubMed] [Google Scholar]
- 18.Polekhina, G., Giddings, K. S., Tweten, R. K. & Parker, M. W. (2004) Acta Crystallogr. D 60, 347-349. [DOI] [PubMed] [Google Scholar]
- 19.Valeva, A., Walev, I., Pinkernell, M., Walker, B., Bayley, H., Palmer, M. & Bhakdi, S. (1997) Proc. Natl. Acad. Sci. USA 94, 11607-11611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu, R. & Sharom, F. J. (1998) Biochemistry 37, 6503-6512. [DOI] [PubMed] [Google Scholar]
- 21.Iwamoto, M., Ohno-Iwashita, Y. & Ando, S. (1990) Eur. J. Biochem. 194, 25-31. [DOI] [PubMed] [Google Scholar]
- 22.Tweten, R. K., Harris, R. W. & Sims, P. J. (1991) J. Biol. Chem. 266, 12449-12454. [PubMed] [Google Scholar]
- 23.Weis, S. & Palmer, M. (2001) Biochim. Biophys. Acta 1510, 292-299. [DOI] [PubMed] [Google Scholar]
- 24.Shepard, L. A., Shatursky, O., Johnson, A. E. & Tweten, R. K. (2000) Biochemistry 39, 10284-10293. [DOI] [PubMed] [Google Scholar]
- 25.Hotze, E. M., Wilson-Kubalek, E. M., Rossjohn, J., Parker, M. W., Johnson, A. E. & Tweten, R. K. (2001) J. Biol. Chem. 276, 8261-8268. [DOI] [PubMed] [Google Scholar]
- 26.Hotze, E. M., Heuck, A. P., Czajkowsky, D. M., Shao, Z., Johnson, A. E. & Tweten, R. K. (2002) J. Biol. Chem. 277, 11597-11605. [DOI] [PubMed] [Google Scholar]
- 27.Alouf, J. E. & Geoffroy, C. (1991) in Sourcebook of Bacterial Toxins, eds. Alouf, J. E. & Freer, J. J. (Academic, London), pp. 147-186.
- 28.Morris, G. M., Goodsell, D. S., Halliday, R. S., Huey, R., Hart, W. E., Belew, R. K. & Olson, A. J. (1998) J. Comp. Chem. 19, 1639-1662. [Google Scholar]
- 29.Saitou, N. & Nei, M. (1987) Mol. Biol. Evol. 4, 406-425. [DOI] [PubMed] [Google Scholar]
- 30.Kraulis, P. J. (1991) J. Appl. Crystallogr. 24, 946-950. [Google Scholar]
- 31.Merritt, E. A. & Bacon, D. J. (1997) Methods Enzymol. 277, 505-524. [DOI] [PubMed] [Google Scholar]
- 32.Esnouf, R. M. (1999) Acta Crystallogr. D 55, 938-940. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}