

Despite recent advances in treatment for patients with chronic lymphocytic leukemia (CLL), the disease remains incurable with approved therapies outside of allogeneic stem cell transplantation. Unfortunately, no single, targetable mutation or protein mediating pathogenesis of the disease has yet been identified, but next generation sequencing has identified multiple pathways that may initiate disease development[1–3]. Additionally, mutations or other genetic changes may arise to circumvent cell death and apoptosis following treatment. Therefore single agent treatment with targeted therapies may not be adequate, and therapies which simultaneously target multiple signaling pathways are actively being pursued.
Alvespimycin (17-Dimethylaminoethylamino-17-demethoxygeldanamycin, 17-DMAG) is a geldanamycin derivative that binds the N-terminal domain of the heat shock protein Hsp90, a molecular chaperone protein important in maintaining enzymatic activity of several client proteins involved in cellular signaling pathways[4]. This binding disrupts the interaction between Hsp90 and its client proteins, including kinases AKT, and IKK-α and IKK-β which through activation of the NF-κB pathway play a critical role in promoting proliferation and survival of CLL cells. In vitro data has demonstrated that alvespimycin is cytotoxic to primary CLL cells. Additionally, it exhibits minimal toxicity towards normal B-, T- and NK-cells[5].
In a prior phase I study of intravenous 17-DMAG in refractory acute myeloid leukemia and chronic myeloid leukemia[6,7], the maximum tolerated dose (MTD) was reached at 24 mg/m2. Two dose limiting toxicities (DLTs) were observed: acute myocardial infarction and elevation of troponin. Both clinical and biologic activity was observed; 2 AML patients achieved complete response with incomplete count recovery, and 1 patient had stable disease.
Based on our previous in vitro data in CLL, we designed a phase 1 study (NCT01126502) to determine the single agent MTD of 17-DMAG in patients with previously treated CLL/SLL as well as to assess pharmacokinetics (PK), preliminary efficacy, and feasibility of measuring pharmacodynamic markers including Hsp90 client proteins AKT, IKK-α and IKK-β. An institutional review board approved the study, and written informed consent was obtained from all patients. Patients ≥ 18 years with CLL/SLL relapsed after ≥ 1 nucleoside analogue containing therapy (or alternative regimen if contraindicated) and requiring therapy by International Workshop on CLL (IWCLL) guidelines[8] were enrolled. Included patients demonstrated serum creatinine ≤ 2.0 mg/dL or creatinine clearance ≥ 50 mL/min, AST/ALT ≤ 2.5 × upper limits of normal, QTc < 500 msec, LVEF > 40% by MUGA, and pulse oximetry > 88% with no symptomatic pulmonary disease. Only patients with an Eastern Cooperative Oncology Group (ECOG) performance status ≤ 2 were included.
Dose escalation proceeded according to a standard 3+3 design with each cohort enrolling 3 to 6 patients at each dose level. Treatment consisted of intravenous 17-DMAG at escalating dose levels (16, 20, 24 mg/m2) given on days 1, 4, 8 and 11 of 21 day cycles. All patients were treated for a minimum of 2 cycles in the absence of progressive disease. An expansion cohort of 12 patients was planned at the MTD but was subsequently abandoned when no objective responses were observed among patients treated in the dose escalation.
Cardiac rhythm disturbances were reported among patients previously treated with 17-DMAG resulting in the requirements of electrocardiograms to be performed at screening, immediately prior to treatment and within 30 minutes of treatment on Day 1 of Cycle 1. Therapy was held for QTc > 500 msec. Electrolytes including potassium, magnesium and calcium were checked and corrected within 24 hours prior to each administration of 17-DMAG. Prophylactic antiviral, antibacterial and antifungal medications and allopurinol were administered at the discretion of the treating physician.
Adverse events were defined and graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events version 4.0. Response assessment occurred after cycles 2 and 6, and was defined according to IWCLL criteria[8]. Patients in each cohort were independently evaluated for DLT during the first cycle of 17-DMAG. DLT was defined as any reversible grade 3–4 non-hematologic toxicity with the exception of transient electrolyte abnormalities that are not life-threatening, fatigue, alopecia, nausea, vomiting or diarrhea; grade 3–4 nausea, vomiting, or diarrhea that occurs despite appropriate prophylaxis treatment; irreversible grade 2 renal, pulmonary, cardiac or neurological toxicity; grade 3 QTc prolongation; hematologic toxicities including grade 4 thrombocytopenia or grade 4 neutropenia lasting 14 or more days in patients with baseline ANC > 1 × 109 and platelets > 75 × 1012; or grade 4 neutropenia associated with fever or infection in patients with ANC < 1 × 109.
Immunoblot analysis was performed as previously described[5]. Antibodies used included IKKα and IKKβ (Imgenex, San Diego, CA), MCL1 and BCL2 (Santa Cruz Biotechnology, Santa Cruz CA), AKT (Cell Signaling, Danvers, MA), and GAPDH (Merck-Millipore, Billerica, MA). Real-time PCR for NF-κB target genes (MCL1 and BCL2) was performed as previously described[5] using commercially available primers (Applied Biosystems, Foster City, CA). Whole blood samples for pharmacokinetic analysis were collected from all patients (see Supplementary Material).
A total of fifteen patients were treated. Baseline characteristics are summarized in Table I. No dose limiting toxicities were observed at any of the dose levels. The most common grade 3–4 adverse events were hematologic and included anemia (67%), neutropenia (53%), thrombocytopenia (20%) and hemolysis (7%). Other grade 3–4 adverse events included pain (20%), electrolyte abnormalities (20%), infection (14%), hypertension (14%), dyspnea (14%), hypoxia (7%), and upper gastrointestinal hemorrhage (7%). All were considered unrelated to treatment. Two patients died while on study, both with complications of progressive disease.
Table I.
Baseline Characteristics for Enrolled Patients (N = 15)
| Characteristic | N (%) |
|---|---|
| Age | |
| Median (range) | 59 (49–76) |
| Gender | |
| Male : Female | 10 : 5 |
| Prior therapies | |
| Median (range) | 5 (1–14) |
| Prior nucleoside analog | 13 (87) |
| Prior allogeneic stem cell transplant | 1 (7) |
| Cytogenetic abnormalities | |
| Normal | 1 (7) |
| Trisomy 12 | 1 (7) |
| Deletion 11q | 1 (7) |
| Deletions 17p | 2 (14) |
| Complex | 10 (67) |
| Treatment Delivery | |
| Dose Level 1 (16 mg/m2) | 5 |
| Dose Level 2 (20 mg/m2) | 3 |
| Dose Level 3 (24 mg/m2) | 7 |
Fourteen of the 15 patients were evaluable for response. Disease progressed in 7 patients prior to finishing 1 month, 4 patients prior to finishing 2 months, and 3 patients prior to finishing 3 months of therapy. Of the 3 patients considered to have stable disease, 2 were treated at dose level 2 and 1 was treated at dose level 3.
Immunoblot analysis of Hsp90 client proteins showed that BCL2 was decreased in 2 of the 3 cases treated at the 16 mg/m2 dose (Figure). We also determined transcript levels of NF-κB target genes BCL2 and MCL1 by real time RT-PCR. There was no change in these genes in 4 cases treated at the 20–24 mg/m2 doses (data not shown). These results suggest that we did not disrupt NF-κB signaling, and that the regulation of BCL2 is likely through direct client protein interaction with Hsp90 or through other factors regulating BCL2. In support of this hypothesis, we investigated proteins involved in NF-κB signaling and found that there was minimal effect on IKKα, IKKβ or AKT protein levels at any dose level. There was a decrease in these proteins in one case (patient 10) enrolled at the 24 mg/m2 dose at cycle 1 day 4; however, the NF-κB target MCL1 was not altered to a large extent in this patient. Together these results suggest we did not achieve complete inhibition of Hsp9 even at the higher dose range.
Figure 1.
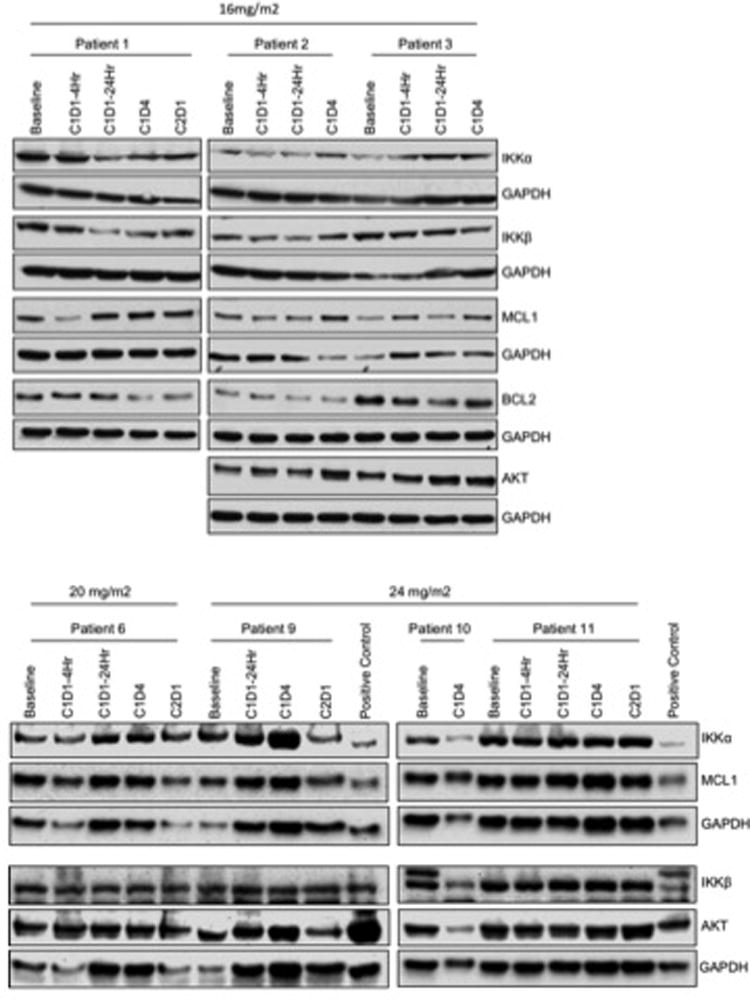
17-DMAG has a variable effect on Hsp90 client proteins and NF-κB targets. Immunoblot analysis for IKKα, IKKβ, BCL2, MCL1 and AKT in B cells isolated from CLL patients treated with 17-DMAG. The correlative data is shown for the 16, 20 and 24 mg/m2 dose levels for all patients with available sample. Blots are probed with GAPDH as a loading control.
While there were no objective responses observed at the highest (24 mg/m2) dose, there were also no DLTs. This suggests that the MTD was not achieved in this study, and a greater effect on client protein regulation may be observed at increased doses. Our in vitro data indicates effective regulation of client proteins at 1 μM for 24 hours in either primary cells or cell lines. PK data (see supplementary material) indicate median Cmax of approximately 600 nM at the 24 mg/m2 dose level which quickly decreases to a median of approximately 100 nM within 4 hours after initiation of treatment. These concentrations are on the low end of the dose range used in previously described in vitro studies, and are unlikely adequate to achieve sustained client protein inhibition. In a previous study in AML, similar results were noted; although increased Hsp70 was evident at the 24 mg/m2 dose, there was no change in p-AKT. We considered the possibility that free concentrations of 17-DMAG were reduced in human serum relative to those evaluated in vitro when using fetal bovine serum in cell culture. However, there was no detectable difference in potency of 17-DMAG when dosing in vitro between systems (data not shown).
Although inhibitors targeting BTK and other signaling pathways in CLL[9] have been effective, CLL that relapses on these treatments follows an aggressive course for which there are few treatment options. Recently, mutations in key proteins in this pathway have been identified that lead to acquired resistance to ibrutinib treatment[10]. Agents such as Hsp90 inhibitors, which simultaneously target several signaling pathways, could be effective in these refractory cases, particularly when the exact mechanism of resistance is unknown. In this regard, preclinical studies are currently ongoing in our lab with AT13387, a resorcinol derivative Hsp90 inhibitor. AT13387 has demonstrated a longer duration of action both in vitro and in xenograft animal models of non-small cell lung cancer[11]. AT13387 has also been shown to be effective in melanoma resistant to BRAF inhibitors[12], suggesting that it may be an attractive treatment option ibrutinib in resistant CLL. An available oral formulation would also facilitate continuous dosing schedules that could prolong client protein suppression in vivo.
Supplementary Material
{kind=link}
{kind=link}
Footnotes
Supplementary material is contained in a separate file.
References
- 1.Landau DA, Carter SL, Stojanov P, et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell. 2013;152:714–726. doi: 10.1016/j.cell.2013.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Puente XS, Pinyol M, Quesada V, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011;475:101–105. doi: 10.1038/nature10113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Quesada V, Conde L, Villamor N, et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet. 2012;44:47–52. doi: 10.1038/ng.1032. [DOI] [PubMed] [Google Scholar]
- 4.Goetz MP, Toft DO, Ames MM, Erlichman C. The Hsp90 chaperone complex as a novel target for cancer therapy. Ann Oncol. 2003;14:1169–1176. doi: 10.1093/annonc/mdg316. [DOI] [PubMed] [Google Scholar]
- 5.Hertlein E, Wagner AJ, Jones J, et al. 17-DMAG targets the nuclear factor-kappaB family of proteins to induce apoptosis in chronic lymphocytic leukemia: clinical implications of HSP90 inhibition. Blood. 2010;116:45–53. doi: 10.1182/blood-2010-01-263756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lancet JGI, Baer M, et al. Phase I, pharmacokinetic (PK) and pharmacodynamic (PD) study of the Hsp-90 inhibitor, KOS-1022 (17-DMAG), in patients with refractory hematological malignancies. Proc Amer Soc Clin Oncol. 2006;24:99s. [Google Scholar]
- 7.Lancet JE, Gojo I, Burton M, et al. Phase I study of the heat shock protein 90 inhibitor alvespimycin (KOS-1022, 17-DMAG) administered intravenously twice weekly to patients with acute myeloid leukemia. Leukemia. 2010;24:699–705. doi: 10.1038/leu.2009.292. [DOI] [PubMed] [Google Scholar]
- 8.Hallek MCB, Catovsky D, Caligaris-Cappio F, Dighiero G, Dohner H, Hillmen P, Keating MJ, Montserrat E, Rai KR, et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: A report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood. 2008;111:5446–5456. doi: 10.1182/blood-2007-06-093906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Byrd JC, Furman RR, Coutre SE, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369:32–42. doi: 10.1056/NEJMoa1215637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Woyach JA, Furman RR, Liu TM, et al. Resistance mechanisms for the Bruton's tyrosine kinase inhibitor ibrutinib. N Engl J Med. 2014;370:2286–2294. doi: 10.1056/NEJMoa1400029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Graham B, Curry J, Smyth T, et al. The heat shock protein 90 inhibitor, AT13387, displays a long duration of action in vitro and in vivo in non-small cell lung cancer. Cancer Sci. 2012;103:522–527. doi: 10.1111/j.1349-7006.2011.02191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smyth T, Paraiso KH, Hearn K, et al. Inhibition of HSP90 by AT13387 Delays the Emergence of Resistance to BRAF Inhibitors and Overcomes Resistance to dual BRAF and MEK Inhibition in Melanoma Models. Mol Cancer Ther. 2014 doi: 10.1158/1535-7163.MCT-14-0452. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.