

Abstract
Purpose:
The advent of genomic diagnostic technologies such as next-generation sequencing has recently enabled the use of genomic information to guide targeted treatment in patients with cancer, an approach known as precision medicine. However, clinical outcomes, including survival and the cost of health care associated with precision cancer medicine, have been challenging to measure and remain largely unreported.
Patients and Methods:
We conducted a matched cohort study of 72 patients with metastatic cancer of diverse subtypes in the setting of a large, integrated health care delivery system. We analyzed the outcomes of 36 patients who received genomic testing and targeted therapy (precision cancer medicine) between July 1, 2013, and January 31, 2015, compared with 36 historical control patients who received standard chemotherapy (n = 29) or best supportive care (n = 7).
Results:
The average progression-free survival was 22.9 weeks for the precision medicine group and 12.0 weeks for the control group (P = .002) with a hazard ratio of 0.47 (95% CI, 0.29 to 0.75) when matching on age, sex, histologic diagnosis, and previous lines of treatment. In a subset analysis of patients who received all care within the Intermountain Healthcare system (n = 44), per patient charges per week were $4,665 in the precision treatment group and $5,000 in the control group (P = .126).
Conclusion:
These findings suggest that precision cancer medicine may improve survival for patients with refractory cancer without increasing health care costs. Although the results of this study warrant further validation, this precision medicine approach may be a viable option for patients with advanced cancer.
INTRODUCTION
Precision cancer medicine involves the detection of tumor-specific somatic mutations, including insertions/deletions (indels), single nucleotide variants, translocations, and copy number alterations, followed by treatment with therapeutics that specifically target identified actionable alterations.1-7 This approach using precision medicine has largely been hampered by the high cost of testing and the extended turnaround times associated with in-depth genomic diagnostic analysis. However, advances in genomic technologies, including next-generation sequencing (NGS) and droplet digital polymerase chain reaction, have now rendered extended genomic analyses of human malignancies technologically and financially feasible for use in the clinic.8-10
Concomitant with these advances in genomic technologies, there have been significant advances in two overlapping areas of cancer research, each with major clinical ramifications; the first is a greater understanding of the underlying genomic alterations and molecular mechanisms of cancer, and the second is development of novel therapeutic agents and biomolecules that exploit specific genomic aberrations in tumors.11-16 These advances are the underpinnings of the new precision cancer medicine clinical paradigm.17
In the precision medicine approach to cancer, the physician and patient use the identification of specific genetic aberrations that affect cancer-related genes to better inform treatment decisions. The underlying rationale is that this personalized diagnostic approach will lead to a clinical recommendation for targeted cancer therapies that will ultimately result in improved clinical outcomes. This approach has been successfully applied to single tumor types with predetermined genomic variants such as EGFR-positive non–small-cell lung cancer (NSCLC)18-20 and BRAF-positive melanoma,14,21,22 whereas previous studies revealed that precision medicine can improve survival in a single cancer type.23 Earlier studies indicated that targeted therapies given to patients whose tumors harbored specific alterations may improve outcomes as measured by tumor responsiveness.24 However, the impact of precision medicine compared with standard therapies on survival and the effect of implementing sophisticated diagnostic technologies such as NGS on the costs of cancer care, remain unknown.
Our precision cancer medicine program was clinically established in a single region of the Intermountain Healthcare delivery system. Patients with advanced, refractory cancer were referred to the precision medicine clinic where they received genomic testing, an in-depth interpretation of the genomic results from a multi-institutional molecular tumor board, and a list of treatment options for implementation at the discretion of the treating oncologist.
We report here the progression-free survival (PFS), total costs, and cost per week of survival associated with the initial cohort of patients who received targeted treatment in the precision cancer medicine program compared with control patients who received standard chemotherapy or best supportive care.
PATIENTS AND METHODS
The Intermountain Healthcare Institutional Review Board approved this study, and all living participants provided written informed consent before enrollment. The Board granted a waiver of consent for decedents.
Study Design
Research objectives
The objective of this retrospective observational study was to compare the outcomes of patients with cancer who were treated with precision cancer targeted therapies with a historical control cohort treated with a nontargeted approach.
Research subjects
Male and female adults with measurable recurrent/metastatic solid tumors for whom standard first-line treatments (proposed by the National Comprehensive Cancer Network [NCCN] guidelines) failed were included in this study. Other inclusion requirements were Eastern Cooperative Oncology Group performance status of 0, 1, or 2 and adequate renal, hepatic, and bone marrow function. Patients who had only brain metastases or whose brain metastases had not been controlled for > 3 months and patients who were participating in a clinical trial with an experimental drug were excluded. Pregnant or breastfeeding women also were excluded.
All patients in the precision medicine group had tumor molecular abnormalities for which the Intermountain Healthcare Multi-Institutional Molecular Tumor Board (MTB) provided an interpretation. Actionable mutations were defined as variants that had been validated in the peer-reviewed literature and for which a targeted therapy was available. The MTB selected treatment options only for actionable mutations for which there was published clinical or preclinical evidence. Patients included in the control group received standard-of-care genomic testing only, without interpretation by the MTB or molecularly targeted therapy beyond the relevant standard of care.
Sample size
A simulation power analysis was performed for a Cox proportional hazards model with 100,000 simulations. In determining the sample size that we used for study, we considered the methodology of Tsimberidou et al.24
Selection of end points
The primary end point was PFS according to radiographic determination of tumor progression and Response Evaluation Criteria in Solid Tumors (RECIST) 1.1. Tumor measurements by computed tomography imaging were obtained before treatment was initiated and every 8 weeks thereafter. Secondary end points included health care–associated cost of care.
Blinding
Clinician researchers were blinded to the identities of those in the control cohort. Cancer registrars selected the control cohort and provided data about the controls to the study statistician (A.M.B.).
Statistical Methods
A Cox proportional hazards model was fit for PFS. Treatment, sex, age, cancer type, and three or more lines of treatment were included in this model. A full likelihood ratio versus a reduced likelihood ratio test was performed comparing the full model to a reduced model containing only treatment. This test showed that the reduced model adequately fits the data, and only treatment is needed in the model (P = .508). Basic demographics as well as two-sample t tests and linear regression models were created to investigate the cost of therapy. Collecting demographic variables and using the Cox model controlled for confounding factors.
Cost Analysis
In calculating patient costs, a payer perspective was adopted. Patient costs were estimated by using standard Intermountain Healthcare payer charges. Only charges incurred between the treatment line start and end dates were included in the total charge estimates for each patient. Patient costs included all amounts for patient treatment, toxicity, patient sequencing, and targeted drug therapy. Treatment costs for both targeted and control patients included all facility-based and clinic-based charges associated with treatment, including chemotherapy, drug, radiology, and laboratory costs. Palliative care costs were limited to daily reimbursement charge rates determined by the Centers for Medicare and Medicaid Services. Toxicity costs included all patient charges associated with treating the adverse effects resulting from treatment. Sequencing costs for target patients were obtained from the test provider and were based upon estimated payer reimbursement rates. Drug cost data were drawn from local specialty pharmacies and drug manufacturers and were based upon estimated payer reimbursement rates, including estimates of any out-of-pocket costs for the patient. A discount rate was not applied to costs to adjust for the time value of money. Given the limited availability of quality-of-life data for control patients, PFS weeks were not quality adjusted. The mean per patient cost per PFS week was calculated by adding the costs per PFS week for each patient and dividing by the total number of patients. Statistical comparisons of costs between precision medicine and control groups were performed by using a two-sided Wilcoxon rank sum test.
Molecular Diagnostic Testing
All samples analyzed were either formalin-fixed paraffin-embedded (FFPE) or fresh. Patient samples were analyzed in a Clinical Laboratory Improvement Amendments–certified laboratory. Genomic analysis included NGS-based oligoselective exon sequencing of 96 cancer-related genes: ABL1, AKT1, ALK, APC, ATM, AURKA, AURKB, AXL, BCL2, BRAF, BRCA1, BRCA2, CCND1, CDH1, CDK2, CDK4, CDK5, CDK6, CDK8, CDK9, CDK12, CDKN2A, CEBPA, CSF1R, CTNNB1, CYP2D6, DDR2, DNMT3A, DPYD, EGFR, EPCAM, ERBB2, ERBB3, ERBB4, ERCC1, ERCC2, ERCC3, ERCC5, ERCC6, EZH2, ESR1, FGFR1, FGFR2, FGFR3, FGFR4, FLT3, GNA11, GNAQ, GNAS, HNF1A, HRAS, IDH1, IDH2, JAK2, JAK3, KDR, KIT, KRAS, MAP2K1, MAP2K2, MAPK1, MET, MLH1, MPL, MRE11, MSH2, MTOR, MSH6, MYC, MUTYH, NOTCH1, NPM1, NRAS, PARP1, PARP2, PDGFRA, PIK3CA, PMS2, PTCH1, PTCH2, PTEN, PTPN11, RB1, RET, RUNX1, SMAD4, SMARCB1, SMO, SRC, STK11, TET2, TP53, UGT1A1, VEGFA, VHL, and WT1. Sample tumor concentration of at least 40% was verified by board-certified anatomic pathologists. Samples were extracted by using the ReliaPrep FFPE gDNA miniprep kit (Promega, Madison, WI) for FFPE samples or the Puregene blood core Kit A (Qiagen, Santa Clarita, CA) for extracting fresh samples. DNA shearing to an average of 500 bp lengths was performed by using an M220 ultrasonicator (Covaris, Woburn, MA). Additional sample preparation, library preparation, and NGS were performed by using the TOMAseq kit (adaptor, extension, and capture sets; TOMA Biosciences, Foster City, CA) according to the manufacturer’s protocol and instructions. Library quantification was performed with a Bio-Rad q200 droplet digital polymerase chain reaction analyzer (Bio-Rad, Hercules, CA). Sequencing was performed on the MiSeq platform (Illumina, San Diego, CA). Data analysis, including curation, interpretation, alignment, and quality checks were implemented by using legacy algorithms, and the variant calling was done using Freebayes. Patient samples were compared with a reference genome, and genetic variants, including copy number alterations, point mutations, frameshift mutations, translocations, and single nucleotide polymorphisms, were identified and reported. Some samples were initially tested by an external laboratory (Caris Biosciences, Foundation Medicine, or TOMA Biosciences), and those with sufficient quantity were subsequently reanalyzed by using the 96-gene panel described previously.
RESULTS
After obtaining informed consent, we evaluated 61 patients who had an actionable mutation and who subsequently received targeted therapy on the basis of the actionable mutation (precision cancer medicine), defined as known variants validated in peer-reviewed literature for which a targeted therapy was available. Given the heterogeneous nature of the treatment cohort in terms of tumor type, age, and sex, we sought to compare their outcomes with the outcomes of control patients who were matched to treatment patients according to tumor type, age, sex, and number of previous lines of treatment. We searched our institutional enterprise data warehouse to identify historical control patients who had received standard therapy between July 2010 and January 2015 and who could be matched according to age, sex, diagnosis, and number of previous lines of treatment with patients who received precision medicine (Fig 1). Of the 61 patients with an actionable mutation who had received precision medicine, 36 had an institutional historical match (25 patients did not have a historical match). We gathered outcomes data from those 36 patients who received precision medicine and the 36 matched patients who received standard therapy, including standard molecular testing, for a total of 72 patients (Fig 1). Table 1 lists patients’ demographic characteristics by treatment type. No significant differences were found between precision medicine and standard therapy groups except for race/ethnicity, which was 100% (n = 36) non-Hispanic white in the precision medicine arm and was 83.3% (n = 30) non-Hispanic white in the control arm, with 2.8% (n = 1) non-Hispanic black, 11.1% (n = 4) white and nonwhite Hispanic, and 2.8% (n = 1) other race/ethnicity. Mean age at time of treatment was 67.8 years for the precision medicine group and 67.0 years for the control group (P = .748). Both groups were 61% male (n = 44). Four precision medicine patients were matched at a later line than their controls resulting in average lines of treatment of 3.1 for the precision medicine group and 2.9 for the control group (P = .168). The cancer types were identically matched for both groups and were comprised of patients with diverse solid tumor types encompassing 10 different histologically distinct cancers. NSCLC was the largest subtype (n = 11; 31%) in both cohorts (Table 1).
FIG 1.

Schematic of the study design delineates the patient population from which the study was conducted. PFS, progression-free survival.
Table 1.
Patient Characteristics

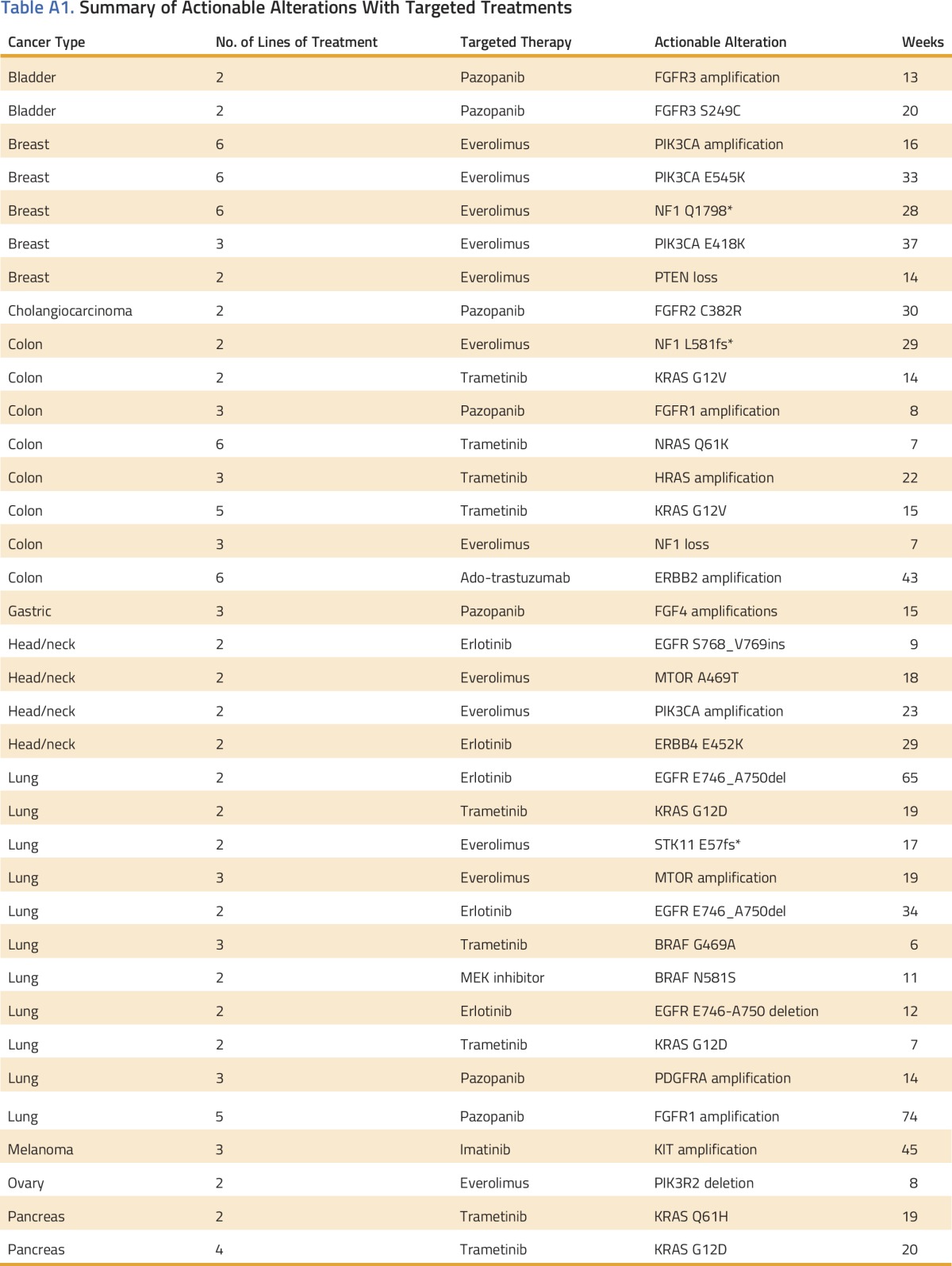
The actionable variant and targeted therapy for each patient in the precision medicine cohort is listed in Appendix Table A1 (online only). Patients in the historical control cohort had appropriate standard molecular testing according to NCCN guidelines at the time of their diagnosis. Three patients in the precision medicine cohort with NSCLC were found to harbor validated EGFR mutations, and they received erlotinib (Table A1); the EGFR mutations were not identified at the time of initial diagnosis. Five patients with breast cancer (with hormone-negative [triple negative, n = 2] or hormone refractory [n = 3] disease) who received a targeted therapy after MTB interpretation, were included in the analysis.
The protocol-specified primary end point of PFS was significantly prolonged in the precision medicine group (Fig 2) compared with the control group (mean PFS, 22.9 v 12.0 weeks, respectively; P = .002). More specifically, precision medicine was associated with a 53% decreased risk of progression (adjusted hazard ratio, 0.47; 95% CI, 0.29 to 0.75; P = .002) when adjusted for age, sex, histologic diagnosis, and number of previous lines of treatment. At the time of study conclusion, four patients (11%) in the precision medicine arm had not yet progressed (Fig 2). A sensitivity analysis suggested that a difference in patient performance status between the two cohorts was unlikely to account for the difference in PFS (Appendix Fig A1, online only). The cohort of 25 patients who did not have a historical institutional match also demonstrated a prolonged PFS compared with the control cohort (19.3 weeks; P = .026).
FIG 2.

The progression-free survival of patients in the standard and precision medicine treatment cohorts were measured and compared over weeks. The fraction of patients surviving without disease progression is plotted against the number of progression-free weeks.
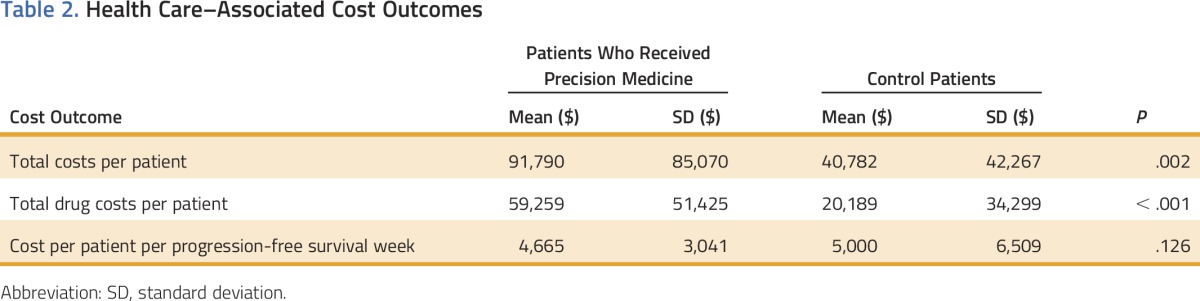
To determine the costs associated with the two treatment approaches, we performed a health care–related cost analysis. We evaluated all of the patients from each cohort and identified 22 matched patient pairs who had received all of their care within the Intermountain Healthcare system and therefore had complete cost data available (Fig 1). An analysis of PFS in this subset of 22 matched patient pairs revealed the same statistically significant PFS improvement in the precision medicine cohort compared with the standard therapy cohort (21.4 v 11.0 weeks; P = .004; Table 2). As expected, total costs per patient during the study period were higher for the precision medicine treatment group than the control group ($91,790 v $40,782 per patient; P = .002; Table 2). Drug costs were the main factor contributing to the higher cost for precision medicine patients ($59,259 v $20,189 per patient; P < .001; Table 2). Patients in the precision medicine group had longer survival times resulting in lower patient costs per PFS week than the control group ($4,665 v $5,000 per week; P = .126) but did not reach a level of significance (Table 2).
Table 2.
Health Care–Associated Cost Outcomes
DISCUSSION
The findings presented here examine survival and the health care–related costs associated with precision cancer medicine in a retrospective cohort of patients. The results suggest a survival benefit for patients who received precision cancer medicine treatment compared with patients who received standard therapy, although the potential contribution of the tumor’s molecular composition to the resulting prolongation in PFS could not be assessed, given that the control cohort tumors did not undergo genomic analysis. A subset analysis of patients who received all care within the Intermountain Healthcare system determined that the costs associated with each cohort were not associated with a per week increase in health care costs. The simultaneous improvement in PFS, without increasing per week costs, suggests that a precision medicine approach may be a feasible option in patients with refractory cancer.
In a previous study, Tsimberidou et al24 evaluated the outcomes associated with a phase I personalized medicine program and found that patients who received therapy on the basis of specific molecular alterations, independent of tumor type, experienced improved survival. Similarly, Kris et al23 reported improved survival in patients with lung cancer when selecting therapies on the basis of oncogenic driver mutations. Although this study, along with those reported by Tsimberidou et al and Kris et al were not randomized trials, they nevertheless suggest that molecularly guided therapies may improve survival in patients with advanced refractory cancer.
Although the improvement in PFS identified in this study partially results from identifying and treating previously known molecularly distinct cancer subtypes such as EGFR-positive lung cancer (n = 3 in the targeted treatment cohort), the majority (n = 33) of cases resulted from targeting distinct molecular alterations in diverse tumor types, regardless of histologic subtype. Many of the durable responses were the result of identifying well-known molecular alterations from a single cancer subtype, such as an activating c-KIT mutation commonly found in gastrointestinal stromal tumor, and targeting that alteration in a histologically different cancer such as melanoma. Similar responses were seen in FGFR1-amplified squamous cell lung cancer, FGFR2-mutant cholangiocarcinoma, and MEK1-activated NSCLC. Although similar responses in some tumor subtypes with these alterations have been reported previously as single case reports or case series,25-27 the findings presented here suggest that genomic profiling of diverse tumor subtypes followed by molecularly targeted treatment may improve outcomes.
The majority of patients in the control cohort were historical, having received their treatment within the same institution (Intermountain Healthcare) within the previous 5 years. The retrospective nature of the control cohort raises the possibility of bias in the analysis. Controlling for the number of previous lines of treatment that a patient received before enrollment in the study helps mitigate the risk of bias but does not completely eliminate the possibility. A statistical sensitivity analysis included in the Appendix (Fig A1, online only) affirms that a difference in performance status between the two cohorts was unlikely to account for the difference in PFS.
The cost associated with implementing novel medical treatment approaches has been historically difficult to measure because of limited availability of data and limitations on data sharing.17 The cost associated with precision cancer medicine remains a primary question for both payers and providers alike. We attempted to address that question by analyzing the costs associated with both study cohorts. The overall costs of treatment, including cost of testing and cost of drug, were higher in the precision medicine cohort, as might be expected in a cohort that experiences an increased survival time. Evaluating the two cohorts on a cost-per-week basis revealed that the two groups were not statistically different, and drug-related costs remained the primary driver of charges for both cohorts. In an era of increasing health care costs and static resources, measuring the value of treatment becomes critical to sustainability. Although the costs of large-scale genomic testing have historically precluded widespread adoption of precision medicine, the equivalence in cost per PFS week between the two cohorts suggests that widespread adoption of precision cancer medicine may no longer be constrained by economic metrics.
A major question surrounding the implementation of precision cancer medicine is its relevance in the community setting in which nearly 85% of patients with cancer treated in the United States receive their care. Developing a model for the clinical implementation of precision medicine in a community setting, therefore, is a necessary step in determining whether this approach warrants further consideration as a viable option for the vast majority of patients with advanced cancer. The survival and cost outcomes reported here were generated entirely in an integrated health care delivery system with patients receiving treatment in a community cancer center and suggest that precision cancer medicine can be applied to the community setting with measurable patient benefit.
ACKNOWLEDGMENT
The authors recognize and appreciate the patients and families who contributed to this study. We appreciate the contributions of Terri Kane, RN, and William Sause, MD, both from Intermountain Healthcare, for their contributions to the study. Supported by Grant No. 5K08CA166512-04 from the National Cancer Institute, National Institutes of Health (L.D.N.), the Conquer Cancer Foundation Young Investigator Award (L.D.N.), the Gastric Cancer Foundation, and the Carl Kawaja Foundation. D.S.H. and L.D.N. had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Appendix
FIG A1.

Statistical sensitivity analysis of performance status (PS) on progression-free survival (PFS). Hazard ratios and 95% CIs were calculated for conditions in which the difference in performance status between precision medicine and control cohorts was large (delta = –2.5) or small (delta = 0). Gamma (0 to 3) represents the relative hazard of death for unmeasured performance status. Hazard ratios in green represent conditions in which targeted treatment causes an increase in PFS. Hazard ratios in black represent conditions in which targeted treatment neither increases nor decreases PFS significantly. Hazard ratios in red represent conditions in which targeted treatment causes a decrease in PFS.
Table A1.
Summary of Actionable Alterations With Targeted Treatments
AUTHOR CONTRIBUTIONS
Conception and design: Derrick S. Haslem, James M. Ford, Lincoln D. Nadauld
Administrative support: Rajendu Srivastava
Provision of study materials or patients: David L. Loughmiller
Collection and assembly of data: Derrick S. Haslem, S. Burke Van Norman, Gail Fulde, Andrew J. Knighton, Tom Belnap, Heather Gilbert, Brian P. Tudor, Karen Lin, Gary R. Stone, Lincoln D. Nadauld
Data analysis and interpretation: Derrick S. Haslem, Andrew J. Knighton, Tom Belnap, Allison M. Butler, Sharanya Raghunath, David Newman, David L. Loughmiller, Pravin J. Mishra, Rajendu Srivastava, Lincoln D. Nadauld
Manuscript writing: All authors
Final approval of manuscript: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
A Retrospective Analysis of Precision Medicine Outcomes in Patients With Advanced Cancer Reveals Improved Progression-Free Survival Without Increased Health Care Costs
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/journal/jop/site/misc/ifc.xhtml.
Derrick S. Haslem
No relationship to disclose
S. Burke Van Norman
No relationship to disclose
Gail Fulde
Stock or Other Ownership: Amgen
Andrew J. Knighton
Employment: UnitedHealth Group
Stock or Other Ownership: UnitedHealth Group
Tom Belnap
No relationship to disclose
Allison M. Butler
No relationship to disclose
Sharanya Raghunath
No relationship to disclose
David Newman
No relationship to disclose
Heather Gilbert
No relationship to disclose
Brian P. Tudor
No relationship to disclose
Karen Lin
No relationship to disclose
Gary R. Stone
No relationship to disclose
David L. Loughmiller
No relationship to disclose
Pravin J. Mishra
No relationship to disclose
Rajendu Srivastava
No relationship to disclose
James M. Ford
Research Funding: Myriad Genetics, Invitae, Varian Medical Systems, Natera
Lincoln D. Nadauld
Stock or Other Ownership: TOMA Biosciences
REFERENCES
- 1.Roychowdhury S, Iyer MK, Robinson DR, et al. Personalized oncology through integrative high-throughput sequencing: A pilot study. Sci Transl Med. 2011;3:111ra121. doi: 10.1126/scitranslmed.3003161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cancer Genome Atlas Research Network Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543–550. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cancer Genome Atlas Network Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fraumeni JF., Jr Constitutional disorders of man predisposing to leukemia and lymphoma. Natl Cancer Inst Monogr. 1969;32:221–232. [PubMed] [Google Scholar]
- 5.Garraway LA, Verweij J, Ballman KV. Precision oncology: An overview. J Clin Oncol. 2013;31:1803–1805. doi: 10.1200/JCO.2013.49.4799. [DOI] [PubMed] [Google Scholar]
- 6.Nadauld LD, Ford JM. Molecular profiling of gastric cancer: Toward personalized cancer medicine. J Clin Oncol. 2013;31:838–839. doi: 10.1200/JCO.2012.47.1714. [DOI] [PubMed] [Google Scholar]
- 7.Forbes SA, Bindal N, Bamford S, et al. COSMIC: Mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011;39:D945–D950. doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Margulies M, Egholm M, Altman WE, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bentley DR, Balasubramanian S, Swerdlow HP, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ley TJ, Mardis ER, Ding L, et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature. 2008;456:66–72. doi: 10.1038/nature07485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lander ES, Linton LM, Birren B, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 12.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 13.Brose MS, Volpe P, Feldman M, et al. BRAF and RAS mutations in human lung cancer and melanoma. Cancer Res. 2002;62:6997–7000. [PubMed] [Google Scholar]
- 14.Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 15.Slamon DJ, Clark GM, Wong SG, et al. Human breast cancer: Correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177–182. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- 16.Tommasi S, Paradiso A, Mangia A, et al. Biological correlation between HER-2/neu and proliferative activity in human breast cancer. Anticancer Res. 1991;11:1395–1400. [PubMed] [Google Scholar]
- 17.Rubin MA. Health: Make precision medicine work for cancer care. Nature. 2015;520:290–291. doi: 10.1038/520290a. [DOI] [PubMed] [Google Scholar]
- 18.Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 19.Paez JG, Jänne PA, Lee JC, et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 20.Pao W, Miller V, Zakowski M, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci USA. 2004;101:13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hodis E, Watson IR, Kryukov GV, et al. A landscape of driver mutations in melanoma. Cell. 2012;150:251–263. doi: 10.1016/j.cell.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Curtin JA, Fridlyand J, Kageshita T, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005;353:2135–2147. doi: 10.1056/NEJMoa050092. [DOI] [PubMed] [Google Scholar]
- 23.Kris MG, Johnson BE, Berry LD, et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA. 2014;311:1998–2006. doi: 10.1001/jama.2014.3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tsimberidou AM, Iskander NG, Hong DS, et al: Personalized medicine in a phase I clinical trials program: The MD Anderson Cancer Center initiative. Clin Cancer Res 18:6373-6383, 2012. [DOI] [PMC free article] [PubMed]
- 25.Borad MJ, Champion MD, Egan JB, et al. Integrated genomic characterization reveals novel, therapeutically relevant drug targets in FGFR and EGFR pathways in sporadic intrahepatic cholangiocarcinoma. PLoS Genet. 2014;10:e1004135. doi: 10.1371/journal.pgen.1004135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liao RG, Jung J, Tchaicha J, et al. Inhibitor-sensitive FGFR2 and FGFR3 mutations in lung squamous cell carcinoma. Cancer Res. 2013;73:5195–5205. doi: 10.1158/0008-5472.CAN-12-3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Conley AP, Koplin S, Caracciollo JT, et al. Dramatic response to pazopanib in a patient with metastatic malignant granular cell tumor. J Clin Oncol. 2014;32:e107–e110. doi: 10.1200/JCO.2012.47.1078. [DOI] [PubMed] [Google Scholar]