

Abstract
We present a simple theory that uses thermodynamic parameters to predict the probability that a protein retains the wild-type structure after one or more random amino acid substitutions. Our theory predicts that for large numbers of substitutions the probability that a protein retains its structure will decline exponentially with the number of substitutions, with the severity of this decline determined by properties of the structure. Our theory also predicts that a protein can gain extra robustness to the first few substitutions by increasing its thermodynamic stability. We validate our theory with simulations on lattice protein models and by showing that it quantitatively predicts previously published experimental measurements on subtilisin and our own measurements on variants of TEM1 β-lactamase. Our work unifies observations about the clustering of functional proteins in sequence space, and provides a basis for interpreting the response of proteins to substitutions in protein engineering applications.
Keywords: mutational robustness, protein evolution, protein stability, directed evolution, β-lactamase
The ability to predict a protein's tolerance to amino acid substitutions is of fundamental importance in understanding natural protein evolution, developing protein engineering strategies, and understanding the basis of genetic diseases. Computational and experimental studies have demonstrated that both protein stability and structure affect a protein's tolerance to substitutions. Simulations have shown that more stable proteins have a higher fraction of folded mutants (1–4) and that some structures are encoded by more sequences than others (5–7). Experiments have demonstrated that proteins can be extremely tolerant to single substitutions; for example, 84% of single-residue mutants of T4 lysozyme (8) and 65% of single-residue mutants of lac repressor (9) were scored as functional. For multiple substitutions, the fraction of functional proteins decreases roughly exponentially with the number of substitutions, although the severity of this decline varies among proteins (10–12). Protein mutagenesis experiments have also underscored the contribution of protein stability to mutational tolerance by finding “global suppressor” substitutions that buffer a protein against otherwise deleterious substitutions by increasing its stability (13, 14).
We unify these diverse experimental and computational results into a simple framework for predicting a protein's tolerance to substitutions. A fundamental measure of this tolerance is the fraction of proteins retaining the wild-type structure after a single random substitution, often called the neutrality (15). We extend this concept to multiple substitutions by defining the m-neutrality as the fraction of proteins that fold to the wild-type structure among all sequences that differ from the wild-type sequence at m residues. Because mutants that fail to fold also generally fail to function, the m-neutrality provides an upper bound to the fraction of proteins with m substitutions that retain biochemical function. We show that a protein's m-neutrality can be accurately predicted from measurable thermodynamic parameters, and that these predictions capture the contributions of both stability and structure to determining a protein's tolerance to substitutions.
Methods
Lattice Protein Model. We performed simulations with lattice proteins (16) of length L = 20 monomers of 20 types corresponding to the natural amino acids. We folded the proteins on a two-dimensional lattice, allowing them to occupy any of the 41,889,578 possible compact or noncompact conformations. The energy of a conformation 𝒞 is the sum of the nonbonded nearest-neighbor interactions
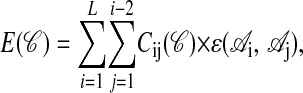 |
where 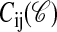 is one if residues i and j are nearest neighbors in conformation 𝒞 and zero otherwise, and
is one if residues i and j are nearest neighbors in conformation 𝒞 and zero otherwise, and 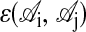 is the interaction energy between residue types
is the interaction energy between residue types 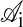 and
and 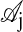 , given in table 5 of ref. 17.
, given in table 5 of ref. 17.
The primary advantage of using lattice proteins is that we can exactly compute the stability of a conformation 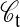 as
as
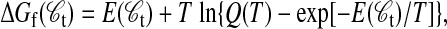 |
where Q(T) is the partition sum.
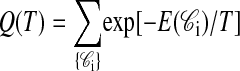 |
over all conformations, made tractable by noting that there are only 910,972 unique contact sets. All simulations were performed at a reduced temperature of T = 1.0.
TEM1 β-Lactamase Mutant Libraries. To examine the effects of mutations on the retention of protein function, we constructed mutant libraries of wild-type and the thermostable M182T variant of TEM1 β-lactamase. The 861-bp genes (a kind gift from Brian Shoichet, Northwestern University School of Medicine, Chicago; ref. 18) were subcloned into the pMON:1A2 plasmid (19) with SacI and HindIII by using PCR primers 5′-GCGGCGGAGCTCATGAGTATTCAACATTTCCGTGTCGC-3′ and 5′-GCGGCGAAGCTTTTACCAATGCTTAATCAGTGAGGCAC-3′ (restriction sites are underlined). We first created a control unmutated library by cutting the gene directly from the plasmid. This unmutated gene was used as the template for a round of error-prone PCR with 100-μl reactions containing 3 ng of template, 0.5 μM each of the above primers, 7 mM MgCl2, 75 μM MnCl2, 200 μM dATP and dGTP, 500 μM dTTP and dCTP, 1× Applied Biosystems PCR buffer without MgCl2, and 5 units of Applied Biosystems TaqDNA polymerase. The PCR conditions were 95°C for 5 min, and then 14 cycles of 30 s each at 95°C, 50°C, and 72°C. The product from this PCR was digested with SacI/HindIII and gel purified, and then used as the template for another identical round of error-prone PCR. This process was repeated to create five libraries with increasing numbers of mutations, which we labeled EP-0 (for the unmutated control) to EP-5 (for the product of the fifth round of error-prone PCR). We quantified the number of doublings for each round by running PCR product versus a known standard on an agarose gel, and found that our protocol consistently yielded 10 doublings.
To measure the fraction of genes in the mutant libraries that still encoded functional proteins, we ligated the genes into the pMON:1A2 plasmid with T4 Quick DNA Ligase in 20-μl reactions containing 50 ng each of gene and plasmid, and then transformed 5 μl of the ligation reactions into 50 μl of XL1-Blue Supercompetent cells from Stratagene. The transformed cells were plated on LB agar plates containing 10 μg/ml kanamycin (selective only for plasmid) and on LB agar plates containing 10 μg/ml kanamycin and 20 μg/ml ampicillin (selective for both plasmid and active TEM1 gene) at a density that gave 100–300 colonies per unselected plate. The fractions functional were computed as the average of at least five pairs of selected/unselected plates, and are shown in Table 1.
Table 1. TEM1 mutant library measurements.
| Round | 〈mnt〉 | 〈maa〉 | WT | M182T |
|---|---|---|---|---|
| 0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.76 ± 0.03 | 0.74 ± 0.04 |
| 1 | 1.3 ± 0.2 | 0.9 ± 0.1 | 0.59 ± 0.03 | 0.68 ± 0.03 |
| 2 | 2.6 ± 0.3 | 1.8 ± 0.2 | 0.47 ± 0.03 | 0.54 ± 0.02 |
| 3 | 3.9 ± 0.4 | 2.7 ± 0.2 | 0.28 ± 0.02 | 0.45 ± 0.04 |
| 4 | 5.2 ± 0.4 | 3.6 ± 0.3 | 0.18 ± 0.01 | 0.28 ± 0.01 |
| 5 | 6.5 ± 0.5 | 4.5 ± 0.4 | 0.13 ± 0.01 | 0.20 ± 0.02 |
Measured fractions of functional proteins in mutant libraries of wild-type (WT) and the thermostable M182T variant (M182T) of TEM1 β-lactamase. The table shows the number of rounds of error-prone PCR, the average number of nucleotide mutations per gene, and the fractions of mutated genes that confer ampicillin resistance in Escherichia coli. Values are shown ± their standard errors.
The mutation frequency in the round-five library was determined by sequencing the first 570 bp of 20 genes each from the unselected wild-type and M182T plates with the sequencing primer 5′-GGTCGATGTTTGATGTTATGGAGC-3′. The wild-type and M182T genes were mutated under identical conditions, and the sequencing found the same nucleotide mutation frequencies for both (0.77 ± 0.08% for wild type and 0.74 ± 0.08% for M18T2, corresponding to 6.6 ± 0.7 and 6.4 ± 0.7 nucleotide mutations per 861-bp gene). For better statistics, the sequencing results for both libraries were combined to give the data in Table 2. No biases in the locations of the mutations were observed. Eleven mutations occurred twice, which is in good agreement with the expectation of eight duplicate mutations if all possible mutations were equiprobable. The per-round mutation frequency was calculated as 0.15 ± 0.03% (1.3 ± 0.3 nucleotide mutations per gene) by assuming that each round of error-prone PCR introduced the same average number of mutations. To confirm this assumption, we sequenced 10 unselected clones each from the wild-type and M182T round-one libraries, and found mutation frequencies of 0.16 ± 0.05% for wild type and 0.19 ± 0.06% for M182T. Standard errors were computed assuming Poisson sampling statistics. More detailed sequencing information is in Table 4, which is published as supporting information on the PNAS web site.
Table 2. TEM1 mutation frequencies.
| Base pairs sequenced | 22,800 |
| Total mutations | 172 |
| Total amino acid substitutions | 120 |
| Mutation frequency, % | 0.75 ± 0.06 |
| Mutations per gene | 6.5 ± 0.5 |
| Amino acid substitutions per gene | 4.5 ± 0.4 |
| Mutation types, % | |
| A → T, T → A | 22 |
| A → C, T → G | 9 |
| A → G, T → C | 42 |
| G → A, C → T | 20 |
| G → C, C → G | 1 |
| G → T, C → A | 3 |
| Frameshift | 3 |
Mutation frequencies for TEM1 β-lactamase mutagenesis determined by sequencing 20 unselected clones each from the round five wild-type and M182T error-prone PCR libraries.
Results
Thermodynamic Framework for Predicting Neutrality. A protein's native structure is thermodynamically stable (20, 21), with typical free energies of folding (ΔGf) between -5 and -15 kcal/mol (22). A mutant sequence folds to the wild-type structure only if the stability of that structure meets some minimal threshold. We call the extra stability of the native structure beyond this minimal threshold 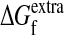 and note that functional proteins always have
and note that functional proteins always have  . We define a protein's m-neutrality as the fraction of sequences with m substitutions that still meet the stability threshold.
. We define a protein's m-neutrality as the fraction of sequences with m substitutions that still meet the stability threshold.
A substitution causes a stability change of
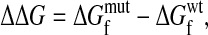 |
where 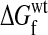 and
and 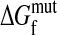 are the wild-type and mutant protein stabilities, respectively. Substitutions tend to be destabilizing: although there are no large collections of ΔΔG measurements for truly random substitutions, in a likely biased collection of >2,000 measured ΔΔG values for single-residue substitutions (23), the mean is 0.9 kcal/mol and the values at the 10th and 90th percentiles are -1.0 and 3.2.
are the wild-type and mutant protein stabilities, respectively. Substitutions tend to be destabilizing: although there are no large collections of ΔΔG measurements for truly random substitutions, in a likely biased collection of >2,000 measured ΔΔG values for single-residue substitutions (23), the mean is 0.9 kcal/mol and the values at the 10th and 90th percentiles are -1.0 and 3.2.
The thermodynamic effects of most substitutions are approximately additive (24–26), meaning that if the stability changes caused by two different single substitutions are ΔΔGa and ΔΔGb, then the stability change due to both substitutions is approximately ΔΔGa + ΔΔGb. If we know the probability distribution p1(ΔΔG) that a single random substitution causes a stability change of ΔΔG, and if we assume that substitutions are additive, then the net effect ΔΔGm of m random substitutions is just the sum of m random variables from the probability distribution p1(ΔΔG). Under this additivity assumption, we can therefore directly calculate the distribution pm(ΔΔGm) for ΔΔGm by performing an m-fold convolution (27) of p1(ΔΔG).
The m-neutrality Pf(m) is simply the the probability that ΔΔGm is not more destabilizing than the extra stability 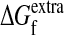 of the wild-type sequence, and can be written as
of the wild-type sequence, and can be written as
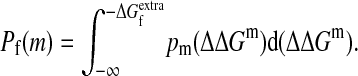 |
[1] |
This formula gives a protein's m-neutrality in terms of its extra stability and the distribution of ΔΔG values for all possible single substitutions.
Lattice Proteins Support Predictions. We tested the ability of this simple framework to predict the fraction of lattice proteins that retained the original structure after random amino acid substitutions. Lattice proteins are highly simplified models of proteins that provide a useful tool for studying protein folding (28–31) and evolution (16, 32) (some example lattice proteins are shown in Fig. 1). We can easily measure the m-neutralities of the lattice proteins by making random amino acid substitutions and seeing whether the sequences still have ΔGf ≤ 0.0. We can also use Eq. 1 to directly predict the m-neutralities because we can exactly compute ΔGf and ΔΔG values.
Fig. 1.

Lattice proteins with different structures but the same stability (ΔGf = -1.0) converge to different exponential declines in m-neutrality. (a) The distributions of ΔΔG for all 380 single amino acid substitutions to the inset lattice proteins. (b) The measured (symbols) and predicted (lines) m-neutralities for the four proteins. Proteins are considered folded if ΔGf ≤ 0.0 for the original native structure. The proteins used for the m-neutrality analyses were generated by adaptive walks from random starting sequences, followed by 2.5 × 105 generations of neutral evolution with a population size of 100 and a per-generation per-residue substitution rate of 5 × 10-5, selecting for sequences with ΔGf ≤ -1.0 and then taking the first sequence generated with a stability within 0.025 of -1.0. The m-neutralities were computed by sampling all mutants for m≤ 2 or 5 × 105 random mutants for m > 2. The predicted m-neutralities were computed according to Eq. 1 by numerically convolving the distribution of single-substitution ΔΔG values using generating functions (27) computed with fast-Fourier transforms and a bin size of 0.01.
Eq. 1 accurately predicted the m-neutralities of all of the lattice proteins we tested. Lattice proteins with different structures have different m-neutralities, even when they have the same ΔGf (Fig. 1). The 1-neutralities of proteins with different structures and the same ΔGf look similar, but for larger values of m some proteins clearly show higher m-neutralities than others. For large m, the m-neutralities of all of the proteins converge to a simple exponential of the form
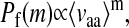 |
where 〈νaa〉 is the average fraction of proteins that are destabilized by a further single random amino acid substitution after several substitutions have already occurred. The underlying reason for the exponential form of this decline is clear: after several substitutions, the distribution of ΔGf among the remaining functional sequences reaches a steady state and each new substitution pushes the same fraction of proteins beyond the stability threshold. The average neutrality 〈νaa〉 is therefore actually the 1-neutrality averaged over all stable sequences with the wild-type structure. Although Pf(m = 1) is similar for all of the protein structures in Fig. 1, the factors that give rise to the different values of 〈νaa〉 for the different structures are present in the distribution of single mutant ΔΔG values, because it is used to predict the m-neutralities for all values of m.
Fig. 2 shows the m-neutralities of proteins with the same structure but different stabilities. After several substitutions, all of the proteins converge to the same value of 〈νaa〉, suggesting that 〈νaa〉 is a generic property of a protein's structure. On the other hand, the response of a protein to the first few substitutions depends strongly on its stability, with more stable proteins exhibiting higher initial m-neutrality. The high initial m-neutrality of stable proteins is readily rationalized in terms of the thermodynamic model: substitutions tend to disrupt a protein's structure by pushing its stability below the minimal threshold, but proteins with an extra stability cushion are buffered against the first few substitutions (33). Proteins that sit on the very margin of the minimal stability threshold exhibit lower 1-neutrality than is predicted by an exponential decline because these proteins are less stable than the average folded protein; thus, surviving sequences will tend to be more stable than the wild-type sequence and therefore be more tolerant to the next substitution.
Fig. 2.

Lattice proteins with the same structure but different stabilities have different 1-neutralities but have the same average neutrality 〈νaa〉. (a) Predicted (lines) and measured (symbols) m-neutralities for proteins with different stabilities and the same structure (III in Fig. 1). (b) Measured values of the 1-neutralities (squares) and average neutralities (circles) for proteins with different stabilities but the same structures (the plots at left and right are for structures I and IV from Fig. 1, respectively). The sequences were generated by finding a sequence with ΔGf = -2.0 by using the procedure described in Fig. 1, and then using this sequence as a starting point for neutral evolution selecting for the indicated target stabilities. The proteins with different stabilities are highly diverged, with average pairwise sequence identities of 15% and 41% for the structures at left and right, respectively. The m-neutralities were computed as in Fig. 1, and 〈νaa〉 was computed as the square root of the 6-neutrality divided by the 4-neutrality.
Real Proteins Support Predictions. Our theory makes two main predictions: first, that the decline in m-neutrality is determined by the ΔΔG values for single amino acid substitutions, and second, that among proteins with the same structure, more stable variants will have higher m-neutralities. We tested these predictions against measurements of the fractions of functional proteins in mutant libraries of subtilisin and variants of TEM1 β-lactamase. Our theory is designed to predict the fraction of proteins that retain the wild-type structure, but the experiments measure the fraction of proteins that retain function. However, because proteins that fail to fold also generally fail to function, our theory provides an upper bound on the fraction of functional proteins. We expect that, for many proteins, this upper bound will closely approximate the actual fraction of proteins that remain functional because mutagenesis studies suggest that most functionally disruptive random substitutions disrupt the structure rather than specifically affect functional residues (13, 34, 35).
To test the ability of our theory to predict the decline in m-neutrality, we used data on the fractions of functional proteins in subtilisin mutant libraries created by Shafikhani et al. (10) (population 6B of table 2 in ref. 10, normalized by the fraction of functional clones in the control libraries) and our own mutant libraries of TEM1 (Table 1). Each mutant library contains a distribution of sequences with different numbers of nucleotide mutations. The form of this distribution is known: the probability that a sequence in a library with an average of 〈mnt〉 nucleotide mutations created by N cycles of PCR with a PCR efficiency of λ will have mnt mutations is
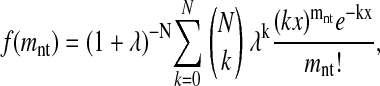 |
where x = 〈mnt〉 (1 + λ)/(Nλ) (36, 37). Subtilisin was mutagenized by using 13 PCR cycles with 10 effective doublings (10), so N is 13 times the number of rounds of error-prone PCR and λ = 0.77. TEM1 was mutagenized by using 14 PCR cycles with 10 effective doublings, so N is 14 times the number of rounds and λ = 0.71. We confirmed that f(mnt) accurately describes the distribution of mutations in our libraries (Fig. 5, which is published as supporting information on the PNAS web site).
The expected fraction of folded sequences in a mutant library is easily calculated from f(mnt) and the probability Pf(mnt) that a sequence is still functional after mnt nucleotide mutations as
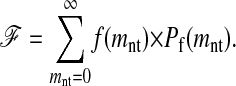 |
We calculated the probability Pf(mnt) that a sequence was still folded after mnt nucleotide mutations by using two existing computer programs for estimating the ΔΔG values for single substitutions to proteins with known structures (Protein Data Bank ID 1IAV for subtilisin and 1BTL for TEM1): Gilis and Rooman's popmusic potential (38) and Serrano and coworkers' foldef potential (39) with van der Waals clash energies. Because the genetic code makes nucleotide mutations more likely to induce some amino acid substitutions than others, and because error-prone PCR introduces a nonrandom distribution of nucleotide mutations, we weighted each ΔΔG value by the probability that it would be induced by a single nucleotide mutation made according to the observed error-prone PCR nucleotide mutation frequencies (given in table 1 of ref. 10 for subtilisin and Table 2 for TEM1). We assigned a ΔΔG of zero to synonymous nucleotide mutations because they do not cause an amino acid substitution, and we assigned a ΔΔG of 25 kcal/mol to frameshift and nonsense mutations because premature truncation is expected to inactivate the protein. We ignored the small number of substitutions for which popmusic failed to calculated a ΔΔG. With this weighted ΔΔG distribution for nucleotide mutations, all we needed to construct Pf(mnt) according to Eq. 1 was the value of 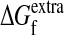 . This value cannot be measured directly because we do not know the minimal stability threshold. However, because
. This value cannot be measured directly because we do not know the minimal stability threshold. However, because 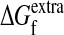 only influences the initial behavior of the m-neutrality and does not affect the limiting decline (Fig. 2), and because we have six data points for each protein, we could do a least-squares fit of
only influences the initial behavior of the m-neutrality and does not affect the limiting decline (Fig. 2), and because we have six data points for each protein, we could do a least-squares fit of 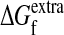 to the data and still test the ability of the theory to predict the decline in the fraction of functional proteins.
to the data and still test the ability of the theory to predict the decline in the fraction of functional proteins.
Fig. 3 shows the measured fractions of functional proteins for subtilisin and wild-type TEM1 versus the theoretical predictions made with popmusic and foldef. The theoretical predictions closely match the measured fractions of functional proteins in all cases, with subtilisin exhibiting slightly higher m-neutralities than TEM1.
Fig. 3.

Theoretical predictions and fractions of functional proteins in mutant libraries of subtilisin (dashed lines) and TEM1 β-lactamase (solid lines) genes. Thick lines show predictions made by using popmusic (38), and thin lines show predictions made by using foldef (39). The TEM1 measurements are from Table 1, normalized by the values from the control unmutated library.
The second major prediction of our theory is that, among proteins with the same structure, more stable variants will exhibit higher initial m-neutralities, but converge to same average neutrality. To test this prediction, we compared the fractions of functional proteins in mutant libraries of wild-type and the M182T variant of TEM1. The M182T variant differs from wild-type by only a single substitution, yet is 2.7 kcal/mol more stable (18), so we predict that it should exhibit a higher fraction of functional proteins at the same level of mutation. Fig. 4 shows the measured fractions functional for wild-type and the M182T variant, as well as the theoretical predictions made with both popmusic and foldef. As predicted, the M182T variant exhibits a higher fraction of functional proteins, and once again the predictions made with both potentials are in good agreement with the experimental measurements.
Fig. 4.

The more stable M182T variant of TEM1 β-lactamase (dashed lines) exhibits a higher fraction of functional mutants relative to wild type (solid lines), as predicted. Thick lines show predictions made by using popmusic (38), and thin lines show predictions made by using foldef (39). The measurements are from Table 1, normalized by the values from the control unmutated library.
To further explore the range of possible neutralities for different proteins, we used ΔΔG values from popmusic to predict the expected average neutralities to both amino acid substitutions (〈νaa〉) and nucleotide mutations (〈νnt〉) for proteins chosen from several different cath (40) protein structure classifications. Because we do not know 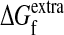 for these proteins, we computed the fraction of proteins expected to be inactivated by the 10th mutation because, after this many mutations, effects of the initial protein stability should be small. Table 3 shows the predicted average neutralities to both random amino acid substitutions and nucleotide mutations made according to the mutation probabilities of our TEM1 mutagenesis. The predicted average neutralities differ considerably, showing that our theory predicts that different proteins can have substantially different neutralities.
for these proteins, we computed the fraction of proteins expected to be inactivated by the 10th mutation because, after this many mutations, effects of the initial protein stability should be small. Table 3 shows the predicted average neutralities to both random amino acid substitutions and nucleotide mutations made according to the mutation probabilities of our TEM1 mutagenesis. The predicted average neutralities differ considerably, showing that our theory predicts that different proteins can have substantially different neutralities.
Table 3. Predicted average neutralities.
| PDB | Protein | cath architecture | Length, bp | 〈νnt〉 | 〈νaa〉 |
|---|---|---|---|---|---|
| 1IAV | Subtilisin | αβ 3-layer sandwich | 269 | 0.65 | 0.55 |
| 1B9C | GFP | β-barrel | 236 | 0.62 | 0.56 |
| 1BTL | TEM1 β-lactamase | αβ 3-layer sandwich | 263 | 0.58 | 0.46 |
| 1RLV | tRNA endonuclease | Not classified | 305 | 0.55 | 0.44 |
| 1HZW | Thymidylate synthase | αβ 2-layer sandwich | 290 | 0.50 | 0.41 |
| 2BNH | Ribonuclease inhibitor | αβ horseshoe | 457 | 0.45 | 0.35 |
| 1HEL | Hen lysozyme | α orthogonal bundle | 129 | 0.43 | 0.38 |
Predictions of the average heutralities of various proteins to both nucleotide mutations (〈νnt〉) and amino acid substitutions (〈νaa〉). The Protein Data Bank ID codes (PDB) and the cath (40) architectures are shown along with the lengths of the protein chains in the PDB structures (in all cases we consider chain A). The average neutralities are computed by calculating the fraction of sequences predicted be inactivated by the 10th mutation or substitution, by using ΔΔG values from popmusic (38) and assuming that the proteins all have the same value of 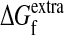 as wild-type TEM1 β-lactamase. The values of 〈νnt〉 are computed assuming that nucleotide mutations are made according to the error-prone PCR mutation frequencies of Table 2.
as wild-type TEM1 β-lactamase. The values of 〈νnt〉 are computed assuming that nucleotide mutations are made according to the error-prone PCR mutation frequencies of Table 2.
Discussion
We have presented a theory for calculating the probability that a protein will retain its structure after random amino acid substitutions, and have confirmed the main theoretical predictions with simulations and experiments. Our theory naturally separates a protein's m-neutrality into components caused by structure and stability. The eventual severity of the exponential decline in m-neutrality with the number of substitutions is a property of a protein's structure. On the other hand, increased stability confers greater tolerance to the first few substitutions, in effect allowing a protein to “take a few hits” before it is pushed into the inevitable structurally determined exponential decline in m-neutrality. This increased tolerance to mutations due to extra stability is probably also the underlying reason for the existence of global suppressor mutations (13, 14) that buffer proteins against otherwise deleterious mutations.
The major assumption underlying our theory is that the thermodynamic effects of substitutions are additive. This assumption is clearly not strictly true because protein residues do interact. Substitutions are most likely to be nonadditive if the mutated residues are in close contact in a protein's structure (24, 25). Because proteins are large, two randomly chosen residues will rarely contact each other, and so although the additivity assumption is certainly violated for some specific combinations of substitutions, it is accurate when averaged over all possible substitutions. When we apply our theory to measurements of the fraction of mutant proteins that retain function, we are making a second assumption by ignoring the possibility that some substitutions may disrupt a protein's function in ways other than affecting its stability. Therefore, for proteins with a high fraction of functional residues, our theory provides only an upper bound on the fraction of functional proteins. However, our theory's remarkable success for both the subtilisin and the TEM1 mutant libraries suggests that this assumption is also valid.
Our theory provides a quantitative rationale for earlier work with lattice proteins on the organization of functional proteins in sequence space. Bornberg-Bauer and Chan (2) proposed that proteins are located in superfunnels in sequence space, with the most stable sequence having the most neutral neighbors; others have reported that folded proteins surround highly stable prototype sequences in sequence space (3, 4, 41), and Shakhnovich and coworkers (1) showed that proteins with a large energy gap between the lowest and second lowest energy conformations are stabilized against mutations. We provide a clear explanation: more stable proteins are able to tolerate more of the possible mutations before unfolding, and so a higher fraction of their neighboring sequences fold.
In addition to these stability-based effects, different protein structures have different inherent designabilities, with more sequences folding into some structures than others (5, 42, 43). Proteins with more designable structures might be expected to show a higher average neutrality because their structures occupy a larger fraction of sequence space. Therefore, the average neutrality 〈νaa〉 provides a quantitative measure of designability that can be estimated with current computational techniques.
Our work suggests a more nuanced approach to experimentally analyzing protein neutralities than has been applied in the past. Loeb and coworkers (11) have performed a careful analysis of the neutralities of several proteins or regions of proteins under the assumption of a strict exponential decline in m-neutrality. However, our work suggests that a protein's m-neutrality can deviate from a strict exponential for the first few substitutions if the protein has a large amount of extra stability, as we show for the M182T variant of TEM1. Experimental mutagenesis studies suggest that, during natural evolution, proteins accumulate mildly destabilizing mutations that are counterbalanced by stabilizing mutations (26). We suggest that it is also important to examine whether some natural proteins have systematically accumulated stabilizing mutations to provide them with additional robustness (15) to amino acid substitutions.
Our work also has applications in protein engineering. Directed evolution involves screening libraries of mutant proteins for new or improved functions (44). Each round of directed evolution typically introduces only one or two amino acid substitutions because the rapid decline in m-neutrality means that higher mutation rates will yield libraries of mostly unfolded proteins. Our work suggests that using highly stable parents for directed evolution should increase the fraction of folded mutants at a given level of substitutions, and it provides a method for predicting which structures will better tolerate large numbers of substitutions.
Supplementary Material
Acknowledgments
We thank Brian Shoichet for providing us with genes for the TEM1 β-lactamase variants, Titus Brown for programming assistance, Michelle Meyer and Eric Zollars for helpful advice and discussions, and two anonymous reviewers for helpful comments. J.D.B. is supported by a Howard Hughes Medical Institute predoctoral fellowship. D.A.D. is supported by the National Institutes of Health, National Research Service Award 5 T32 MH19138 from the National Institute of Mental Health. C.A. and C.O.W. were supported in part by the National Science Foundation Grant DEB-9981387.
Author contributions: J.D.B. and F.H.A. designed research; J.D.B. performed research; J.S. assisted with research; J.D.B., C.O.W., and D.A.D. analyzed data; J.D.B. wrote the paper; and C.A. and F.H.A. provided guidance.
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Broglia, R. A., Tiana, G, Roman, H. E., Vigezzi, E. & Shakhnovich, E. (1999) Phys. Rev. Lett. 82, 4727-4730. [Google Scholar]
- 2.Bornberg-Bauer, E. & Chan, H. S. (1999) Proc. Natl. Acad. Sci. USA 96, 10689-10694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wingreen, N. S., Li, H. & Tang, C. (2004) Polymer 45, 699-705. [Google Scholar]
- 4.Xia, Y. & Levitt, M. (2004) Proteins 55, 107-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li, H, Helling, R, Tang, C. & Wingreen, N. (1996) Science 273, 666-669. [DOI] [PubMed] [Google Scholar]
- 6.Shakhnovich, E. I. & Gutin, A. M. (1991) J. Theor. Biol. 149, 537-546. [DOI] [PubMed] [Google Scholar]
- 7.Govindarajan, S. & Goldstein, R. A. (1996) Proc. Natl. Acad. Sci. USA 93, 3341-3345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rennell, D, Bouvier, S. E., Hardy, L. W. & Poteete, A. R. (1991) J. Mol. Biol. 222, 67-87. [DOI] [PubMed] [Google Scholar]
- 9.Markiewicz, P, Kleina, L. G., Cruz, C, Ehret, S. & Miller, J. H. (1994) J. Mol. Biol. 240, 421-433. [DOI] [PubMed] [Google Scholar]
- 10.Shafikhani, S, Siegel, R. A., Ferrari, E. & Schellenberger, V. (1997) BioTechniques 23, 304-310. [DOI] [PubMed] [Google Scholar]
- 11.Guo, H. H., Choe, J. & Loeb, L. A. (2004) Proc. Natl. Acad. Sci. USA 101, 9205-9210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Daugherty, P. S., Chen, G, Iverson, B. L. & Georgiou, G. (1999) Proc. Natl. Acad. Sci. USA 97, 2029-2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shortle, D. & Lin, B. (1985) Genetics 110, 539-555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Poteete, A. R., Rennell, D, Bouvier, S. E. & Hardy, L. W. (1997) Protein Sci. 6, 2418-2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wilke, C. O. & Adami, C. (2003) Mutat. Res. 522, 3-11. [DOI] [PubMed] [Google Scholar]
- 16.Chan, H. S. & Bornberg-Bauer, E. (2002) Appl. Bioinform. 1, 121-144. [PubMed] [Google Scholar]
- 17.Miyazawa, S. & Jernigan, R. L. (1985) Macromolecules 18, 534-552. [Google Scholar]
- 18.Wang, X, Minasov, G. & Shoichet, B. K. (2002) J. Mol. Biol. 320, 85-95. [DOI] [PubMed] [Google Scholar]
- 19.Meyer, M. M., Silberg, J. J., Voigt, C. A., Endelman, J. B., Mayo, S. L., Wang, Z. G. & Arnold, F. H. (2003) Protein Sci. 12, 1686-1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anfinsen, C. B. (1973) Science 181, 223-230. [DOI] [PubMed] [Google Scholar]
- 21.Ellis, R. J., Dobson, C. M. & Hartl, U. (1998) Trends Biochem. Sci. 23, 468-468. [DOI] [PubMed] [Google Scholar]
- 22.Fersht, A. R. (1999) Structure and Mechanism in Protein Science (Freeman, New York).
- 23.Bava, K. A., Gromiha, M. M., Uedaira, H, Kitajimi, K. & Sarai, A. (2004) Nucleic Acids Res. 32, D120-D121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wells, J. A. (1990) Biochemistry 29, 8509-8517. [DOI] [PubMed] [Google Scholar]
- 25.Zhang, X. J., Baase, W. A., Shoichet, B. K., Wilson, K. P. & Matthews, B. W. (1995) Protein Eng. 8, 1017-1022. [DOI] [PubMed] [Google Scholar]
- 26.Serrano, L, Day, A. G. & Fersht, A. R. (1993) J. Mol. Biol. 233, 305-312. [DOI] [PubMed] [Google Scholar]
- 27.van Kampan, N. G. (1992) Stochastic Processes in Physics and Chemistry (Elsevier, Amsterdam).
- 28.Dill, K. A., Bromberg, S, Yue, K, Fiebig, K. M., Yee, D. P., Thomas, P. D. & Chan, H. S. (1995) Protein Sci. 4, 561-602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hinds, D. A. & Levitt, M. (1994) J. Mol. Biol. 243, 668-682. [DOI] [PubMed] [Google Scholar]
- 30.Shakhnovich, E. I. & Gutin, A. M. (1993) Proc. Natl. Acad. Sci. USA 90, 7195-7199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Socci, N. D., Onuchic, J. N. & Wolynes, P. G. (1998) Proteins 32, 136-158. [PubMed] [Google Scholar]
- 32.Xia, Y. & Levitt, M. (2004) Curr. Opin. Struct. Biol. 14, 202-207. [DOI] [PubMed] [Google Scholar]
- 33.Axe, D. D. (2004) J. Mol. Biol. 341, 1295-1315. [DOI] [PubMed] [Google Scholar]
- 34.Loeb, D. D., Swanstrom, R, Everitt, L, Manchester, M, Stamper, S. E. & Hutchison, C. A., III (1989) Nature 340, 397-400. [DOI] [PubMed] [Google Scholar]
- 35.Pakula, A. A., Young, V. B. & Sauer, R. T. (1986) Proc. Natl. Acad. Sci. USA 83, 8829-8833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sun, F. (1995) J. Comput. Biol. 2, 63-86. [DOI] [PubMed] [Google Scholar]
- 37.Drummond, D. A. & Arnold, F. H. (2004) arXiv: q-bio.QM/0411041.
- 38.Gilis, D. & Rooman, M. (2000) Protein Eng. 13, 849-856. [DOI] [PubMed] [Google Scholar]
- 39.Guerois, R, Nielsen, J. E. & Serrano, L. (2002) J. Mol. Biol. 320, 369-387. [DOI] [PubMed] [Google Scholar]
- 40.Orengo, C. A., Michie, A. D., Jones, S, Jones, D. T., Swindells, M. B. & Thornton, J. M. (1997) Structure (London) 5, 1093-1108. [DOI] [PubMed] [Google Scholar]
- 41.Taverna, D. M. & Goldstein, R. A. (2002) J. Mol. Biol. 315, 479-484. [DOI] [PubMed] [Google Scholar]
- 42.Wolynes, P. G. (1996) Proc. Natl. Acad. Sci. USA 93, 14249-14255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.England, J. L. & Shakhnovich, E. I. (2003) Phys. Rev. Lett. 90, 218101. [DOI] [PubMed] [Google Scholar]
- 44.Arnold, F. H. (1998) Acc. Chem. Res. 31, 125-131. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.