

Abstract
Chlorophyll d-producing cyanobacteria are a recently described group of phototrophic bacteria that is a major focus of photosynthesis research, previously known only from marine environments in symbiosis with eukaryotes. We have discovered a free-living member of this group from a eutrophic hypersaline lake. Phylogenetic analyses indicated these strains are closely related to each other but not to prochlorophyte cyanobacteria that also use an alternative form of chlorophyll as the major light-harvesting pigment. We have also demonstrated that these bacteria acquired a fragment of the small-subunit rRNA gene encoding a conserved hairpin in the bacterial ribosome from a proteobacterial donor at least 10 million years before the present. Thus, our most widely used phylogenetic marker can be a mosaic of sequence fragments with widely divergent evolutionary histories.
Keywords: cyanobacteria, lateral gene transfer
Chlorophyll (Chl) d-producing cyanobacteria are a recently discovered group of oxygenic photoautotrophs that are unique in their use of this far-red light-absorbing structural relative of Chl a as the major photosynthetic pigment (1, 2). The photosynthetic apparatus of these bacteria is structurally and functionally distinct (3, 4), and its study has had a major impact on current perspectives regarding both the mechanisms and evolutionary origins of oxygenic photosynthesis (e.g., refs. 5 and 6).
Research on photosynthesis with Chl d has thus far been limited to a single strain, Acaryochloris marina MBIC-11017, and the extent of the diversity of this group is unknown. Chl d-producing organisms have been reported from only two marine habitats. A. marina MBIC-11017 was isolated from a colonial ascidian from low-nutrient tropical western Pacific coastal waters (1), whereas strain Awaji was obtained from epiphytic growth on a red alga (2). Here, we report the isolation of a strain of Chl d-producing cyanobacteria from the Salton Sea, a moderately hypersaline (41-45 g·liter-1) lake in California. In contrast to the nutrient-poor environment where this group was first discovered, the Salton Sea is eutrophic, because nearly all of its inflow is derived from agricultural and municipal waste-water (7).
The evolutionary origin of photosynthesis with Chl d was not resolved in a previous phylogenetic analysis (8). Because these bacteria are superficially similar to prochlorophyte cyanobacteria in their use of an alternative form of Chl as the major pigment and the absence of the phycobilisome light-harvesting complex found in most cyanobacteria, it is of interest to determine whether Chl d-producing cyanobacteria were derived from a prochlorophyte lineage. Our small-subunit (SSU) rRNA gene phylogenies indicated that these Chl d-producing strains are closely related and provided evidence that the closest relative of this group is not a prochlorophyte. Additionally, Miyashita et al. (8) discovered that the variable 1 region of the SSU rRNA gene (V1) (Escherichia coli nucleotide positions 61-106) in A. marina strain MBIC-11017 is anomalously long for a cyanobacterium and is identical to the V1 DNA of certain proteobacteria. They noted that this similarity could potentially be explained either by lateral gene transfer (LGT) from a foreign donor or by convergent evolution (8). Motivated by our own observation that the SSU rRNA gene of our strain also contains this anomalous sequence, we used molecular evolutionary approaches grounded in a maximum likelihood statistical framework to show that the V1 DNA in Chl d-producing cyanobacteria and the RNA hairpin it encodes were acquired relatively recently by LGT from a β-proteobacterial donor. This demonstrates that ribosome-encoding DNA can be transferred in nature between distant relatives that last shared a common ancestor >2.5 billion years ago.
Materials and Methods
Strain Isolation and Maintenance. Strain CCMEE 5410 was isolated from epilithic microbial mat collected in January 1999 from rip rap rock forming a dike at the southern shore of the Salton Sea (33° 08.57′ N and 115° 39.32′ W; ref. 9). The positive enrichment in IOBG11 medium [BG11 with 24 g·liter-1 Instant Ocean salt (Aquarium Systems, Mentor, OH)] had been inoculated with 1 ml of 105-fold diluted sample homogenate. Upon streak-dilution plating, a majority of colonies were lime-green, and the strain isolated from this plate was considered to be a clone after multiple rounds of plating. Growth conditions were 23°C and 50 μmol photons m-2·s-1. The strain has been deposited in the University of Oregon Culture Collection of Microorganisms from Extreme Environments.
Pigment Analysis. Cultures of strain CCMEE 5410 and A. marina strain MBIC-11017 were grown in IOBG11 medium under 15 μmol photons m-2·sec-1 of continuous fluorescent light. Approximately 3 mg of dried cells was suspended in 1 ml of acetone/methanol (7:3, vol/vol) containing 200 mM hydroquinone, sonicated for 2 min, and briefly dark-incubated on ice. After centrifugation, the supernatant was transferred to a test tube and stored in the dark at 4°C. Pigment extracts were applied onto a Beckman System Gold HPLC system (Beckman Coulter) with a 250 × 4.6-mm Phenomenex (Belmont, CA) C-18 reverse-phase column (00G-4097-EO), and Chl and carotenoid peaks were detected at 420 nm. Pigments were eluted with an acetone/water elution gradient at a sample flow rate of 1 ml·min-1. Absorption spectra of HPLC fractions were used for pigment identification and quantitation with published extinction coefficients. The isotopic mass of each fraction was determined by MALDI-TOF mass spectroscopy with an Applied Biosystems Mass Spec Voyager DE-STR.
Transmission Electron Microscopy. Exponential phase cells were fixed overnight in 3% glutaraldehyde/0.1 M sodium cacodylate, pH 7.0, followed by 2% osmium tetroxide in the same buffer for 2 h, and embedded in Spurr's resin. Sections were cut with a LKB Ultrotome, poststained on a grid with 5% uranyl acetate and lead citrate, and imaged with a Phillips (Eindhoven, The Netherlands) CM12 electron microscope.
DNA Extraction, Amplification, and Sequencing. Protocols for genomic DNA isolation and thermal cycling were as described (10). The nearly complete 16S rRNA gene sequence was amplified as two fragments spanning E. coli positions 8-1334 and 359-1528 with primer sets AGAGTTTGATCMTGGCTCAGG/CTTCAYGYAGGCGAGTTGCAGC and GGGGAATYTTCCGCAATGGG/AAAGGAGGTGATCCAGCC. Fragments were sequenced bidirectionally on an Applied Biosystems 3700.
Phylogeny Reconstruction. Sequences were aligned as described (10). Phylogenies were reconstructed from 1,412 nucleotides of SSU rRNA gene sequence data by maximum likelihood, parsimony, and neighbor-joining methods with paup*, Ver. 4.0 (11). The substitution model used for likelihood analysis (general time reversible and gamma) was selected by Akaike information criterion (AIC) as the best model by the program modeltest (12). After the starting tree was obtained by random stepwise addition, heuristic searches for likelihood and parsimony were performed by using the tree-bisection-reconnection branch-swapping algorithm. Phylogenies were bootstrapped with either 100 (likelihood) or 1,000 (parsimony, neighbor-joining) replicates. rRNA secondary structure models were developed by using models available at the Comparative RNA Web Site (www.rna.icmb.utexas.edu).
Analyzing V1 DNA Sequence Length Distributions. SSU rRNA gene sequences including the V1 region (E. coli positions 61-106) were obtained from GenBank for 531 cyanobacterial and 355 β-proteobacterial laboratory isolates. Sequences for each group were separately aligned with clustalw, Ver. 1.83 (13). For each sequence, the length of V1 DNA was quantified as the number of nucleotides (alignment gaps excluded) between E. coli positions 60 and 107. This alignment was anchored by conserved sequences flanking V1, ensuring accurate determination of V1 length. Negative binomial distributions were fit separately by maximum likelihood to cyanobacterial and β-proteobacterial data (sas, Ver. 8, SAS Institute, Cary, NC). AIC was used to statistically compare likelihood models (14). The model with the lowest AIC score is the best model, and the probability of each model is given by e0.5δi, where Δi is the difference in AIC score between model i and the best model.
Testing the Molecular Clock Hypothesis. Sequence data used to reconstruct phylogenies above were analyzed with hyphy, Ver. 0.96 (www.hyphy.org). All tests assumed the general timereversible and gamma substitution model. molecularclock.bf uses a likelihood ratio test to test the null hypothesis that sequence divergence over the entire phylogeny can be described by a global molecular clock. It compares two nested models, one constrained to clock-like behavior and the other unconstrained. Significance of the test is given by its asymptotic P value. Similarly, localmolclock.bf performs a likelihood ratio test for each subtree of the phylogeny to evaluate whether sequence divergence of any of these subtrees conforms to a local clock.
Estimating Sequence Divergence in the Chl d/Synechococcus Strain IR11 Clade. Branch lengths (in units of substitutions per site) and their standard errors were estimated for the two nodes in this clade by maximum likelihood with the program baseml in the paml package (http://abacus.gene.ucl.ac.uk/software/paml.html) under a discrete gamma, general time-reversible model as above, with a local molecular clock imposed. Standard errors were estimated by the curvature method.
Results and Discussion
Discovery of a Free-Living Chl d-Producing Cyanobacterium from a Eutrophic Saline Lake. We discovered a Chl d-producing cyanobacterium as part of a multidisciplinary effort to characterize the limnology and biology of the Salton Sea (9). The strain was isolated from an epilithic microbial mat community collected at 0.25-m depth from bare rip rap jetty rock along the southern shore of the lake between two of its major tributaries, the New and Alamo Rivers. At the time of our collection, the salinity of the water column was 34‰, ≈80% of full lake salinity, indicating prevalence of lake water at the site with only a minor influence of freshwater influent.
The lake and its tributaries are characterized by high levels of nitrogen and a high N:P ratio, indicating that phosphorus is the limiting nutrient for growth (15). At the time of our collection, nitrogen levels in southern basin lake water were 2.5 mg·l-1 total N, primarily consisting of organic-N (1.1 mg·l-1) and ammonia-N (1.2 mg·l-1); total P was estimated to be 0.1 mg·l-1 (15). The high level of ammonia-N in the lake is indicative of frequent reducing conditions, most notably manifested as periodic fish kills during episodes of anoxia and sulfide accumulation throughout the entire water column. Levels of dissolved organic carbon are also high, with an average concentration for 1999 of 40.9 mg·l-1 (15).
During 1999, the 1% light transmittance depth throughout the basin ranged from 1.8 to 4.8 m (15). Dinoflagellates and diatoms dominate the phytoplankton, and Chl a concentrations as high as 560 μg·l-1 have been measured throughout the near-shore southern basin (16). Chl a selectively absorbs red light, enriching the relative amount of far-red wavelengths in transmitted light, which can be efficiently harvested by Chl d.
Several morphological types of cyanobacteria, but no eukaryotic algae, were observed in the microbial mat sample by light and epifluorescent microscopy. Unicells conforming to the dimensions of strain CCMEE 5410 (1.5-2 μm for long axis) and larger (≈3-μm diameter) phycocyanin-rich coccoid unicells were the most abundant cyanobacteria. Three types of oscillatorian (Subsection III) cyanobacteria were in lower relative abundance: a phycoerythrin-containing member of the genus Leptolyngbya; a motile phycoerythrin-lacking Geitlerinema; and a third corresponding morphologically to Phormidium foveolarum Gomont (9). All of these types have been isolated in laboratory culture, and two of the filamentous strains are described in detail elsewhere (9).
Only unicells were present in the most diluted (105-fold) successful enrichment, confirming their numerical abundance in the community. The majority of colonies observed after plating were lime-green, and strain CCMEE 5410 was isolated from one of these. The absorption spectrum of a methanol extract of this strain's pigments resembled that of A. marina MBIC-11017 (Fig. 1A), indicating that strain CCMEE 5410 produces large amounts of Chl d. HPLC analysis and quantitation by absorption spectroscopy of the major pigments of these strains further confirmed that both are very similar in terms of the kind and amount of Chls and carotenoids produced (Fig. 1B; Table 1). Isotopic masses of the Chl HPLC fractions, determined by MALDI-TOF mass spectroscopy, were likewise indicative of Chl d (894.5 Da) and Chl a (892.5 Da) (data not shown). Transmission electron micrographs further showed that strain CCMEE 5410 is a prokaryote and that, like A. marina (1), its peripherally stacked thylakoid membranes lack phycobilisomes, the light-harvesting antennae used by most cyanobacteria (Fig. 2).
Fig. 1.

Pigment analysis of Chl d-producing strains. (A) Absorption spectra of 90% methanol extracts from strain CCMEE 5410 (solid) and A. marina MBIC-11017 (dashed); peaks at 448 and 698 nm are characteristic of Chl d. (B) HPLC chromatograms for strains CCMEE 5410 (solid) and MBIC-11017 (dashed).
Table 1. Pigment molar ratios in Chl d-producing strains.
| Ratio | CCMEE 5410 | MBIC-11017 |
|---|---|---|
| Chl a/Chl d | 0.025 ± 0.0013 | 0.016 ± 0.0030 |
| Zeaxanthin/Chl d | 0.24 ± 0.020 | 0.16 ± 0.015 |
| α-Carotene/Chl d | 0.35 ± 0.021 | 0.25 ± 0.026 |
±SE; average of five experiments.
Fig. 2.

TEM showing the thylakoid membranes of strain CCMEE 5410. Phycobilisomes were not observed. (Bar, 100 nm.)
The major insight of our discovery is that we have extended the known niche of Chl d-producing cyanobacteria to a eutrophic hypersaline lake and have shown that these organisms are not restricted to intimate ecological associations with macroscopic eukaryotes. At the macroscale, the Salton Sea environment is dramatically different from the relatively pristine waters from which the symbiotic forms were isolated. However, details of the actual microenvironmental conditions, in particular redox and nutrient status, at the interface between the symbiotic forms and their hosts were not reported. Further investigation of these associations would resolve whether the frequent reducing conditions, availability of dissolved organic carbon and reduced N, and/or the relatively far-red enriched light field in the Salton Sea reflect fundamental characteristics of the realized niches of these bacteria. If so, additional forms of free-living Chl d-producing bacteria may occur in other eutrophic habitats where these conditions prevail.
Phylogeny of Chl d-Producing Cyanobacteria. The phylogenetic position of A. marina MBIC-11017 was not resolved in a previous analysis (8). We reconstructed SSU rRNA gene phylogenies to determine whether Chl d-producing cyanobacteria form an evolutionarily coherent clade and to attempt to identify their closest relative that does not produce this pigment. Nearly complete sequences of the SSU rRNA gene from strains CCMEE 5410 and MBIC-11017 (1,496 and 1,466 bp, respectively) are 99.2% identical. In maximum likelihood, parsimony, and neighbor-joining SSU rRNA gene phylogenies, the strains cluster as sister taxa within the cyanobacteria and form a moderate to strongly bootstrap-supported clade with Synechococcus IR11, a strain isolated from a Japanese tidepool (Fig. 3). The latter primarily harvests light with phycobiliproteins (unpublished data). This suggests that these bacteria were not derived from a lineage of prochlorophytes, which also mainly harvest light with an alternative Chl (Chl b).
Fig. 3.

Maximum likelihood phylogeny of the cyanobacteria inferred from the 16S rRNA gene (excluding E. coli positions 61-106), rooted with outgroups E. coli and B. subtilis. Chl d-containing strains are in bold. Scale is 0.1 substitutions per site. Letters indicate nodes with >50% bootstrap support for likelihood, neighbor-joining, and parsimony phylogenies (Inset).
Unusual V1 in both Chl d Strains. Strains CCMEE 5410 and MBIC-11017 (8) both possess an identical V1 region of the SSU rRNA gene (E. coli positions 61-106, excluded during phylogeny reconstruction above) that is 14-18 nt longer than all other cyanobacterial sequences in our original dataset (Fig. 4). Instead, β-proteobacterial V1s yielded the highest expectation values in a blast similarity search for this region, and the V1 sequence of the Chl d strains was completely identical to that of two clinical isolates, Bordetella strain 61717 and Alcaligenes strain LMG 5906 (Fig. 4). In contrast, nucleotide identities of the Chl d strains at variable sites in V1 flanking DNA (E. coli positions 38, 43, 50, and 108) are characteristic of other cyanobacteria. Because we sequenced this gene directly from a population of PCR products and detected no sequence heterogeneity, we are confident that no conventional cyanobacterial copies of this gene exist in the strain CCMEE 5410 genome.
Fig. 4.

Alignment of the V1 region of the 16S rRNA gene (large box) and flanking sequence spanning E. coli positions 35-111. Invariant nucleotides are indicated by an asterisk. Also shown are the tetraloop (L) and bulge motifs (B) of the V1-encoded RNA hairpin. The identical V1s of Chl d-containing strains and two β-proteobacteria are in bold, with differences in flanking sequence between them indicated in the small boxes.
Evidence for LGT of V1. The cyanobacteria and β-proteobacteria diverged from their most recent common ancestor at least 2.5 (and perhaps >3.5) billion years ago based on geological, geochemical, and paleontological evidence (17-19). The domain Bacteria exhibits >5-fold variation in V1 length (www.rna.icmb.utexas.edu). If most of this variation is partitioned between, rather than within, bacterial phyla, however, then phyla may have idiosyncratic V1 length signatures that facilitate the identification of sequences of foreign origin as outliers. We determined V1 length for the SSU rRNA gene sequences of 531 cyanobacterial laboratory isolates deposited in GenBank. The identical V1 of the Chl d strains, 44 nt in length, is a major outlier of the cyanobacterial distribution (Fig. 5). In contrast, the distribution of the V1 sequences of 355 β-proteobacterial laboratory isolates largely does not overlap the cyanobacterial distribution but does contain the value observed for the Chl d strains. To estimate whether V1 DNA of the Chl d strains could have possibly evolved by chance during cyanobacterial evolution, we used AIC (14) to statistically compare two alternative models (LGT and chance) for the origin of V1. For both models, negative binomial distributions were fit by maximum likelihood to V1 length data for cyanobacteria and β-proteobacteria, respectively. Data for the Chl d strains were assumed to belong to the cyanobacterial distribution in the chance model and to the β-proteobacterial distribution (i.e., and subsequently acquired by a cyanobacterium) in the LGT model. The LGT model fit the data much better than the chance model, indicated by its lower AIC score (-149,642 vs. -149,627) and the large difference in score between models (Δi = 23.12). The likelihoods of the chance and LGT models (given by e0.5Δi), are 9.5 × 10-6 and 1, respectively. The probability that the chance model is correct is 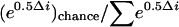 , or <10-5. That is, there is no statistical support for the hypothesis of chance convergence, but there is strong support for LGT.
, or <10-5. That is, there is no statistical support for the hypothesis of chance convergence, but there is strong support for LGT.
Fig. 5.

Frequency distributions of length (in nucleotides) of the V1 segment of the SSU rRNA gene for 531 cyanobacterial (black) and 355 β-proteobacterial sequences (white).
Secondary structure models of mature rRNAs are accurately inferred from their encoding gene sequences (www.rna.icmb.utexas.edu) and also support a LGT origin of the V1 DNA in these strains (Fig. 6). V1 encodes the spur of the ribosome, a conserved hairpin in the 5′ domain of the bacterial SSU rRNA (20). The inferred secondary structure for the Chl d strains bears little similarity to other cyanobacterial spurs (Fig. 6). For example, these strains and β-proteobacteria share several bulge motifs that have no structural equivalent in other cyanobacteria, including an AG bulge at E. coli positions 71-72 and a G bulge at position 100 (Figs. 4 and 6). To explain this as convergence, one must invoke the independent evolution of structural novelties unique to the cyanobacterial radiation. Furthermore, not only is the spur of the Chl d strains identical to those of Bordetella 6717 and Alcaligenes LMG5906, but it also exhibits only slight differences from many other β-proteobacterial spurs (Fig. 6).
Fig. 6.

Maximum likelihood phylogeny for the SSU rRNA gene (excluding V1) for representative cyanobacteria (green), β-proteobacteria (blue), and γ-proteobacterium E. coli (red). Bootstrap values for 1,000 pseudoreplicated datasets are indicated. Scale is 0.1 substitutions per site. Also shown are the inferred secondary structural models for the distal end of the V1-encoded ribosomal spur (E. coli positions 68-101). Structures of strains CCMEE 5410, MBIC-11017, Bordetella 6717, and Alcaligenes LMG5906 are identical (blue, boxed).
Using Sequence Divergence Times to Address When V1 Was Acquired. Based on our phylogenetic information, the acquisition of the V1 DNA in strains CCMEE 5410 and MBIC-11017 is constrained to the time period (i.e., the branch on the phylogeny) after these strains last shared a common ancestor with Synechococcus IR11 (Node B in Fig. 3) but before their divergence from each other (Node A in Fig. 3). Using published rates for SSU rRNA gene evolution, we can estimate dates for these key divergence events that define the evolutionary window within which LGT occurred, provided that divergence of this locus in the Chl d strains/Synechococcus IR11 clade can be described by a molecular clock (i.e., if the amount of evolution from the root of this clade to each tip is constant). For the cyanobacterial sequence data (excluding V1) and their inferred branching order in the likelihood phylogeny in Fig. 3, we used maximum likelihood to implement a general time-reversible model of nucleotide substitution with among-site rate heterogeneity, either with or without an imposed clock. For the entire tree, as well as for every possible subtree defined by the internal nodes of the phylogeny, pairs of clock and no-clock models were compared with likelihood ratio tests. Whereas the tree as a whole clearly does not obey a global molecular clock (-2ΔL = 1995, P < 0.0001), sequence evolution of the clade comprising the Chl d strains and Synechococcus strain IR11 does conform to a local clock, indicated by lack of a statistical difference between the likelihoods of the clock and no-clock models (-2ΔL = 0.13, P = 0.72).
We next estimated the timing of divergence at the nodes that bound the LGT event using the fossil record calibrated SSU rRNA gene clock for a group of aphid endosymbionts (genus Buchnera) (21). The clock's rate of 0.01-0.02 nucleotide substitutions per site per 50 million years [sub/site per 50 mega-annum (Ma)] agrees well with a more general estimate for Bacteria of 0.01 sub/site per 50 Ma (22). Under our local clock model above, estimated sequence divergence (±SE) is 0.005 ± 0.0014 substitutions per site after the bifurcation at Node A (Fig. 3) and 0.044 ± 0.0056 per site after the split at Node B. For a rate of 0.02 sub/site per 50 Ma, the divergence times at Nodes A and B are estimated to be 12.5 ± 3.50 and 109.7 ± 14.00 Ma ago, respectively. Divergence time estimates using simpler evolutionary models that did not necessarily assume among-site rate heterogeneity were qualitatively similar (data not shown). Estimates are doubled if one assumes the slower rate, thereby dating the acquisition of β-proteobacterial DNA by the ancestor of the Chl d strains between ≈10 and 200 Ma ago. Because these bacteria are the only organisms known to produce Chl d (2), these dates also estimate the time frame during which the use of this novel pigment evolved, which suggests that it occurred relatively late during cyanobacterial diversification.
Implications of LGT for Phylogeny Reconstruction and Structural Innovation. Hybrid ribosomes combining protein-RNA or RNA-RNA components from different and in some cases distantly related bacteria have been successfully constructed in the laboratory (23-26). However, results of LGT of rrn operons between enterobacteria in the laboratory (27), in which recipients suffered a substantial reduction in fitness, are consistent with the expectation that such events are typically deleterious, and it has remained controversial whether highly conserved loci including the SSU rRNA gene belong to a genomic core that is recalcitrant to LGT in nature (28-34). Still, possible examples of LGT of SSU rRNA-encoding DNA in nature have been reported based on patterns of rrn sequence heterogeneity, but these are limited to relatively closely related organisms, including halophilic Archaea (35), rhizobia (36), and certain actinomycetes (37-39). Our conclusion that an ancestor of extant Chl d-producing cyanobacteria acquired SSU rRNA-encoding DNA from a proteobacterium by LGT is therefore unprecedented for this locus in nature with respect to the astonishing evolutionary distance between donor and recipient lineages.
Because traditional phylogenetic methods ignore recombination, the SSU rRNA gene is assumed to be immune to LGT during phylogeny reconstruction. Simulations show, however, that even low levels of recombination within a gene can have dramatic effects on both the shape and topology of phylogenies, particularly if large divergent fragments are exchanged (40, 41). In the present case, LGT was readily detected due to the evolutionary distance between donor and recipient lineages, and the affected nucleotides were removed before phylogenetic analysis. Because the amount of DNA exchanged was small, inclusion of V1 DNA in the maximum likelihood analysis did not alter the topology of the phylogeny and had negligible impact on estimates of tree parameters (data not shown). Still, because the probability of genetic exchange is generally considered to increase with the relatedness of donor and recipient, and the size of exchanged fragments can be large (42), this example of intragenic transfer between distant relatives raises the possibility that DNA exchange within this gene may be more frequent among more closely related microorganisms than is currently recognized. Left undetected, cryptic LGT events involving larger and more similar DNA fragments could substantially affect the inference of both phylogenetic relationships and evolutionary tree parameters.
LGT within the SSU rRNA gene also represents a possible mechanism for introducing structural novelties into the ribosome that are unlikely to evolve by the mutational process alone. The fixation and long-term maintenance of this hybrid gene in Chl d-producing cyanobacteria suggest that it has been favored by natural selection. Although a function for the spur has yet to be identified, the importance of this RNA hairpin is suggested by both its evolutionary conservation in bacterial ribosomes and its large conformational displacement during the translation cycle after the binding of elongation factor G (43). An exciting challenge will be to determine whether this LGT event in fact has had consequences for ribosome function in these organisms.
Acknowledgments
We thank D. Pedersen for assisting with GenBank data acquisition, E. Crone for statistical advice, S. Hurlbert for useful discussion and for sharing data before publication, C. Bothun for microscopy assistance, M. Waters and W. Yeoung for culturing support and N. Kuring for landsat images. We also thank S. Arnold, R. Capaldi, R. DeSalle, J. S. Lodmell, M. Saks, G. Sprague, J. Thornton, and two anonymous reviewers for comments on an earlier version of the manuscript. This research was supported by National Science Foundation Grant MCB-0354738 (to S.R.M.), Environmental Protection Agency Grant R86552-01 through the Salton Sea Authority (to A.M.W.), Office of Naval Research Grant N0014-99-1-0177 (to A.M.W.), and National Aeronautical and Space Administration Grant NNG04GK59G (to R.E.B.).
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: Chl, chlorophyll; SSU, small subunit; LGT, lateral gene transfer; V1, variable 1 region of SSU rRNA gene; AIC, Akaike information criterion.
Data deposition: The sequence reported in this paper has been deposited in the GenBank database (accession no. AY790243).
References
- 1.Miyashita, H., Ikemoto, H., Kurano, N., Adachi, K., Chihara, M. & Miyachi, S. (1996) Nature 383, 402. [Google Scholar]
- 2.Murakami, A., Miyashita, H., Iseki, M., Adachi, K. & Mimuro, M. (2004) Science 303, 1633. [DOI] [PubMed] [Google Scholar]
- 3.Miyachi, S., Strassdat, K., Miyashita, H. & Senger, H. (1997) Z. Naturforsch. C52, 636-638. [Google Scholar]
- 4.Hu, Q., Miyashita, H., Iwasaki, I., Kurano, N., Miyachi, S., Iwaki, M. & Itoh, S. (1998) Proc. Natl. Acad. Sci. USA 95, 13319-13323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blankenship, R. E. & Hartman, H. (1998) Trends Biochem. Sci. 23, 94-97. [DOI] [PubMed] [Google Scholar]
- 6.Green, B. R. (2001) Proc. Natl. Acad. Sci. USA 98, 2119-2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cohen, M. J., Morrison, J. I. & Glenn, E. P. (1999) in Pacific Institute for Studies in Development, Environment, and Security Report (Pacific Institute for Studies in Development, Environment, and Security, Oakland, CA), p. 64.
- 8.Miyashita, H., Ikemoto, H., Kurano, N., Miyachi, S. & Chihara, M. (2003) J. Phycol. 39, 1247-1253. [Google Scholar]
- 9.Wood, A. M., Miller, S. R., Li, W. K. W. & Castenholz, R. W. (2002) Hydrobiologia 473, 77-92. [Google Scholar]
- 10.Miller, S. R. & Castenholz, R. W. (2000) Appl. Environ. Microbiol. 66, 4222-4229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Swofford, D. L. (2003) paup*, Phylogenetic Analysis Using Parsimony (*and Other Methods) (Sinauer, Sunderland, MA), Ver. 4.
- 12.Posada, D. & Crandall, K. A. (1998) Bioinformatics 14, 817-818. [DOI] [PubMed] [Google Scholar]
- 13.Thompson, J., Higgins, D. & Gibson, T. (1994) Nucleic Acids Res. 22, 4673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johnson, J. B. & Omland, K. S. (2004) Trends Ecol. Evol. 19, 101-108. [DOI] [PubMed] [Google Scholar]
- 15.Holdren, G. C. & Montaño, A. (2002) Hydrobiologia 473, 1-21. [Google Scholar]
- 16.Reifel, K. M., Swan, B. K., Olivo, E., Watts, J. M., Trees, C. C. & Hurlbert, S. H. Hydrobiologia, in press.
- 17.Holland, H. D. (1994) in Early Life on Earth, ed. Bengston, S. (Columbia Univ. Press, New York), pp. 237-244.
- 18.Summons, R. E., Jahnke, L. L., Hope, J. M. & Logan, G. A. (1999) Nature 400, 554-557. [DOI] [PubMed] [Google Scholar]
- 19.Schopf, J. W. (2000) in The Ecology of Cyanobacteria, eds. Whitton, B. A. & Potts, M. (Kluwer, Dordrecht, The Netherlands), pp. 13-35.
- 20.Wimberley, B. T., Brodersen, D. E., Clemons, W. M., Jr., Morgan-Warren, R. J., Carter, A. P., Vonrhein, C., Hartsch, T. & Ramakrishnan, V. (2000) Nature 407, 327-339. [DOI] [PubMed] [Google Scholar]
- 21.Moran, N. A., Munson, M. A., Baumann, P. & Ishikawa, H. (1993) Proc. R. Soc. London Ser. B 253, 167-171. [Google Scholar]
- 22.Ochman, H. & Wilson, A. C. (1987) J. Mol. Evol. 26, 74-86. [DOI] [PubMed] [Google Scholar]
- 23.Bellemare, G., Vigne, R. & Jordan, B. R. (1973) Biochimie 55, 29-35. [DOI] [PubMed] [Google Scholar]
- 24.Daya-Grosjean, L., Geissler, M., Stöffler, G. & Garret, R. A. (1973) FEBS Lett. 37, 17-20. [DOI] [PubMed] [Google Scholar]
- 25.Nomura, M., Traub, P. & Bechmann, H. (1968) Nature 219, 793-799. [DOI] [PubMed] [Google Scholar]
- 26.Wrede, P. & Erdmann, V. A. (1973) FEBS Lett. 33, 315-319. [DOI] [PubMed] [Google Scholar]
- 27.Asai, T., Zaporojets, D., Squires, C. & Squires, C. L. (1999) Proc. Natl. Acad. Sci. USA 96, 1971-1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Doolittle, W. F. (1999) Science 284, 2124-2128. [DOI] [PubMed] [Google Scholar]
- 29.Brown, J. R. (2003) Nat. Rev. Genet. 4, 121-132. [DOI] [PubMed] [Google Scholar]
- 30.Eisen, J. (2000) Curr. Opin. Genet. Dev. 10, 606-611. [DOI] [PubMed] [Google Scholar]
- 31.Nesbo, C. L., Boucher, Y. & Doolittle, W. F. (2001) J. Mol. Evol. 53, 340-350. [DOI] [PubMed] [Google Scholar]
- 32.Jain, R., Rivera, M. C. & Lake, J. A. (1999) Proc. Natl. Acad. Sci. USA 96, 3801-3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Makarova, K. S., Aravind, L., Galperin, M. Y., Grishin, N. V., Tatusov, R. L., Wolf, Y. I. & Koonin, E. V. (1999) Genome Res. 9, 608-628. [PubMed] [Google Scholar]
- 34.Snel, B., Bork, P. & Huynen, M. A. (1999) Nat. Genet. 21, 108-110. [DOI] [PubMed] [Google Scholar]
- 35.Mylvaganam, S. & Dennis, P. P. (1992) Genetics 130, 399-410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van Berkum, P., Terefework, Z., Paulin, L., Suomalainen, S., Lindström, K. & Eardly, B. D. (2003) J. Bacteriol. 185, 2988-2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ueda, K., Kido, Y., Yoshida, T. & Kataoka, M. (1999) J. Bacteriol. 181, 78-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yap, W. H., Zhang, Z. & Wang, Y. (1999) J. Bacteriol. 181, 5201-5209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang, Y. & Zhang, Z. (2000) Microbiology 146, 2845-2854. [DOI] [PubMed] [Google Scholar]
- 40.Schierup, M. H. & Hein, J. (2000) Genetics 156, 879-891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Posada, D. & Crandall, K. A. (2002) J. Mol. Evol. 54, 396-402. [DOI] [PubMed] [Google Scholar]
- 42.Ochman, H. (2001) Curr. Opin. Genet. Dev. 11, 616-619. [DOI] [PubMed] [Google Scholar]
- 43.Gao, H., Sengupta, J., Valle, M., Korostelev, A., Eswar, N., Stagg, S. M., Van Roey, P., Agrawal, R. K., Harvey, S. C., Sali, A., et al. (2003) Cell 113, 789-801. [DOI] [PubMed] [Google Scholar]