

Abstract
Purpose
Type 2 diabetes mellitus (T2DM) is becoming increasingly prevalent worldwide. Epidemiologic data suggest that T2DM is associated with an increased incidence and mortality from many cancers. The purpose of this review is to discuss the links between diabetes and cancer, the effects of various antidiabetic medications on cancer incidence and mortality, and the effects of anticancer therapies on diabetes.
Design
This study is a review of preclinical and clinical data regarding the effects of antidiabetic medications on cancer incidence and mortality and the effects of anticancer therapies on glucose homeostasis.
Results
T2DM is associated with an increased risk and greater mortality from many cancer types. Metformin use has been associated with a decrease in cancer incidence and mortality, and there are many ongoing randomized trials investigating the effects of metformin on cancer-related outcomes. However, data regarding the association of other antidiabetes medications with cancer incidence and mortality are conflicting. Glucocorticoids, hormone-based therapies, inhibitors that target the phosphatidylinositol 3-kinase-Akt-mammalian target of rapamycin pathway, and insulin-like growth factor 1 receptor–targeted therapy have been associated with high rates of hyperglycemia. These agents mediate their deleterious metabolic effects by reducing insulin secretion and increasing insulin resistance in peripheral tissues.
Conclusion
Studies must be performed to optimize cancer screening strategies in individuals with T2DM. A greater understanding of the mechanisms that link diabetes and cancer are needed to identify targets for therapy in individuals with diabetes who develop cancer. Data from clinical studies are needed to further elucidate the effects of antidiabetic medications on cancer incidence and progression. As several anticancer therapies alter glucose homeostasis, physicians need to be aware of these potential effects. Careful patient screening and monitoring during treatment with these agents is necessary.
DIABETES MELLITUS AND CANCER
Epidemiology
Type 2 diabetes.
Type 2 diabetes mellitus (T2DM) is a chronic progressive disease that is characterized by years of insulin resistance and hyperinsulinemia preceding the development of hyperglycemia. The prediabetes phase may predate the diagnosis of T2DM by up to 10 years. This potentially modifiable phase is frequently associated with the metabolic syndrome, a condition that comprises abdominal obesity, impaired glucose tolerance or impaired fasting glucose, dyslipidemia with elevated triglycerides (TGs) and low HDL cholesterol, and hypertension.1 The prevalence of T2DM and obesity has now reached epidemic proportions worldwide. A number of large-scale epidemiologic studies and meta-analyses, including a recent umbrella review of meta-analyses and observational studies, have shown a consistent increase in site-specific cancer incidence among patients with T2DM.2 This includes a two- to three-fold increase in the incidence rate of pancreatic cancer, a two-fold increased risk for hepatobiliary cancers, a 20% increased risk for breast cancer, a two-fold increased risk for endometrial cancer, and a 50% increased incidence of colorectal cancer.2 Of note, prostate cancer incidence has been found to be consistently lower among men with diabetes.2 It has also been demonstrated that patients with T2DM have excess mortality for a number of cancers, including a 30% to 40% increase with pancreatic, a 2.5-fold increase with liver, a 30% increase with endometrial, a 15% to 30% increase with breast cancer, and a 20% to 50% increase with colorectal cancer.2-5
Type 1 diabetes.
In contrast to T2DM, type 1 DM (T1DM) is characterized by hyperglycemia and insulin deficiency as a result of the autoimmune destruction of pancreatic β-cells. It constitutes 5% to 15% of all cases of diabetes. Whereas some studies suggest that T1DM is associated with an increased risk of overall and site-specific cancer incidence and mortality,5 a review of epidemiologic evidence has noted no significant link in case-control studies and mixed results in cohort studies.6 Most recently, an analysis of five nationwide T1DM registries found that T1DM was associated with a nonsignificant overall elevated incidence of cancer in the total population and a 7% increase in incidence rate among women only. However, cancer incidence was increased for stomach, liver, pancreas, endometrium, and kidney cancers and was reduced for prostate cancer.7 These cancers were mostly detected within the first year after diagnosis of T1DM, which suggests a detection bias in those with newly diagnosed T1DM and that long-term hyperglycemia or insulin administration were not contributing to cancer risk. As patients in studies on T1DM tend to be younger than those in studies of T2DM, further studies with longer follow-up time are needed to understand if T1DM is truly associated with an increase in cancer risk.
Potential Mechanisms Linking Diabetes and Cancer
A number of factors have been proposed to contribute to the increased risk of cancer development and mortality in the setting of obesity and T2DM. These include hyperglycemia, insulin resistance, hyperinsulinemia, increased insulin-like growth factor-1 (IGF-1) levels, dyslipidemia, inflammatory cytokines, increased leptin, and decreased adiponectin.8 These mechanisms are summarized in Figures 1 and 2.
Fig 1.

Systemic effects of type 2 diabetes and insulin resistance that potentially promote tumor development and progression. Insulin resistance in metabolic tissues, such as fat, liver, and skeletal muscle, result in increased production of insulin from pancreatic β-cells, which leads to circulating hyperinsulinemia. Pancreatic β-cells eventually decompensate and hyperglycemia develops. Hyperglycemia also develops as a result of increased hepatic glucose production secondary to insulin resistance in the liver and decreased uptake into skeletal muscle and adipose tissue. Endogenous insulin acting on the liver increases insulin-like growth factor-1 (IGF-1) synthesis and leads to decreased concentrations of IGF-binding proteins (IGFBPs) 1 and 2, thus potentially increasing local concentration of bioavailable IGF-1. Adipose tissue inflammation occurs with insulin resistance with production of cytokines and changes in the circulating concentrations of adipokines, such as increased leptin and decreased adiponectin. Excess adiposity may lead to increased local aromatization of androgens to estrogens, which together with a decrease in the hepatic production of sex hormone–binding globulin (SHBG) caused by insulin resistance in the liver, may lead to an increase in levels of bioavailable estrogen. Insulin resistance is also associated with lipid abnormalities, including elevated triglycerides (TGs) and decreased HDL cholesterol. IL, interleukin; TNF, tumor necrosis factor.
Fig 2.

Effects of hyperinsulinemia on the tumor cell microenvironment and intracellular signaling that contribute to tumor growth and progression. The schematic depicts the potential direct and indirect effects of insulin on tumor growth. Insulin and insulin-like growth factors 1 and 2 (IGF-1 and IGF-2) bind to the two insulin receptor (IR) isoforms (IR-A, IR-B), IGF-1 receptor (IGF-1R), and IR/IGF-1R hybrid receptors (IR-A/IGF-1R, IR-B/IGF-1R) with different affinities. Solid arrows indicate strong affinity for the receptor, and dashed arrows represent weak affinity for the receptor. IGF-binding proteins (IGFBP) 1 and 2 are decreased by insulin and may result in increased bioavailable IGF-1 and IGF-2. Binding of insulin to IR primarily activates the phosphoinositide 3-kinase (PI3K)–Akt–mammalian target of rapamycin (mTOR) signaling pathway. Binding of IGF-1 and IGF-2 to IGF-1R stimulates the PI3K-Akt-mTOR and Ras-Raf-MAPK pathways. Increased local production of estrogen may occur as a result of increased expression of aromatase, activating estrogen receptor α in tumor cells. Inflammation in the tissue microenvironment may also be increased by hyperinsulinemia, which leads to local cytokine production and activation the Jak-Stat signaling pathway in the tumor. Erk, extracellular regulated kinase; IL, interleukin; IRS1/2, insulin receptor substrate; TNF, tumor necrosis factor.
Several studies have reported an increased incidence of breast, endometrial, and colorectal cancers that is most significant within the first months after T2DM diagnosis9 and even in the prediabetes phase.10,11 These results suggest that hyperinsulinemia, rather than hyperglycemia, in diabetes is associated with an increased risk of cancer. Endogenous hyperinsulinemia and high C-peptide levels—a marker of insulin secretion—are associated with breast and colorectal cancer progression, recurrence, and mortality.12,13
Insulin is a key member of the IGF family, which consists of the ligands, IGF-1, IGF-2, and insulin, and the tyrosine kinase receptors, insulin receptor (IR) and IGF-1 receptor (IGF-1R; Fig 2). In addition, there are six IGF-binding proteins (IGFBP) that bind to IGF-1 and IGF-2, but not insulin. These binding proteins protect IGFs from degradation, but IGFs bound to IGFBP are not free to bind to the receptors. IGF-1 levels are higher in overweight individuals,14 and high insulin levels inversely correlate with IGFBP-1 and -2 levels, which potentially leads to more free IGF-1 at tissue and cellular levels. A number of epidemiologic studies have shown that higher IGF-1 levels in the normal population correlate with an increased risk of breast, lung, prostate, and colorectal cancers.15 Furthermore, IGF-1R is overexpressed in a number of cancers, including liver, colorectal, breast, and prostate.15 Although insulin and IR signaling was traditionally thought to activate metabolic pathways, it has emerged that insulin and IR signaling can also have mitogenic effects. There are two subtypes of IR, named IR-A and IR-B. Whereas IR-B has mostly metabolic effects and is highly expressed in metabolic tissues, including the liver, IR-A is mainly expressed in fetal and cancer tissues.16 Both insulin and IGF-1 bind to the IR-A isoform, although the affinity of IGF-1 for IR-A is 60-fold lower than human insulin16 (Fig 2). As a result of the structural similarity of IGF-1R and IR, hybrid receptors also exist, which are made up of one half of an IR and one half of an IGF-1R (IR-A/IGF-1R and IR-B/IGF-1R). IGF-1, but not insulin, binds to these hybrid receptors.16 Unlike normal tissues, human breast cancers do not downregulate IR in the presence of hyperinsulinemia.17 In fact, IR expression is frequently upregulated relative to IGF-1R in breast cancer.18 Therefore, in cancers that overexpress IGF-1R or IR, chronically elevated endogenous insulin and/or IGF-1 levels such as are present in the setting of prediabetes may lead to mitogenic signaling and increased tumor growth and metastasis.
DIABETES PHARMACOTHERAPY AND CANCER
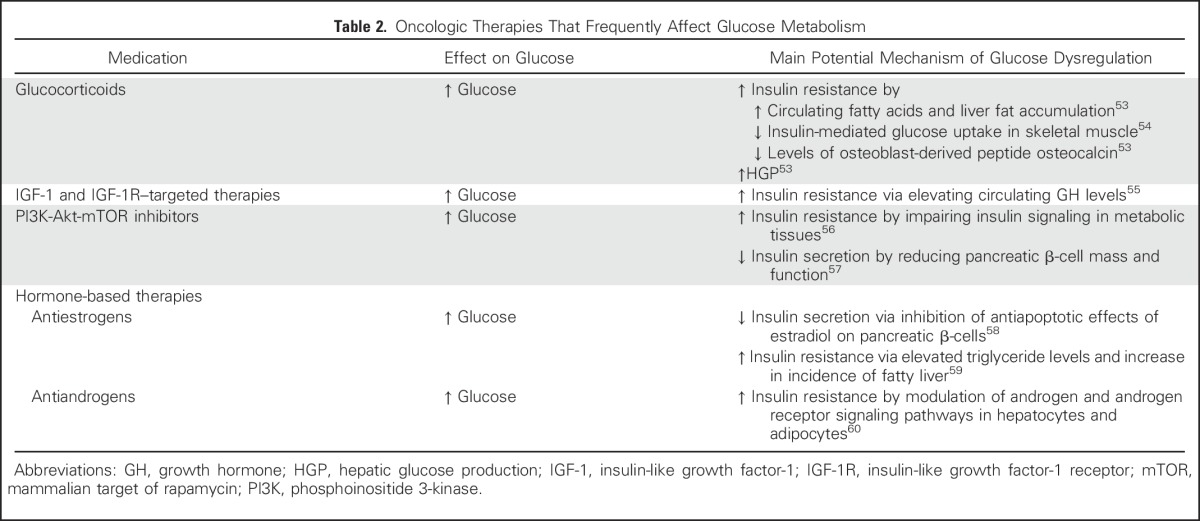
T2DM can be controlled by either oral or injectable hypoglycemic medications, whereas T1DM requires insulin treatment (Table 1). A number of studies have examined how medication used to treat diabetes may increase or decrease the risk of cancer development and/or mortality (Table 1). More recently, there has been a greater interest in the effects of cancer therapy on insulin resistance and hyperglycemia (Table 2).
Table 1.
Mechanisms of Diabetes Medications and Potential Risks and Benefits in Oncology Patients
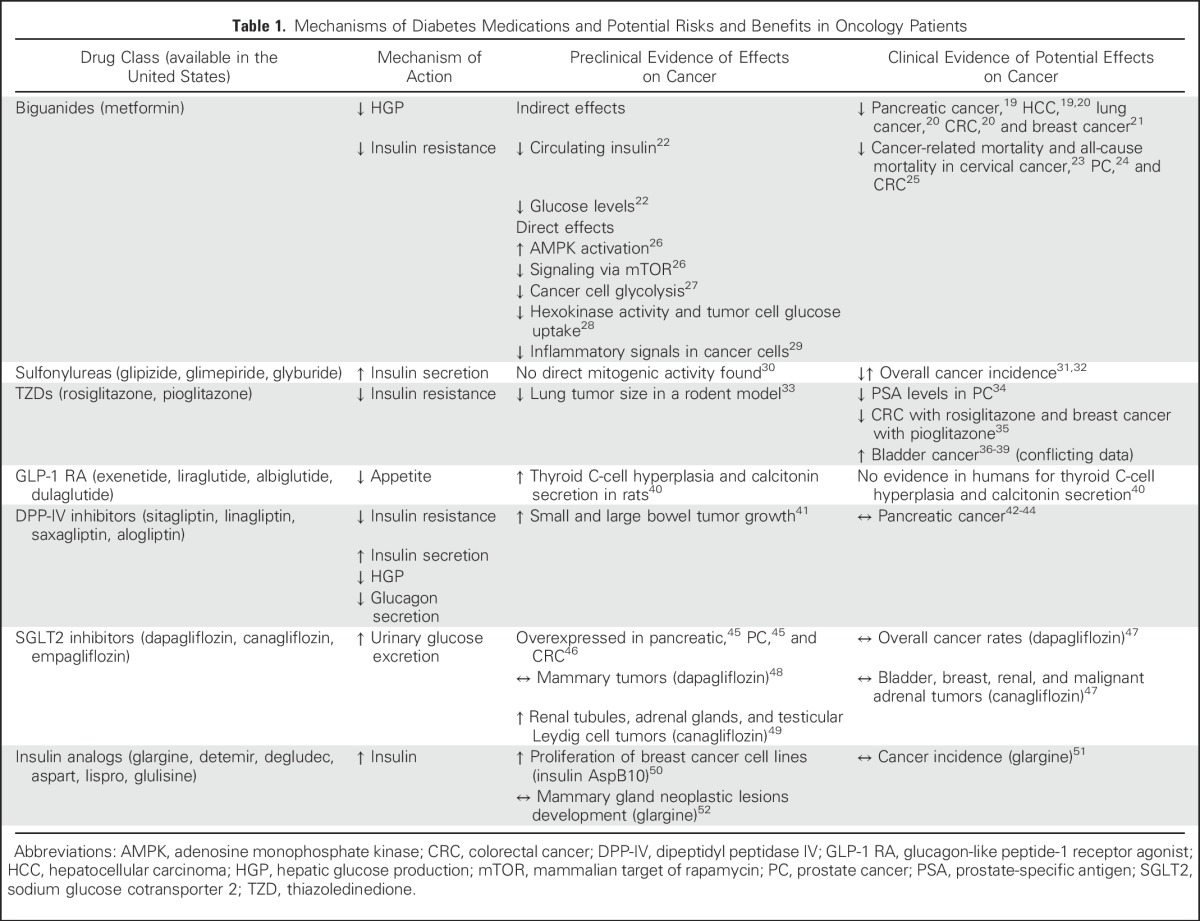
Table 2.
Oncologic Therapies That Frequently Affect Glucose Metabolism
Metformin and Cancer
Metformin is an oral biguanide that is well established as the first-line treatment of T2DM. Metformin lowers blood glucose by promoting hepatic adenosine monophosphate kinase phosphorylation, which thus inhibits hepatic gluconeogenesis and, secondarily, decreases circulating insulin levels22 (Table 1). Increased adenosine monophosphate kinase phosphorylation has been proposed to be cancer protective.61 There are two main hypotheses to explain the antineoplastic effects of metformin: indirect and direct mechanisms, as shown in Table 1. To date, most in vitro studies and animal models that demonstrate the antineoplastic effects of metformin involve doses that are substantially higher than those indicated for treatment of T2DM.62 Therefore, it is unclear if metformin can exert direct effects on tumors in patients.
Meta-analyses of epidemiologic studies have supported the proposed association between metformin use and lower incidence of pancreatic,19 hepatocellular,19,20 lung,20 colorectal,20 and breast cancer21 in patients with diabetes; however, as metformin is a first-line agent for diabetes and until recently was contraindicated in the US in patients with mild-moderate renal impairment, patients on metformin therapy alone are likely to have fewer comorbidities, which may confound these results.63 Other observational studies and meta-analyses have failed to report a lower incidence of prostate, lung, breast, and colorectal cancers among metformin users.64 The role of metformin in cancer-specific mortality has also been extensively studied. In a retrospective study of women with T2DM and cervical cancer, metformin use was associated with a 21% lower rate of cancer-related mortality and all-cause mortality.23 Metformin use was also associated with a 24% lower rate of prostate cancer–specific mortality and all-cause mortality among men with T2DM.24 A meta-analysis of observational studies found that metformin was associated with a 35% reduction in all-cause and cancer-specific mortality for colon cancer, but no significant mortality reduction was reported for breast or prostate cancer.25 Furthermore, a Canadian population-based retrospective analysis showed no significant association between cumulative duration of metformin use and overall as well as cancer-specific mortality among patients with breast cancer and recently diagnosed T2DM.65 As metformin has few adverse effects and is inexpensive, its use as an antineoplastic agent is intriguing. Currently, there are more than 200 ongoing trials,66 many of which are randomized control phase II and III trials investigating the effects of metformin on cancer-related outcomes, including all-cause mortality and cancer-related mortality. The largest of these studies is the MA.32 study,67 which is a phase III randomized trial of metformin versus placebo examining recurrence and survival in early-stage breast cancer. Other smaller trials are assessing the effects of metformin on several end points, including tumor proliferation markers, mTOR activity, and clinical end points, such as disease progression and changes in serum cancer biomarkers, among patients with prostate, hepatobiliary, lung, and colorectal cancer. The results of these trials will help guide clinicians in selecting the appropriate patients who may benefit from metformin as an adjuvant antineoplastic agent.
Thiazolidinediones and Cancer
There are two thiazolidinediones (TZDs) in current clinical use, rosiglitazone and pioglitzone. TZDs are peroxisome proliferator-activated receptor-γ (PPAR-γ) agonists. PPAR-γ is a transcription factor that is predominantly expressed in adipocytes. Activation of PPAR-γ by TZDs leads to adipocyte differentiation, increased adiponectin production, increased glucose uptake, and decreased inflammatory cytokines, which leads to improved insulin sensitivity and decreased circulating insulin levels.68
The role of TZDs in cancer treatment and prevention is uncertain (Table 1). TZDs have been shown to be associated with approximately 20% to 40% lower prostate-specific antigen levels among patients with prostate cancer34 and regression of lung tumor size in a rodent model.33 In a meta-analysis of randomized trials, rosiglitazone was associated with a significantly lower risk of colon cancer and pioglitazone was associated with a significant reduction in breast cancer.35 Other studies have failed to corroborate their antineoplastic effect in colorectal and breast cancer.69,70 Furthermore, there are concerns of increased incidence of bladder cancer associated with pioglitazone use,36-39 which may be related to a detection bias, but should be addressed by well-designed clinical studies.
Incretin-Based Drugs and Cancer
Incretin-based therapies are a relatively new class of medications to treat T2DM. Incretin therapies fall into two broad groups, glucagon-like peptide-1 (GLP-1) receptor agonists (GLP-1RAs) and dipeptidyl peptidase-4 (DPP4) inhibitors. GLP-1 is a hormone secreted from intestinal l-cells in response to food intake and activates the GLP-1 receptor.71 Circulating GLP-1 is rapidly degraded by the enzyme DPP4.72 In individuals with T2DM, the incretin effect is blunted, which makes it desirable to administer GLP-1RA or increase the activity of endogenous GLP-1 by inhibiting its degradation by using DPP4 inhibitors. These incretin-based therapies work by several mechanisms to reduce blood glucose (Table 1). Several drugs from both classes have been approved for treatment of T2DM. In 2011, an analysis by the US Food and Drug Administration (FDA) Adverse Events Reporting reported increased rates of pancreatic cancer with incretin-based drugs compared with other hypoglycemic agents.73 However, the FDA Adverse Events Reporting report had several methodologic shortcomings, including reporting biases and lack of information regarding other well-established risk factors for pancreatic cancer. A recent analysis of FDA and European Medicine Agency databases provided no compelling evidence for an increased risk of pancreatic neoplasia with incretin-based therapy.44 Large-scale clinical trials that evaluated the efficacy and safety of DPP4 inhibitors did not identify an increased risk for pancreatic cancer.42,43 Concerns have also been raised regarding the effects of GLP-1RA on thyroid C-cell hyperplasia and the development of medullary thyroid cancer; however, whereas GLP-1RA has been shown to increase calcitonin secretion and stimulate C-cell hyperplasia in rodents, a similar effect was not found for humans.40 The effects of incretin therapy on colonic epithelia are not entirely clear: it was previously suggested that GLP-1RA and DPP4 inhibitors have the capacity to decrease growth and survival of colon cancer cells74; however, more recent preclinical data indicate that GLP-1RA agonists may promote small and large bowel tumor growth.41 Of interest, an increased risk of colorectal cancer has been reported after bariatric surgery, which has been proposed to be the result of increased incretin secretion.75
As these incretin-based agents are increasingly used in the treatment of T2DM, their potential antineoplastic and procarcinogenic effects must be further addressed in clinical studies.
Insulin Analogs and Cancer
Recombinant human insulin (HI) analogs have different alterations in the insulin amino acid sequence, which results in variable pharmacokinetics and tumor-promoting properties. Insulin AspB10 is an insulin analog that is not in clinical use and has been shown to increase proliferation in breast cancer cell lines.76 Whereas it was initially hypothesized that its mitogenic effect is mediated by its higher affinity to IGF-1R than HI, it has relatively low affinity to IGF-1R compared with IGF-1.50 Preclinical studies have demonstrated that mitogenic effects of AspB10 may instead be mediated by its greater affinity for IR and prolonged phosphorylation of IR and the downstream signaling cascade.50 Theoretically, insulin glargine, a long-acting insulin analog, is more likely to have increased mitogenic action compared with HI as a result of its higher affinity to IGF-1R than HI; however, studies in rodents have failed to demonstrate any significant effect of glargine injections on mammary gland tumor development.52 Epidemiologic studies have raised concerns for increased cancer rates among insulin users, particularly for insulin glargine users76; however, methodologic limitations have been identified in these studies,77 and meta-analyses have shown inconsistent results.76,77 To date, the only prospective randomized trial that assessed the effect on insulin glargine among dysglycemic patients reported a neutral association with cancer incidence.51
In summary, whereas in vitro studies have raised concerns regarding the mitogenic effects of insulin analogs, particularly glargine, the relevance of these in vitro studies to humans is questionable. There is no compelling evidence that any insulin analog in clinical use increases cancer risk or cancer-related mortality.
Sulfonylureas and Cancer
Sulfonylureas (SUs) are among the oldest drug classes available for the treatment of T2DM. SUs increase insulin secretion from pancreatic β-cells.78 Although SUs have been in clinical use for many years, their associations with cancer remains uncertain. In an observational study, treatment with the SU glibenclamide was associated with a 20% to 25% increased risk of overall cancer incidence, but with no single cancer type.31 However, others reported either a neutral or protective effect of SU on overall cancer incidence.32 No direct tumorigenic activity of glibenclamide has been found in preclinical studies.30
Sodium-Glucose Cotransporter Type 2 Inhibitors and Cancer
Sodium-glucose cotransporter type 2 (SGLT2) inhibitors block renal glucose reabsorption, thereby causing glycosuria,47 lowering blood glucose levels. There are currently three FDA-approved SGLT2 inhibitors for the treatment of T2DM. SGLT2 has been found to be overexpressed in pancreatic,45 prostate,45 and colon cancer,46 and SGLT2 inhibitors were found to reduce tumor growth and increase survival of pancreatic adenocarcinoma in a rodent model.45
Concerns have been raised with regard to dapagliflozin use and its association with increased rates of bladder and breast cancer79; however, observational studies found no overall difference in rates of malignancies between patients treated with dapagliflozin and either placebo or other hypoglycemic medications.47 In preclinical studies, rats exposed to high-dose dapagliflozin did not exhibit an increased incidence of mammary tumors.48 Human database analyses found that the overall incidence of bladder, breast, renal, and malignant adrenal tumors was not increased in patients treated with the SGTL2 inhibitor, canagliflozin, compared with nonusers.47 Whereas canagliflozin has been associated with increased neoplasms of renal tubules, adrenal glands, and testicular Leydig cell tumors among rats,49 the proposed underlying mechanism is canagliflozin-induced carbohydrate malabsorption and disrupted calcium homeostasis, which are not observed in humans who are exposed to canagliflozin.49 As SGLT2 inhibitors are relatively new in clinical practice, their effects on cancer incidence and mortality should be further delineated in large-scale, controlled studies with longer durations of follow-up.
CANCER PHARMACOTHERAPY–INDUCED INSULIN RESISTANCE AND HYPERGLYCEMIA
Glucocorticoid-Induced Insulin Resistance and Hyperglycemia
Glucocorticoids (GCs) are a cornerstone of treatment of virtually all hematologic malignancies and are also used in several solid tumors, such as prostate cancer and Kaposi’s sarcoma.80 In addition, GCs are commonly used as supportive treatment of brain metastases–induced cerebral edema, emesis, pain, and cancer-related cachexia.80 As many as 30% of patients treated with GCs will develop hyperglycemia, with even higher rates in individuals with chronic exposure, on higher doses, and receiving more potent GCs.81,82 In a retrospective analysis of patients with multiple myeloma, GC-induced hyperglycemia was found to be a predictor of lower overall survival.83 It has also been shown in vitro that hyperglycemia decreases the antineoplastic effects of GCs, whereas hypoglycemia enhances GC-mediated cell death.56 The mechanisms of GC-induced hyperglycemia are shown in Table 2. Further studies are needed to determine if individuals who develop insulin resistance and hyperglycemia as a result of GC therapy have a worse prognosis than those who do not.
Hormone-Based Therapy–Induced Insulin Resistance and Hyperglycemia
Androgens are an important determinant of male body composition, promoting lean mass and suppressing visceral adiposity.60 Epidemiologic studies and meta-analyses have shown that low testosterone levels in men are associated with the development of insulin resistance and metabolic syndrome.84,85 Androgen-deprivation therapy (ADT), accomplished by either surgical or chemical castration, is the backbone of treatment of advanced prostate cancer. Insulin resistance has been reported as early as 3 months after starting ADT treatment—proposed mechanisms are shown in Table 2.60 In addition, men who undergo long-term ADT show significantly higher rates of insulin resistance and T2DM than do controls.60 Selective androgen receptor modulators are an emerging class of drugs that display tissue-selective activation of androgen receptors and have been shown to improve lean body mass and physical function in clinical trials.60 Compared with traditional ADT, selective androgen receptor modulators may provide metabolic benefits to men with prostate cancer.
Estradiol also plays a central role in metabolic homeostasis. It has beneficial effects on insulin resistance in preclinical studies86 and in humans.87 Selective estrogen modulators (SERMs) are an important drug class in the treatment of estrogen receptor (ER)–positive breast cancer. SERMs exhibit either antagonist or agonist effects on the ER in a tissue-specific pattern.59 Tamoxifen is one of the first-generation SERMs, and it acts as an ER antagonist in mammary tissue and as a partial agonist in the endometrium. In clinical studies, tamoxifen has been shown to have detrimental metabolic effects, including increased T2DM incidence,88 lower insulin sensitivity, and higher TG levels59 (Table 2).
Raloxifene, another SERM, has fewer adverse metabolic effects than tamoxifen.59 New-generation SERMs, such as bazedoxifene—mainly used in the treatment of menopausal symptoms—are associated with an improved metabolic profile, including attenuated weight gain, lower TG and LDL, higher HDL, lower fasting glucose, and superior insulin sensitivity compared with older-generation SERMs.59
Given the potential for hormonal-based therapy–induced metabolic derangements, patients should be educated regarding lifestyle modification and should be screened for diabetes or prediabetes before starting and during hormonal-based treatment.
Phosphoinositide 3-Kinase-Akt-Mammalian Target of Rapamycin Inhibitors–Induced Insulin Resistance and Hyperglycemia
The phosphoinositide 3-kinase (PI3K)-Akt-mammalian target of rapamycin (mTOR) signaling pathway plays a pivotal role in cell proliferation, differentiation, and survival.89 Mutations along this pathway are frequently found in human cancers. Multiple agents have been developed that target specific components of this signaling cascade.89
mTOR inhibitors, which were initially developed as immunosuppressors, are increasingly used as antineoplastic therapy.90 In randomized control trials, hyperglycemia is frequency reported in patients, ranging between 12% and 50%.90 Clinical trials have shown that PI3K inhibitors are associated with hyperglycemia in > 30% of patients.56,89 PI3K and Akt inhibitors are currently at different stages of clinical development.89 The specificity for PI3K and Akt isoforms differs between drugs and seems to affect their propensity to induce insulin resistance and hyperglycemia.56 The PI3K-Akt-mTOR pathway is a major insulin and IR signaling pathway (Fig 2), and mTOR complex 1 is also a positive regulator of pancreatic β-cell mass and function.57 Therefore, PI3K-Akt-mTOR inhibition not only increases insulin resistance, but may also impair insulin secretion (Table 2).
Given the high risk of insulin resistance and hyperglycemia associated with these medications, a 2012 Task Force of the National Cancer Institute Investigational Drug Steering Committee convened an interdisciplinary expert panel and issued a consensus statement regarding screening, monitoring, and management of hyperglycemia in patients treated with PI3K-Akt-mTOR inhibitors.91 It is recommended that all patients who are treated with these agents undergo baseline screening assessment for prediabetes or diabetes with fasting glucose and/or hemoglobin A1c and be monitored periodically thereafter.91 The threshold for starting treatment in nondiabetic patients who are hyperglycemic depends on whether the hyperglycemia is transient, that is, resolves before a subsequent dose, and also on the level of hyperglycemia. The steering committee recommends that if a patient’s fasting glucose is 125 to 160 mg/dL, once daily home glucose monitoring should be initiated; if 160 to 500 mg/dL, home glucose monitoring should be performed twice daily; and if > 500 mg/dL, referral should be made to an endocrinologist or diabetes specialist.91
IGF-1R–Targeted Therapy–Induced Insulin Resistance and Hyperglycemia
Binding of IGF-1 and IGF-2 to the IGF-1R has been shown to stimulate a number of oncologic pathways, including the PI3K-Akt-mTOR and MAPK pathways92 (Fig 2). On the basis of the proposed importance of the IGF-1 and IGF-1R signaling pathway in carcinogenesis, it was postulated that IGF-1R blockade may be an effective antineoplastic therapy. Several anti–IGF-1R therapeutic agents have been developed, including monoclonal antibodies and small-molecule tyrosine kinase inhibitors (TKIs); however, whereas early trials showed promising results, subsequent larger clinical studies have failed to show significant benefit in many cancers.92 An important factor that underlies the failure of some of these drugs in clinical trials is potentially a result of increased circulating IGF-1, which leads to activation of IR, particularly the IR-A isoform, and may be more sensitive to IGF-1 in the presence of IGF-1R blockade.92 This effect may be more common with monoclonal antibodies that target IGF-1R rather than TKIs, as—because of the similarity between the tyrosine kinase domains of IR and IGF-1R—TKIs block activation of both receptors. In addition, IGF-1R blockade also disrupts the normal inhibitory feedback at the hypothalamic-pituitary level, which results in increased circulating growth hormone levels, which may activate the growth hormone receptor on tumors, in addition to exacerbating insulin resistance and further increasing circulating IGF-1 levels55 (Table 2).
Hyperglycemia is a frequently reported adverse effect in many patients who are treated with IGF-1R inhibitors.55 The overall incidence of severe hyperglycemia can be as high as 20% to 25%.56 The highest incidence of hyperglycemia has been reported for the combination of anti–IGF-1R and mTOR inhibitors, reaching almost 72% in a single study.55 Patients with preexisting glucose intolerance and concomitant corticosteroid treatment seem to be more prone to develop hyperglycemia.55 Whether the development of metabolic adverse effects from these agents is important in therapeutic resistance—or development of other adverse events—remains to be determined.
CONCLUSION
With the escalating prevalence of diabetes, the significantly increased risk of cancer and cancer-related mortality is causing great concern internationally. The possible role of metformin as an antineoplastic agent is being extensively studied and we await the outcomes of large-scale randomized prospective studies. There is currently no definitive evidence for a mitogenic effect of any antidiabetic therapeutics; however, given the novelty of some of these agents and conflicting study results, future well-designed prospective trials are needed to further elucidate their neoplastic or antineoplastic effects.
Many antineoplastic agents have been found to have deleterious metabolic effects, although it is unclear if these effects alter response to therapy or cancer-related mortality. Current recommendations advocate screening for diabetes and prediabetes in patients who begin therapy with an agent that is known to have deleterious metabolic effects, with periodic monitoring. In all patients with T2DM, without contraindications, metformin is recommended as first-line therapy. Hemoglobin A1c goals in patients with T2DM and cancer have been traditionally based on the burden of diabetes treatment, preventing symptomatic hyperglycemia, and the predicted life expectancy of the patient. However, research is needed to determine if more stringent management of insulin resistance and hyperglycemia in patients with cancer improves their response to oncology treatments and overall survival.
Footnotes
Supported by The 2013 Dr. Pinchas Borenstein Talpiot Medical Leadership Program (to G.S.) and by the National Institutes of Health, National Cancer Institute Grants No. 5R01CA171558, 5R01CA128799 (to D.L.), and 5K08CA190770 (to E.J.G.).
Authors’ disclosures of potential conflicts of interest are found in the article online at www.jco.org. Author contributions are found at the end of this article.
AUTHOR CONTRIBUTIONS
Conception and design: Gadi Shlomai, Derek LeRoith, Emily Jane Gallagher
Collection and assembly of data: Gadi Shlomai, Derek LeRoith, Emily Jane Gallagher
Data analysis and interpretation: All authors
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Type 2 Diabetes Mellitus and Cancer: The Role of Pharmacotherapy
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or jco.ascopubs.org/site/ifc.
Gadi Shlomai
No relationship to disclose
Brian Neel
No relationship to disclose
Derek LeRoith
Leadership: Endocrine Fellows Foundation, American Association of Clinical Endocrinologists Board
Consulting or Advisory Role: AstraZeneca
Research Funding: Sanofi
Emily Jane Gallagher
Travel, Accommodations, Expenses: Sanofi
REFERENCES
- 1.Alberti KG. Screening and diagnosis of prediabetes: Where are we headed? Diabetes Obes Metab. 2007;9(Suppl 1):12–16. doi: 10.1111/j.1463-1326.2007.00764.x. [DOI] [PubMed] [Google Scholar]
- 2.Tsilidis KK, Kasimis JC, Lopez DS, et al. Type 2 diabetes and cancer: Umbrella review of meta-analyses of observational studies. BMJ. 2015;350:g7607. doi: 10.1136/bmj.g7607. [DOI] [PubMed] [Google Scholar]
- 3.Barone BB, Yeh HC, Snyder CF, et al. Long-term all-cause mortality in cancer patients with preexisting diabetes mellitus: A systematic review and meta-analysis. JAMA. 2008;300:2754–2764. doi: 10.1001/jama.2008.824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Campbell PT, Newton CC, Patel AV, et al. Diabetes and cause-specific mortality in a prospective cohort of one million U.S. adults. Diabetes Care. 2012;35:1835–1844. doi: 10.2337/dc12-0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harding JL, Shaw JE, Peeters A, et al. Cancer risk among people with type 1 and type 2 diabetes: Disentangling true associations, detection bias, and reverse causation. Diabetes Care. 2015;38:264–270. doi: 10.2337/dc14-1996. [DOI] [PubMed] [Google Scholar]
- 6.Gordon-Dseagu VL, Shelton N, Mindell JS. Epidemiological evidence of a relationship between type-1 diabetes mellitus and cancer: a review of the existing literature. Int J Cancer. 2013;132:501–508. doi: 10.1002/ijc.27703. [DOI] [PubMed] [Google Scholar]
- 7.Carstensen B, Read SH, Friis S, et al. Cancer incidence in persons with type 1 diabetes: A five-country study of 9,000 cancers in type 1 diabetic individuals. Diabetologia. 2016;59:980–988. doi: 10.1007/s00125-016-3884-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Giovannucci E, Harlan DM, Archer MC, et al. Diabetes and cancer: A consensus report. Diabetes Care. 2010;33:1674–1685. doi: 10.2337/dc10-0666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Singh S, Earle CC, Bae SJ, et al. Incidence of diabetes in colorectal cancer survivors. J Natl Cancer Inst. 2016;108:djv402. doi: 10.1093/jnci/djv402. [DOI] [PubMed] [Google Scholar]
- 10.Lipscombe LL, Goodwin PJ, Zinman B, et al. Increased prevalence of prior breast cancer in women with newly diagnosed diabetes. Breast Cancer Res Treat. 2006;98:303–309. doi: 10.1007/s10549-006-9166-3. [DOI] [PubMed] [Google Scholar]
- 11.Onitilo AA, Stankowski RV, Berg RL, et al. Breast cancer incidence before and after diagnosis of type 2 diabetes mellitus in women: Increased risk in the prediabetes phase. Eur J Cancer Prev. 2014;23:76–83. doi: 10.1097/CEJ.0b013e32836162aa. [DOI] [PubMed] [Google Scholar]
- 12.Ahern TP, Hankinson SE, Willett WC, et al. Plasma C-peptide, mammographic breast density, and risk of invasive breast cancer. Cancer Epidemiol Biomarkers Prev. 2013;22:1786–1796. doi: 10.1158/1055-9965.EPI-13-0375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jenab M, Riboli E, Cleveland RJ, et al. Serum C-peptide, IGFBP-1 and IGFBP-2 and risk of colon and rectal cancers in the European Prospective Investigation into Cancer and Nutrition. Int J Cancer. 2007;121:368–376. doi: 10.1002/ijc.22697. [DOI] [PubMed] [Google Scholar]
- 14.Crowe FL, Key TJ, Allen NE, et al. A cross-sectional analysis of the associations between adult height, BMI and serum concentrations of IGF-I and IGFBP-1 -2 and -3 in the European Prospective Investigation into Cancer and Nutrition (EPIC) Ann Hum Biol. 2011;38:194–202. doi: 10.3109/03014460.2010.507221. [DOI] [PubMed] [Google Scholar]
- 15.Yu H, Rohan T. Role of the insulin-like growth factor family in cancer development and progression. J Natl Cancer Inst. 2000;92:1472–1489. doi: 10.1093/jnci/92.18.1472. [DOI] [PubMed] [Google Scholar]
- 16.Belfiore A, Frasca F, Pandini G, et al. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr Rev. 2009;30:586–623. doi: 10.1210/er.2008-0047. [DOI] [PubMed] [Google Scholar]
- 17.Mulligan AM, O’Malley FP, Ennis M, et al. Insulin receptor is an independent predictor of a favorable outcome in early stage breast cancer. Breast Cancer Res Treat. 2007;106:39–47. doi: 10.1007/s10549-006-9471-x. [DOI] [PubMed] [Google Scholar]
- 18.Harrington SC, Weroha SJ, Reynolds C, et al. Quantifying insulin receptor isoform expression in FFPE breast tumors. Growth Horm IGF Res. 2012;22:108–115. doi: 10.1016/j.ghir.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Decensi A, Puntoni M, Goodwin P, et al. Metformin and cancer risk in diabetic patients: A systematic review and meta-analysis. Cancer Prev Res (Phila) 2010;3:1451–1461. doi: 10.1158/1940-6207.CAPR-10-0157. [DOI] [PubMed] [Google Scholar]
- 20.Gandini S, Puntoni M, Heckman-Stoddard BM, et al. Metformin and cancer risk and mortality: A systematic review and meta-analysis taking into account biases and confounders. Cancer Prev Res (Phila) 2014;7:867–885. doi: 10.1158/1940-6207.CAPR-13-0424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Col NF, Ochs L, Springmann V, et al. Metformin and breast cancer risk: A meta-analysis and critical literature review. Breast Cancer Res Treat. 2012;135:639–646. doi: 10.1007/s10549-012-2170-x. [DOI] [PubMed] [Google Scholar]
- 22.Zhou G, Myers R, Li Y, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Han K, Pintilie M, Lipscombe LL, et al. Association between metformin use and mortality after cervical cancer in older women with diabetes. Cancer Epidemiol Biomarkers Prev. 2016;25:507–512. doi: 10.1158/1055-9965.EPI-15-1008. [DOI] [PubMed] [Google Scholar]
- 24.Margel D, Urbach DR, Lipscombe LL, et al. Metformin use and all-cause and prostate cancer-specific mortality among men with diabetes. J Clin Oncol. 2013;31:3069–3075. doi: 10.1200/JCO.2012.46.7043. [DOI] [PubMed] [Google Scholar]
- 25.Lega IC, Shah PS, Margel D, et al. The effect of metformin on mortality following cancer among patients with diabetes. Cancer Epidemiol Biomarkers Prev. 2014;23:1974–1984. doi: 10.1158/1055-9965.EPI-14-0327. [DOI] [PubMed] [Google Scholar]
- 26.El-Mir MY, Nogueira V, Fontaine E, et al. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem. 2000;275:223–228. doi: 10.1074/jbc.275.1.223. [DOI] [PubMed] [Google Scholar]
- 27.Fendt SM, Bell EL, Keibler MA, et al. Metformin decreases glucose oxidation and increases the dependency of prostate cancer cells on reductive glutamine metabolism. Cancer Res. 2013;73:4429–4438. doi: 10.1158/0008-5472.CAN-13-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Salani B, Marini C, Rio AD, et al. Metformin impairs glucose consumption and survival in Calu-1 cells by direct inhibition of hexokinase-II. Sci Rep. 2013;3:2070. doi: 10.1038/srep02070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hirsch HA, Iliopoulos D, Struhl K. Metformin inhibits the inflammatory response associated with cellular transformation and cancer stem cell growth. Proc Natl Acad Sci USA. 2013;110:972–977. doi: 10.1073/pnas.1221055110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Núñez M, Medina V, Cricco G, et al. Glibenclamide inhibits cell growth by inducing G0/G1 arrest in the human breast cancer cell line MDA-MB-231. BMC Pharmacol Toxicol. 2013;14:6. doi: 10.1186/2050-6511-14-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tuccori M, Wu JW, Yin H, et al. The use of glyburide compared with other sulfonylureas and the risk of cancer in patients with type 2 diabetes. Diabetes Care. 2015;38:2083–2089. doi: 10.2337/dc15-1358. [DOI] [PubMed] [Google Scholar]
- 32.Yang X, So WY, Ma RC, et al. Use of sulphonylurea and cancer in type 2 diabetes—The Hong Kong Diabetes Registry. Diabetes Res Clin Pract. 2010;90:343–351. doi: 10.1016/j.diabres.2010.08.022. [DOI] [PubMed] [Google Scholar]
- 33.Girnun GD, Chen L, Silvaggi J, et al. Regression of drug-resistant lung cancer by the combination of rosiglitazone and carboplatin. Clin Cancer Res. 2008;14:6478–6486. doi: 10.1158/1078-0432.CCR-08-1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mueller E, Smith M, Sarraf P, et al. Effects of ligand activation of peroxisome proliferator-activated receptor gamma in human prostate cancer. Proc Natl Acad Sci USA. 2000;97:10990–10995. doi: 10.1073/pnas.180329197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Monami M, Dicembrini I, Mannucci E. Thiazolidinediones and cancer: Results of a meta-analysis of randomized clinical trials. Acta Diabetol. 2014;51:91–101. doi: 10.1007/s00592-013-0504-8. [DOI] [PubMed] [Google Scholar]
- 36.Azoulay L, Yin H, Filion KB, et al. The use of pioglitazone and the risk of bladder cancer in people with type 2 diabetes: Nested case-control study. BMJ. 2012;344:e3645. doi: 10.1136/bmj.e3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lewis JD, Ferrara A, Peng T, et al. Risk of bladder cancer among diabetic patients treated with pioglitazone: Interim report of a longitudinal cohort study. Diabetes Care. 2011;34:916–922. doi: 10.2337/dc10-1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lewis JD, Habel LA, Quesenberry CP, et al. Pioglitazone use and risk of bladder cancer and other common cancers in persons with diabetes. JAMA. 2015;314:265–277. doi: 10.1001/jama.2015.7996. [DOI] [PubMed] [Google Scholar]
- 39.Piccinni C, Motola D, Marchesini G, et al. Assessing the association of pioglitazone use and bladder cancer through drug adverse event reporting. Diabetes Care. 2011;34:1369–1371. doi: 10.2337/dc10-2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rosol TJ. On-target effects of GLP-1 receptor agonists on thyroid C-cells in rats and mice. Toxicol Pathol. 2013;41:303–309. doi: 10.1177/0192623312472402. [DOI] [PubMed] [Google Scholar]
- 41.Koehler JA, Baggio LL, Yusta B, et al. GLP-1R agonists promote normal and neoplastic intestinal growth through mechanisms requiring Fgf7. Cell Metab. 2015;21:379–391. doi: 10.1016/j.cmet.2015.02.005. [DOI] [PubMed] [Google Scholar]
- 42.Gokhale M, Buse JB, Gray CL, et al. Dipeptidyl-peptidase-4 inhibitors and pancreatic cancer: A cohort study. Diabetes Obes Metab. 2014;16:1247–1256. doi: 10.1111/dom.12379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Scirica BM, Bhatt DL, Braunwald E, et al. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N Engl J Med. 2013;369:1317–1326. doi: 10.1056/NEJMoa1307684. [DOI] [PubMed] [Google Scholar]
- 44.Singh S, Chang HY, Richards TM, et al. Glucagonlike peptide 1-based therapies and risk of hospitalization for acute pancreatitis in type 2 diabetes mellitus: A population-based matched case-control study. JAMA Intern Med. 2013;173:534–539. doi: 10.1001/jamainternmed.2013.2720. [DOI] [PubMed] [Google Scholar]
- 45.Scafoglio C, Hirayama BA, Kepe V, et al. Functional expression of sodium-glucose transporters in cancer. Proc Natl Acad Sci USA. 2015;112:E4111–E4119. doi: 10.1073/pnas.1511698112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Saito T, Okada S, Yamada E, et al. Effect of dapagliflozin on colon cancer cell [Rapid Communication] Endocr J. 2015;62:1133–1137. doi: 10.1507/endocrj.EJ15-0396. [DOI] [PubMed] [Google Scholar]
- 47.Lin HW, Tseng CH. A review on the relationship between SGLT2 inhibitors and cancer. Int J Endocrinol. 2014;2014:719578. doi: 10.1155/2014/719578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reilly TP, Graziano MJ, Janovitz EB, et al. Carcinogenicity risk assessment supports the chronic safety of dapagliflozin, an inhibitor of sodium-glucose co-transporter 2, in the treatment of type 2 diabetes mellitus. Diabetes Ther. 2014;5:73–96. doi: 10.1007/s13300-014-0053-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.De Jonghe S, Proctor J, Vinken P, et al. Carcinogenicity in rats of the SGLT2 inhibitor canagliflozin. Chem Biol Interact. 2014;224:1–12. doi: 10.1016/j.cbi.2014.09.018. [DOI] [PubMed] [Google Scholar]
- 50.Gallagher EJ, LeRoith D. Obesity and diabetes: The increased risk of cancer and cancer-related mortality. Physiol Rev. 2015;95:727–748. doi: 10.1152/physrev.00030.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bordeleau L, Yakubovich N, Dagenais GR, et al. The association of basal insulin glargine and/or n-3 fatty acids with incident cancers in patients with dysglycemia. Diabetes Care. 2014;37:1360–1366. doi: 10.2337/dc13-1468. [DOI] [PubMed] [Google Scholar]
- 52.Stammberger I, Essermeant L. Insulin glargine: A reevaluation of rodent carcinogenicity findings. Int J Toxicol. 2012;31:137–142. doi: 10.1177/1091581811431111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ferris HA, Kahn CR. New mechanisms of glucocorticoid-induced insulin resistance: Make no bones about it. J Clin Invest. 2012;122:3854–3857. doi: 10.1172/JCI66180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hwang JL, Weiss RE. Steroid-induced diabetes: A clinical and molecular approach to understanding and treatment. Diabetes Metab Res Rev. 2014;30:96–102. doi: 10.1002/dmrr.2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ma H, Zhang T, Shen H, et al. The adverse events profile of anti-IGF-1R monoclonal antibodies in cancer therapy. Br J Clin Pharmacol. 2014;77:917–928. doi: 10.1111/bcp.12228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ariaans G, de Jong S, Gietema JA, et al. Cancer-drug induced insulin resistance: Innocent bystander or unusual suspect. Cancer Treat Rev. 2015;41:376–384. doi: 10.1016/j.ctrv.2015.02.007. [DOI] [PubMed] [Google Scholar]
- 57.Rachdi L, Balcazar N, Osorio-Duque F, et al. Disruption of Tsc2 in pancreatic beta cells induces beta cell mass expansion and improved glucose tolerance in a TORC1-dependent manner. Proc Natl Acad Sci USA. 2008;105:9250–9255. doi: 10.1073/pnas.0803047105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Le May C, Chu K, Hu M, et al. Estrogens protect pancreatic beta-cells from apoptosis and prevent insulin-deficient diabetes mellitus in mice. Proc Natl Acad Sci USA. 2006;103:9232–9237. doi: 10.1073/pnas.0602956103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xu B, Lovre D, Mauvais-Jarvis F. Effect of selective estrogen receptor modulators on metabolic homeostasis. Biochimie. 2016;124:92–97. doi: 10.1016/j.biochi.2015.06.018. [DOI] [PubMed] [Google Scholar]
- 60.Yu IC, Lin HY, Sparks JD, et al. Androgen receptor roles in insulin resistance and obesity in males: The linkage of androgen-deprivation therapy to metabolic syndrome. Diabetes. 2014;63:3180–3188. doi: 10.2337/db13-1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tsilidis KK, Capothanassi D, Allen NE, et al. Metformin does not affect cancer risk: A cohort study in the U.K. Clinical Practice Research Datalink analyzed like an intention-to-treat trial. Diabetes Care. 2014;37:2522–2532. doi: 10.2337/dc14-0584. [DOI] [PubMed] [Google Scholar]
- 62.Pollak M. Overcoming drug development bottlenecks with repurposing: Repurposing biguanides to target energy metabolism for cancer treatment. Nat Med. 2014;20:591–593. doi: 10.1038/nm.3596. [DOI] [PubMed] [Google Scholar]
- 63.Suissa S, Azoulay L. Metformin and the risk of cancer: Time-related biases in observational studies. Diabetes Care. 2012;35:2665–2673. doi: 10.2337/dc12-0788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kowall B, Stang A, Rathmann W, et al. No reduced risk of overall, colorectal, lung, breast, and prostate cancer with metformin therapy in diabetic patients: Database analyses from Germany and the UK. Pharmacoepidemiol Drug Saf. 2015;24:865–874. doi: 10.1002/pds.3823. [DOI] [PubMed] [Google Scholar]
- 65.Lega IC, Austin PC, Gruneir A, et al. Association between metformin therapy and mortality after breast cancer: A population-based study. Diabetes Care. 2013;36:3018–3026. doi: 10.2337/dc12-2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. ClinicalTrials.gov: Home–ClinicalTrials.gov. https://clinicaltrials.gov/
- 67.Goodwin PJ, Stambolic V, Lemieux J, et al. Evaluation of metformin in early breast cancer: A modification of the traditional paradigm for clinical testing of anti-cancer agents. Breast Cancer Res Treat. 2011;126:215–220. doi: 10.1007/s10549-010-1224-1. [DOI] [PubMed] [Google Scholar]
- 68.Rangwala SM, Lazar MA. Peroxisome proliferator-activated receptor gamma in diabetes and metabolism. Trends Pharmacol Sci. 2004;25:331–336. doi: 10.1016/j.tips.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 69.Burstein HJ, Demetri GD, Mueller E, et al. Use of the peroxisome proliferator-activated receptor (PPAR) gamma ligand troglitazone as treatment for refractory breast cancer: A phase II study. Breast Cancer Res Treat. 2003;79:391–397. doi: 10.1023/a:1024038127156. [DOI] [PubMed] [Google Scholar]
- 70.Yee LD, Williams N, Wen P, et al. Pilot study of rosiglitazone therapy in women with breast cancer: Effects of short-term therapy on tumor tissue and serum markers. Clin Cancer Res. 2007;13:246–252. doi: 10.1158/1078-0432.CCR-06-1947. [DOI] [PubMed] [Google Scholar]
- 71.Baggio LL, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology. 2007;132:2131–2157. doi: 10.1053/j.gastro.2007.03.054. [DOI] [PubMed] [Google Scholar]
- 72.Mizutani S, Sumi S, Narita O, et al. Purification and properties of human placental dipeptidyl peptidase IV. Nippon Sanka Fujinka Gakkai Zasshi. 1985;37:769–775. [PubMed] [Google Scholar]
- 73.Elashoff M, Matveyenko AV, Gier B, et al. Pancreatitis, pancreatic, and thyroid cancer with glucagon-like peptide-1-based therapies. Gastroenterology. 2011;141:150–156. doi: 10.1053/j.gastro.2011.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Koehler JA, Kain T, Drucker DJ. Glucagon-like peptide-1 receptor activation inhibits growth and augments apoptosis in murine CT26 colon cancer cells. Endocrinology. 2011;152:3362–3372. doi: 10.1210/en.2011-1201. [DOI] [PubMed] [Google Scholar]
- 75.Derogar M, Hull MA, Kant P, et al. Increased risk of colorectal cancer after obesity surgery. Ann Surg. 2013;258:983–988. doi: 10.1097/SLA.0b013e318288463a. [DOI] [PubMed] [Google Scholar]
- 76.Bronsveld HK, ter Braak B, Karlstad Ø, et al. Treatment with insulin (analogues) and breast cancer risk in diabetics: A systematic review and meta-analysis of in vitro, animal and human evidence. Breast Cancer Res. 2015;17:100. doi: 10.1186/s13058-015-0611-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wu JW, Filion KB, Azoulay L, et al. Effect of long-acting insulin analogs on the risk of cancer: A systematic review of observational studies. Diabetes Care. 2016;39:486–494. doi: 10.2337/dc15-1816. [DOI] [PubMed] [Google Scholar]
- 78.Pasello G, Urso L, Conte P, et al. Effects of sulfonylureas on tumor growth: A review of the literature. Oncologist. 2013;18:1118–1125. doi: 10.1634/theoncologist.2013-0177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ptaszynska A, Cohen SM, Messing EM, et al. Assessing bladder cancer risk in type 2 diabetes clinical trials: The dapagliflozin drug development program as a ‘case study’. Diabetes Ther. 2015;6:357–375. doi: 10.1007/s13300-015-0128-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pufall MA. Glucocorticoids and cancer. Adv Exp Med Biol. 2015;872:315–333. doi: 10.1007/978-1-4939-2895-8_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hickish T, Astras G, Thomas P, et al. Glucose intolerance during adjuvant chemotherapy for breast cancer. J Natl Cancer Inst. 2009;101:537. doi: 10.1093/jnci/djp025. [DOI] [PubMed] [Google Scholar]
- 82.Ellis ME, Weiss RB, Korzun AH, et al. Hyperglycemic complications associated with adjuvant chemotherapy of breast cancer. A cancer and leukemia group B (CALGB) study. Am J Clin Oncol. 1986;9:533–536. doi: 10.1097/00000421-198612000-00013. [DOI] [PubMed] [Google Scholar]
- 83.Wu W, Merriman K, Nabaah A, et al. The association of diabetes and anti-diabetic medications with clinical outcomes in multiple myeloma. Br J Cancer. 2014;111:628–636. doi: 10.1038/bjc.2014.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bosco C, Crawley D, Adolfsson J, et al. Quantifying the evidence for the risk of metabolic syndrome and its components following androgen deprivation therapy for prostate cancer: A meta-analysis. PLoS One. 2015;10:e0117344. doi: 10.1371/journal.pone.0117344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Laaksonen DE, Niskanen L, Punnonen K, et al. Testosterone and sex hormone-binding globulin predict the metabolic syndrome and diabetes in middle-aged men. Diabetes Care. 2004;27:1036–1041. doi: 10.2337/diacare.27.5.1036. [DOI] [PubMed] [Google Scholar]
- 86.Ben-Shmuel S, Scheinman EJ, Rashed R, et al. Ovariectomy is associated with metabolic impairments and enhanced mammary tumor growth in MKR mice. J Endocrinol. 2015;227:143–151. doi: 10.1530/JOE-15-0310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nuutila P, Knuuti MJ, Mäki M, et al. Gender and insulin sensitivity in the heart and in skeletal muscles. Studies using positron emission tomography. Diabetes. 1995;44:31–36. doi: 10.2337/diab.44.1.31. [DOI] [PubMed] [Google Scholar]
- 88.Lipscombe LL, Fischer HD, Yun L, et al. Association between tamoxifen treatment and diabetes: A population-based study. Cancer. 2012;118:2615–2622. doi: 10.1002/cncr.26559. [DOI] [PubMed] [Google Scholar]
- 89.Geuna E, Roda D, Rafii S, et al. Complications of hyperglycaemia with PI3K-AKT-mTOR inhibitors in patients with advanced solid tumours on phase I clinical trials. Br J Cancer. 2015;113:1541–1547. doi: 10.1038/bjc.2015.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Vergès B, Cariou B. mTOR inhibitors and diabetes. Diabetes Res Clin Pract. 2015;110:101–108. doi: 10.1016/j.diabres.2015.09.014. [DOI] [PubMed] [Google Scholar]
- 91.Busaidy NL, Farooki A, Dowlati A, et al. Management of metabolic effects associated with anticancer agents targeting the PI3K-Akt-mTOR pathway. J Clin Oncol. 2012;30:2919–2928. doi: 10.1200/JCO.2011.39.7356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Beckwith H, Yee D. Minireview: Were the IGF signaling inhibitors all bad? Mol Endocrinol. 2015;29:1549–1557. doi: 10.1210/me.2015-1157. [DOI] [PMC free article] [PubMed] [Google Scholar]