

Abstract
Catalysts that promote carbohydrate degradation are of wide potential application, but the use of either enzyme glycosidases or small-molecule catalysts in biological systems raises significant challenges. Herein we demonstrate a novel strategy for the design of synthetic agents that mimic natural glycosidases and address current problems for biological use. The strategy is illustrated by application to the development of potential blood substitutes for rare Bombay blood that is characterized by a deficiency of H-antigen. Metallopeptides with 16 to 20 amino acids have been constructed as artificial fucosidases that exhibit selective carbohydrate cleavage reactivity toward L-fucose rather than D-glucose. Selective fucose cleavage from the H-antigen saccharide promotes efficient removal of H-antigen on erythrocytes and fulfils the conversion of regular human O-type erythrocytes to potential blood substitutes for rare Bombay blood.
Keywords: Carbohydrates, Antigens, Enzymes, Peptides, Bioinorganic chemistry
COMMUNICATION
Metallopeptides were designed as artificial fucosidases that can induce selective cleavage of L-fucose relative to D-glucose. Selective cleavage of fucose from H-trisaccharide promotes the removal of H-antigen from human erythrocytes, which can potentially convert O-type erythrocytes to substitutes for the rare Bombay phenotype.
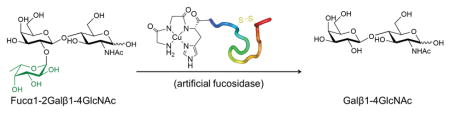
The development of tools for catalytic degradation of carbohydrates has a broad range of potential application.[1] Glycosidases are enzymes responsible for the selective degradation of carbohydrates, however, their selectivity toward specific carbohydrate substrates is a double-edged sword.[2] The presence of some residues adjacent to the target carbohydrate can severely influence or even suppress enzyme activity.[3] For example, an α-fucosidase from bacteroides thetaiotaomicron can hydrolyze pNP-fucoside, but exhibits no activity towards other fucose-containing disaccharides or trisaccharides.[4] Additional problems for protein-based glycosidases include the limited options for natural protein families with a potential immune response in vivo, and the requirement for non-neutral pH.[5] As an alternative, both small-molecule catalysts and heterogeneous catalysts have been developed to promote hydrolysis of carbohydrates, however, harsh conditions that involve non-neutral pH, the use of organic solvents, and negligible discrimination between different carbohydrate substrates undermines their potential application in biological systems.[6] Herein we demonstrate a strategy to design synthetic agents as selective artificial glycosidases that degrade carbohydrates in a controlled and targeted manner. The design of our artificial glycosidases includes a targeting moiety that directs binding toward the substrate of interest, and an independent carbohydrate-degrading moiety that mediates oxidative chemistry on substrates under physiological conditions. The isolation of the catalytic domain from the binding domain ensures greater flexibility for the catalytic apparatus and improved selectivity of the artificial glycosidases toward a broader range of substrates containing the target sugar.
By application of this strategy we have designed artificial fucosidases to produce potential blood substitutes for a rare blood type. While recent studies have described protein-based glycosidases that convert A-blood and B-blood to universal O-blood, there are few efforts to develop rare blood substitutes due to the lack of economic incentives.[7] Blood transfusions for patients with rare blood groups are perceived to be a significant challenge in transfusion medical practice due to the scarce resource of rare blood.[8] Type 2 H-antigen (H2, or CD173) is the terminal polysaccharide residue of glycolipids or glycoproteins on erythrocytes (red blood cells) with the sequence Fucα1-2Galβ1-4GlcNAcβ-.[9] The deficiency of H2-antigens on the erythrocyte surface is known as the Bombay phenotype (also Oh, or h/h).[9–10] The prevalence of this phenotype is rare (1 of 10,000 in India, and 1 per million in Europe).[10–11] Serum in patients carrying the Bombay phenotype contains antibody against H2-antigen, therefore, transfusion of regular human O blood to patients with Bombay phenotype can lead to a severe and fatal transfusion reaction.[12] Herein, synthetic metallopeptides were designed as artificial fucosidases to remove fucose from H2-antigens on human erythrocytes and produce Bombay blood substitutes.
The cleavage domain, composed of an ATCUN (amino terminal Cu(II)- and Ni(II)-binding) motif (GGH), was incorporated into a fucose-selective binding domain derived from odorranalectin (OL: YASPKCFRYPNGVLACT) or its truncated form (tOL: KCFRYPNGVLACT).[13] In addition, two C-terminal amidated analogues were also designed, since reduction of negative charge may enhance the interaction between the peptides and negatively-charged surface of erythrocytes.[14] L-fucose binding affinity to the designed peptides was studied by use of isothermal titration calorimetry (ITC). CuGGH-tOL-NH2 displays a significant binding affinity to L-fucose with a KD of 57.8 μM (Figure S3), consistent with the reported KD (54.7 μM) of odorranalectin.[13] Interestingly, the binding of the copper-free GGH-tOL-NH2 to L-fucose exhibits a negative ΔH in contrast to the endothermic binding pattern of its copper-bound analogue (Figure S3). This change of ΔH can be ascribed to the potential for hydroxyl coordination to Cu2+ from fucose, with binding reflecting the entropic loss of water molecules.[15] Nevertheless, titration of L-fucose into other peptides, either in copper-bound form or copper-free form, resulted in a negligible enthalpy response that precluded calorimetric evaluation of binding affinity. Alternatively, surface plasmon resonance was also used to measure the fucose binding affinity of all peptides (Table 1 and Figure S4). In fact, the removal of the first four residues YASP from full-length odorranalectin improves the fucose-binding affinity of peptides by ~18–26%, while amidation of C-terminus significantly improves the binding affinity by ~60–71%. Therefore, the fucose-binding affinity should mainly arise from domain K5 to T17 of odorranalectin, while the N-terminal YASP residues and the C-terminal carboxylate contribute little to carbohydrate binding affinity.
Table 1.
Binding affinity of metallopeptides to L-fucose
| CuGGH-tOL-NH2 | CuGGH-tOL-OH | CuGGH-OL-NH2 | CuGGH-OL-OH | |
|---|---|---|---|---|
| KD (μM)[a] | 61.3 ± 7.5 | 105 ± 17 | 77.6 ± 6.9 | 124 ± 7 |
Dissociation constants were measured by use of surface plasmon resonance.
Carbohydrate cleavage reactivity for metallopeptides was evaluated by use of chromogenic substrates linked to p-nitrophenolate (pNP). A physiologically relevant coreagent (ascorbate or hydrogen peroxide) is required to stimulate the redox chemistry of the Cu-ATCUN motif and promote the formation of metal-bound oxygen species.[16] The latter abstract hydrogen from the carbohydrate ring, which eventually leads to glycosidic bond cleavage.[17] All the metallopeptides with a fucose-targeting domain exhibit a dominant preference for L-fucose cleavage relative to the D-glucose substrate (Table 2 and Figure S5), while the latter is abundant in blood and is commonly used for blood preservation.[18] The lower KM for the fucose substrate, relative to the glucose substrate, demonstrates selectivity arising from the fucose-binding domain. The larger kcat for the metallopeptides with truncated odorranalectin CuGGH-tOL-NH2 and CuGGH-tOL-OH can be ascribed to the shorter distance between the copper center and the fucose-binding site, relative to their analogues CuGGH-OL-NH2 and CuGGH-OL-OH. CuGGH lacking the fucose-targeting domain exhibits no discrimination between L-fucose and D-glucose. These results demonstrate that the targeting domain is a prerequisite for selective L-fucose cleavage activity of these artificial fucosidases.
Table 2.
Michaelis–Menten parameters for carbohydrate cleavage.
| Peptide sequence | pNP-α-L-fucoside[a] | pNP-β-D-glucoside[a] | ||||
|---|---|---|---|---|---|---|
|
| ||||||
| KM (mM) | kcat (min−1) | kcat/KM (M−1min−1) | KM (mM) | kcat(min−1) | kcat/KM (M−1min−1) | |
| CuGGH-tOL-NH2 | 0.36 ± 0.08 | 1.20 ± 0.17 | 3360 ± 310 | 2.25 ± 0.62 | 0.51 ± 0.08 | 245 ± 34 |
| CuGGH-tOL-OH | 0.51 ± 0.07 | 0.84 ± 0.03 | 1680 ± 240 | 2.94 ± 0.31 | 0.93 ± 0.28 | 376 ± 25 |
| CuGGH-OL-NH2 | 0.35 ± 0.02 | 0.56 ± 0.08 | 1630 ± 310 | 2.16 ± 0.08 | 1.07 ± 0.13 | 514 ± 37 |
| CuGGH-OL-OH | 0.42 ± 0.06 | 0.44 ± 0.05 | 1380 ± 300 | 2.12 ± 0.17 | 0.32 ± 0.08 | 150 ± 31 |
| CuGGH | 1.17 ± 0.13 | 1.49 ± 0.12 | 1270 ± 36 | 0.72 ± 0.18 | 1.40 ± 0.34 | 1930 ± 120 |
Reactions were performed with 5 μM metallopeptides, 1 mM ascorbate and 1 mM H2O2 in 50 mM sodium phosphate buffer (pH 7.4) at 37°C.
Some protein-based fucosidases have been discovered to induce cleavage to pNP-fucose, while lower enzyme activity was observed with polysaccharides as a result of their substrate selectivity.[4,19] To confirm the fucose cleavage reactivity of our artificial fucosidases towards polysaccharides, blood group type 2 H-trisaccharide, Fucα1-2Galβ1-4GlcNAc (exact mass = 529.2) was used as a model to represent the saccharide antigens of human erythrocytes (Figure 1a). The cleavage products were separated from the intact trisaccharide by use of an amino (NH2) column and then analyzed by mass spectrometry (MS). CuGGH-tOL-NH2 exhibits efficient cleavage of fucose from H-trisaccharide in the presence of ascorbate and peroxide, where the diminishing MS response of the H-trisaccharide indicates its disappearance (Figure 1b). In contrast to protein-based fucosidases, our artificial fucosidases can display cleavage reactivity towards a more diverse group of fucose substrates by virtue of their smaller size and distinct mode of action. Namely, it can overcome the incomplete cleavage issue of protein-based fucosidases.[20] Following removal of fucose from H-trisaccharide, a disaccharide product Galβ1-4GlcNAc (exact mass = 383.1) was identified by MS that corresponds to the saccharide antigen of the Bombay phenotype (Figure 1c). A shorter retention time (~14.5 min) confirms the existence of this disaccharide, since the amino column exhibits weaker affinity to disaccharides relative to trisaccharides. Consistent with the results of pNP-fucose cleavage, both CuGGH-tOL-NH2 and CuGGH-tOL-OH, each with a shorter distance between the fucose-binding domain and copper center exhibit more robust cleavage reactivity towards H-trisaccharide, relative to the longer CuGGH-OL-NH2 and CuGGH-OL-OH analogues (Figure 1d). CuGGH also displays cleavage reactivity, but is significantly less reactive relative to other metallopeptides containing a fucose-binding domain, most likely reflecting nonselective cleavage.
Figure 1.

Cleavage of type 2 H-trisaccharide monitored by LC-ESI-MS. (a) Removal of fucose from the type 2 H-trisaccharide (Fucα1-2Galβ1-4GlcNAc) forms the disaccharide Galβ1-4GlcNAc. (b) and (c) A 25 μM solution of H-trisaccharide was incubated with 50 μM CuGGH-tOL-NH2, 1 mM ascorbate and 1 mM H2O2 at 37°C for 0, 15, 30, 60, 90, and 120 min. Extracted ion chromatogram (EIC) at m/z 530.2 ± 0.1 (b) and m/z 384.1 ± 0.1 (c). (d) Time-dependant cleavage of 25 μM H-trisaccharide by 50 μM metallopeptides, 1 mM ascorbate and 1 mM H2O2 at 37°C.
H2-antigen on the cell surface was quantitated by use of a FITC-labelled antibody (Figure 2a). In the presence of ascorbate, each of the artificial fucosidases significantly reduced the FITC intensity, indicating the removal of H2-antigen from erythrocytes. We propose that copper redox chemistry mediated by the metallopeptides promotes selective fucose cleavage from the H2-antigens and induces the regression of H2-antigen to the precursor saccharides corresponding to Bombay blood. In fact, H2-antigens remain intact in the absence of either copper or ascorbate, confirming that the copper redox chemistry is the origin of antigen removal (Figure 2b). All artificial fucosidases with a requisite targeting domain were observed to significantly remove H2-antigen with EC50’s of ~ 8–11 μM (Figure 2c, 2d and S6). Metallopeptides with an amidated C-terminus display improved reactivity toward H2-antigen with an enhancement factor ~18–31%, relative to their analogues with a C-terminal carboxylate. Amidation of the C-terminus may reduce the repulsive interactions between the peptides and the negatively-charged surface of the erythrocytes, however, the role of YASP residues remains unclear, because the removal of YASP residues did not improve H2-antigen reactivity. In fact, the microenvironment on the erythrocyte surface, and interactions between YASP residues and glycoproteins/glycolipids may also influence the reactivity of the designed metallopeptides. Additionally, removal of H2-antigen by CuGGH was also observed, although less efficiently, yielding an EC50 of ~25 μM as a consequence of the lack of a targeting domain (Figure 2d and S6). Accordingly, the selective fucose cleavage reactivity of our artificial fucosidases should be the origin of H2-antigen removal. The addition of L-fucose inhibits the binding of artificial fucosidases to cell surface H2-antigen, thereby suppressing cleavage (Figure 2e). The addition of D-glucose also impairs the fucosidase activity through nonselective binding, whereas weaker inhibitory effects were observed relative to L-fucose. When saccharides were applied to CuGGH, the absence of discrimination between the inhibitory effects of L-fucose and D-glucose confirms that the removal of H2-antigen by CuGGH arises from non-selective cleavage chemistry (Figure 2f).
Figure 2.

Removal of H2-antigen from erythrocytes. a) Flow cytometric analysis of H2 antigen on the surface of human red blood cells (RBC). After erythrocytes were incubated with 20 μM CuGGH-tOL-NH2 and 200 μM ascorbate for 4 h, immunofluorescent staining was performed with a FITC-labelled anti-H2 antibody. b) Relative amount of H2 antigen on erythrocytes, after incubation with or without peptide or ascorbate. Solutions containing 200 μM ascorbate and 20 μM CuGGH-tOL-NH2 were used. c) Relative amount of H2 antigen on erythrocytes after incubation with various amount of CuGGH-tOL-NH2. d) The half-maximal effective concentration (EC50) for H2-antigen removal by artificial fucosidases in the presence of 200 μM ascorbate. e) and f) Inhibitory effect of saccharides. Human erythrocytes were incubated with 20 μM CuGGH-tOL-NH2 (e), or CuGGH (f), with the indicated concentration of saccharides, and 200 μM ascorbate.
In conclusion, we have established a convenient strategy to design relatively short metallopeptides (16 to 20 amino acids) as chimeric fucosidases. All metallopeptides exhibit selective carbohydrate cleavage of L-fucose rather than D-glucose. Fucose cleavage from human blood group type 2 H-trisaccharides promotes the regression of H-trisaccharide to a disaccharide product corresponding to the antigen of Bombay blood. We expect that this strategy can be adopted to develop glycosidases against other saccharide substrates of interest, and that the artificial glycosidases developed from our strategy will also offer diverse applications complimentary to conventional protein-based glycosidase. Fucose cleavage of blood group H2-antigen by artificial fucosidases leads to the deletion of H2-antigen from the erythrocyte surface. The treated erythrocytes may potentially be applied as a substitute for rare Bombay blood, and so our results represent the first step to address this problem.
Experimental Section
Experimental details, metallopeptide characterization, ITC and SPR binding traces, and H2-antigen removal profiles are described in supporting information.
Supplementary Material
Acknowledgments
This work was supported by a grant from the National Institutes of Health [HL093446]. Z.Y. was supported by the Ohio State University Pelotonia Fellowship Program.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.Kren V, Thiem J. Chem Soc Rev. 1997;26:463–473. [Google Scholar]
- 2.Cao HN, Walton JD, Brumm P, Phillips GN. J Biol Chem. 2014;289:25624–25638. doi: 10.1074/jbc.M114.583286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Katayama T, Sakuma A, Kimura T, Makimura Y, Hiratake J, Sakata K, Yamanoi T, Kumagai H, Yamamoto K. J Bacteriol. 2004;186:4885–4893. doi: 10.1128/JB.186.15.4885-4893.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sulzenbacher G, Bignon C, Nishimura T, Tarling CA, Withers SG, Henrissat B, Bourne Y. J Biol Chem. 2004;279:13119–13128. doi: 10.1074/jbc.M313783200. [DOI] [PubMed] [Google Scholar]
- 4.Sakurama H, Tsutsumi E, Ashida H, Katayama T, Yamamoto K, Kumagai H. Biosci Biotechnol Biochem. 2012;76:1022–1024. doi: 10.1271/bbb.111004. [DOI] [PubMed] [Google Scholar]
- 5.a) Sauerborn M, Brinks V, Jiskoot W, Schellekens H. Trends Pharmacol Sci. 2010;31:53–59. doi: 10.1016/j.tips.2009.11.001. [DOI] [PubMed] [Google Scholar]; b) Intra J, Perotti ME, Pavesi G, Horner D. Gene. 2007;392:34–46. doi: 10.1016/j.gene.2006.11.002. [DOI] [PubMed] [Google Scholar]; c) Johnson SW, Alhadeff JA. Comp Biochem Physiol B Biochem Mol Biol. 1991;99:479–488. doi: 10.1016/0305-0491(91)90327-a. [DOI] [PubMed] [Google Scholar]
- 6.a) Striegler S, Dunaway NA, Gichinga MG, Barnett JD, Nelson AGD. Inorg Chem. 2010;49:2639–2648. doi: 10.1021/ic9014064. [DOI] [PubMed] [Google Scholar]; b) Cabiac A, Guillon E, Chambon F, Pinel C, Rataboul F, Essayem N. Appl Catal A. 2011;402:1–10. [Google Scholar]; c) Rinaldi R, Meine N, vom Stein J, Palkovits R, Schuth F. ChemSusChem. 2010;3:266–276. doi: 10.1002/cssc.200900281. [DOI] [PubMed] [Google Scholar]; d) Fukuoka A, Dhepe PL. Angew Chem Int Ed. 2006;45:5161–5163. doi: 10.1002/anie.200601921. [DOI] [PubMed] [Google Scholar]
- 7.a) Liu QYP, Sulzenbacher G, Yuan HP, Bennett EP, Pietz G, Saunders K, Spence J, Nudelman E, Levery SB, White T, Neveu JM, Lane WS, Bourne Y, Olsson ML, Henrissat B, Clausen H. Nat Biotechnol. 2007;25:454–464. doi: 10.1038/nbt1298. [DOI] [PubMed] [Google Scholar]; b) Kwan DH, Constantinescu I, Chapanian R, Higgins MA, Kotzler MP, Samain E, Boraston AB, Kizhakkedathu JN, Withers SG. J Am Chem Soc. 2015;137:5695–5705. doi: 10.1021/ja5116088. [DOI] [PubMed] [Google Scholar]
- 8.Woodfield G, Poole J, Nance ST, Daniels G. Immunohematology. 2004;20:244–248. [PubMed] [Google Scholar]
- 9.Kelly RJ, Ernst LK, Larsen RD, Bryant JG, Robinson JS, Lowe JB. Proc Natl Acad Sci USA. 1994;91:5843–5847. doi: 10.1073/pnas.91.13.5843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dean L. Blood Groups and Red Cell Antigens. NCBI; Bethesda, Md: 2005. [Google Scholar]
- 11.Balgir RS. Indian J Hum Genet. 2007;13:109–113. doi: 10.4103/0971-6866.38985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shahshahani HJ, Vahidfar MR, Khodaie SA. Asian J Transfus Sci. 2013;7:86–87. doi: 10.4103/0973-6247.106754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li JX, Wu HB, Hong J, Xu XQ, Yang HL, Wu BX, Wang YP, Zhu JH, Lai R, Jiang XG, Lin DH, Prescott MC, Rees HH. Plos One. 2008;3 [Google Scholar]
- 14.Jan KM, Chien S. J Gen Physiol. 1973;61:638–654. doi: 10.1085/jgp.61.5.638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cowan JA. Inorg Chem. 1991;30:2740–2747. [Google Scholar]
- 16.a) Ross MJ, Bradford SS, Cowan JA. Dalton Trans. 2015;44:20972–20982. doi: 10.1039/c5dt02837j. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Fidai I, Hocharoen L, Bradford S, Wachnowsky C, Cowan JA. J Biol Inorg Chem. 2014;19:1327–1339. doi: 10.1007/s00775-014-1190-x. [DOI] [PubMed] [Google Scholar]
- 17.a) Yu Z, Han ML, Cowan JA. Angew Chem Int Ed. 2015;54:1901–1905. doi: 10.1002/anie.201410434. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Bradford SS, Ross MJ, Fidai I, Cowan JA. ChemMedChem. 2014;9:1275–1285. doi: 10.1002/cmdc.201400070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.a) Cahill GF, Ashmore J, Renold AE, Hastings AB. Am J Med. 1959;26:264–282. doi: 10.1016/0002-9343(59)90316-x. [DOI] [PubMed] [Google Scholar]; b) Gibson JG, Rees SB, Mcmanus TJ, Scheitlin WA. Am J Clin Path. 1957;28:569–578. doi: 10.1093/ajcp/28.6.569. [DOI] [PubMed] [Google Scholar]
- 19.a) Goso Y, Ishihara K, Sugawara S, Hotta K. Comp Biochem Physiol B Biochem Mol Biol. 2001;130:375–383. doi: 10.1016/s1096-4959(01)00442-0. [DOI] [PubMed] [Google Scholar]; b) Rodriguez-Diaz J, Monedero V, Yebra MJ. Appl Environ Microbiol. 2011;77:703–705. doi: 10.1128/AEM.01906-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ashida H, Miyake A, Kiyohara M, Wada J, Yoshida E, Kumagai H, Katayama T, Yamamoto K. Glycobiology. 2009;19:1010–1017. doi: 10.1093/glycob/cwp082. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.