

Abstract
With the recent progress in imaging technologies for assessment of structural damage in glaucoma, a debate has emerged on whether these measurements can be used as valid surrogate endpoints in clinical trials evaluating new therapies for the disease. A discussion of surrogates should be grounded on knowledge acquired from their use in other areas of medicine as well as regulatory requirements. This article reviews the conditions for valid surrogacy in the context of glaucoma clinical trials and critically evaluates the role of biomarkers such as IOP and imaging measurements as potential surrogates for clinically relevant outcomes. Valid surrogate endpoints must be able to predict a clinically relevant endpoint, such as loss of vision or decrease in quality of life. In addition, the effect of a proposed treatment on the surrogate must capture the effect of the treatment on the clinically relevant endpoint. Despite its widespread use in clinical trials, no proper validation of IOP as a surrogate endpoint has yet been conducted for any class of IOP-lowering treatments. Although strong evidence has accumulated about imaging measurements as predictors of relevant functional outcomes in glaucoma, there is still insufficient evidence to support their use as valid surrogate endpoints. However, imaging biomarkers could potentially be used as part of composite endpoints in glaucoma trials, overcoming weaknesses of the use of structural or functional endpoints in isolation. Efforts should be taken to properly design and conduct studies that can provide proper validation of potential biomarkers in glaucoma clinical trials.
Keywords: biomarkers, surrogate endpoints, glaucoma, optical coherence tomography
The past decades have seen the fast development of advanced imaging techniques for the structural evaluation of ocular tissues affected by glaucoma, such as optical coherence tomography (OCT) among others. With the development of such technologies, an important debate has emerged on whether they can provide meaningful biomarkers that could be incorporated as relevant endpoints in clinical trials evaluating new therapies for the disease. Such debate needs to be grounded on knowledge and evidence of the utilities, advantages, and limitations of using biomarkers in clinical trials, especially when these biomarkers are being proposed as potential surrogate outcomes for clinically relevant endpoints.
In the design of clinical trials, fundamental consideration needs to be given as to the primary endpoints used in the study. For phase III trials, the primary endpoint is a clinical event that is relevant to the patient, that is, an outcome that the patient is directly aware. These endpoints are referred to “true” endpoints. As all drugs have safety risks, the only reason a patient would want to take a drug is if it results in a benefit that is detectable by the patient or if it decreases the risk of developing an unwanted condition or disease complication. Therefore, primary endpoints should be a direct measure of these unwanted conditions or disease complications. In the case of glaucoma, true endpoints would be significant loss of vision with decrease in quality of vision or quality of life or development of disability. However, as glaucoma is generally a slowly progressive disease, clinical trials designed to directly observe such endpoints would generally be impractical and expensive. In this situation, an attractive solution is to replace those true endpoints by a biomarker that can be measured more easily or more frequently and that will show earlier changes as the result of disease and response to therapy.1
In principle, many different body measurements may qualify as biomarkers for a particular disease, including blood tests, genetic, metabolic data, and imaging measurements.2 Blood pressure, glucose level, and radiologic measurements of tumor size are all examples of biomarkers. Similarly, intraocular pressure (IOP) measurements or OCT measurements of retinal nerve fiber layer thickness or neuroretinal rim area are examples of potential biomarkers related to glaucoma. Biomarkers may be used for several different purposes, such as disease risk stratification, prevention, screening, diagnosis, classification, and prognosis. However, although many biomarkers can be associated with a disease and have a wide array of uses, only a few potentially qualify as surrogate endpoints to be used in clinical trials. To qualify as a surrogate endpoint, a biomarker needs to demonstrate significant ability to predict a clinically relevant outcome as well as the effect of treatment on this outcome.3–5
It should be noted that although a validated surrogate endpoint allows prediction of a clinically important outcome, the surrogate itself does not measure a clinical benefit.6 For example, blood pressure has long been considered as an acceptable surrogate endpoint for morbidity and mortality in patients taking antihypertensive medication.7 This is based on epidemiologic evidence showing that lowering blood pressure significantly reduces the risk of the “true” cardiovascular-related endpoints such as myocardial infarction and stroke. However, lowering blood pressure does not in itself represent a direct clinical benefit measurable by the patient. In the case of glaucoma, IOP has been generally used as a surrogate endpoint. However, IOP does not in itself measure a clinical benefit. Its use as a surrogate has been based on the evidence that lowering IOP prevents subsequent progression of functional damage in the disease. However, the use of IOP as a surrogate has many limitations, as I discuss next.
The use of validated surrogates in clinical trials may offer several advantages. Because surrogates are usually laboratory measurements or imaging biomarkers, they make it easier to quantify comparisons among treatment interventions compared to, for example, subjective questionnaires assessing quality of life. The use of surrogates also enables shorter and less expensive trials as it is generally less expensive and takes less time to see the effect of the intervention on the surrogate rather than on the true clinical endpoint. In fact, measuring IOP changes is easier than following glaucoma patients over time and monitoring loss of vision. From a practical standpoint, shortening the duration of a clinical trial also limits possible problems with noncompliance and missing data, which are more likely in longer studies, therefore increasing the effectiveness and reliability of research. The use of surrogates may also allow observation of a greater number of endpoints during follow-up than what would be achieved with observation of true endpoints, reducing sample size requirements.
A history of success on the use of surrogate endpoints has been seen recently in the medical field.8 The escalade of the AIDS epidemic and the pressure for an accelerated evaluation of new therapies have led first to the use of CD4 blood count and later of HIV viral loads as surrogate endpoints that replaced the clinical events and overall survival. In spite of some concerns, the use of surrogates led to accelerated approval of highly active antiretroviral therapy drugs. However, despite their attractiveness, the use of surrogate endpoints has the potential to cause harm.9–12 Unless fully validated, surrogates may waste resources and provide ambiguous evidence and not measure what one really wants to study.13 The main potential disadvantage of surrogates is that positive treatment effects on surrogates do not necessarily automatically translate into benefits to health.14
When Is a Biomarker a Valid Surrogate Endpoint?
From a regulatory perspective, a biomarker is not considered an acceptable surrogate endpoint for determination of the efficacy of a new drug unless it has been empirically shown to function as a valid indicator of clinical benefit.15 The International Conference on Harmonization Guidelines on Statistical Principles for Clinical Trials state that “In practice, the strength of the evidence for surrogacy depends upon (i) the biological plausibility of the relationship, (ii) the demonstration in epidemiologic studies of the prognostic value of the surrogate for the clinical outcome and (iii) evidence from clinical trials that treatment effects on the surrogate correspond to effects on the clinical outcome.”16
It is a common misconception to accept that if a biomarker is correlated with the true clinically relevant outcome it can be used as a surrogate endpoint. However, as noted by Fleming and DeMets,9 “a correlate does not a surrogate make.” Correlation is a necessary, but not sufficient, condition for surrogacy. As pointed out earlier, it is essential to demonstrate that the effect of the intervention on the surrogate endpoint is a reliable predictor of the effect of the intervention on the true clinically significant endpoint—a much stronger condition than correlation. For example, the prostate-specific antigen is a useful biomarker for prostate cancer, but generally unreliable as an indicator of treatment response.17
Prentice4 formulates a set of operational criteria for validating a surrogate endpoint. To be valid, first a surrogate endpoint must be statistically correlated to the clinical endpoint, and second, an intervention's “net effect” on the clinical endpoint should be fully captured by the intervention's effect on the surrogate endpoint. The net effect is the aggregate effect accounting for all mechanisms of action of the intervention. Although the first criterion is generally easy to verify, the second is not. In fact, inappropriate validation of the second condition in early attempts to use surrogates led to harmful conclusions for some disease conditions. One of the best known cases of inappropriate use of surrogates is the approval of the U.S. Food and Drug Administration on the use of the following three drugs: eicanide, flecainide, and moricizine. These drugs were approved based on ventricular arrhythmia suppression used as a surrogate endpoint in phase III trials. It was believed that as ventricular arrhythmias are associated with an almost fourfold increased risk of death from cardiac complications, these drugs would reduce the death rate. After approval, more than 200,000 people eventually took these drugs each year, despite the lack of data evaluating their effect on mortality rates. A large clinical trial (Cardiac Arrhythmia Suppression Trial)18 subsequently conducted after the drugs had been approved showed that although these drugs reduced arrhythmias, they paradoxically increased the risk of death from other causes when compared with placebo. In this particular example, the surrogate endpoint (arrhythmia) did not capture the effect of treatment on the true clinical endpoint (death).
An ideal surrogate endpoint is the one in which all mechanisms of action of the disease to the true endpoint are mediated through the surrogate endpoint.9 Specifically, the surrogate is the only causal pathway in the disease process, and the intervention's entire effect on the true endpoint is mediated through its effect on the surrogate. Such ideal surrogate endpoints, however, are not known at present. Even widely accepted surrogates in other areas of medicine such as blood pressure or HIV viral load do not explain the full effect of treatments on the true endpoints. In practice, successful surrogates have been shown to explain only part of the treatment effect, and several statistical methodologies have been developed to quantify this effect.6
There are many different scenarios in which a treatment may significantly affect a biomarker while not providing a meaningful effect on the true endpoint. For example, if the biomarker does not lie in the biological pathway by which the disease process actually influences the occurrence of the clinical endpoint, then affecting the biomarker might not affect the clinical endpoint. Invalid surrogacy may also result when the proposed surrogate endpoint lies in only one of multiple pathways by which the disease may affect the true clinically relevant endpoint. If the intervention does not actually affect all pathways, then the effect of treatment on the true endpoints could be over- or underestimated by the effect on the candidate surrogate. Finally, the intervention might actually affect the true endpoint by unintended mechanisms of action that are independent of the disease process, leading to unexpected results, such as in the cases of the anti-arrhythmic drugs discussed previously.
Validation of a proposed surrogate should be based on biological plausibility and on in-depth empirical evidence. Ideally, one should have a comprehensive understanding of the causal pathways of the disease process and of the intervention's mechanisms of action. The proper development of surrogate endpoints may require conducting a trial with a given treatment while analyzing the true and surrogate endpoints. However, such a trial is what one wanted to avoid in first place. Once a surrogate is validated for a treatment belonging to a certain given class of agents, it can generally be applied to assess other treatments of the same class. However, although surrogacy may be valid when considering treatments of the same class, the validation does not necessarily extrapolate to treatments of different classes.19
Is IOP a Valid Surrogate Endpoint in Glaucoma?
The role of IOP as a risk factor for development and progression of glaucoma is unquestionable. There is strong evidence from several clinical trials to support higher mean IOP as a risk factor for development of glaucoma as well as for progression of disease in individuals with manifest glaucoma. In the Ocular Hypertension Treatment Study (OHTS), European Glaucoma Prevention Study (EGPS), Early Manifest Glaucoma Trial (EMGT), Advanced Glaucoma Imaging Study (AGIS), the Canadian Glaucoma Study, and United Kingdom Glaucoma Treatment Study (UKGTS) each mm Hg of increased mean IOP was associated with an increased risk for progression of 10%–25%.20 However, although IOP is the most important known risk factor for glaucoma, it is clearly an imperfect correlate for the clinically relevant outcomes of the disease. It is know that many patients may develop glaucoma or progressive disease despite relatively low pressures.21 Most patients with high IOP also never develop functional signs of glaucoma despite being followed for many years.22 Although previous clinical trials have established statistically significant relationships between IOP levels and the risk of progressive disease, most studies lack an analysis of the strength of such relationships.
The ability to lower IOP has been used as basis for regulatory approval of new treatments. Drugs in this category are approved based on their proposed labelling to lower intraocular pressure, not to treat glaucoma. However, even though such treatments are approved on the grounds of their effect on IOP (and not technically on glaucoma), the fundamental idea behind it is that as the treatments lower IOP they would be beneficial in preventing vision loss in glaucoma. However, surprisingly, no proper validation of such surrogacy of IOP has ever been conducted for any class of IOP-lowering medications.
The knowledge that IOP is predictive of future visual field loss does not necessarily imply that an IOP-lowering drug will prevent visual loss from glaucoma. A drug could successfully lower IOP, but at the same time have unintended detrimental effects on the clinically relevant outcome by some other mechanism of action. These detrimental effects could offset the benefits caused by IOP lowering resulting in no net benefit or even harm. Conversely, a drug that may have relatively less effect on IOP lowering, could have greater effects in preventing visual field loss by acting through an additional IOP-independent mechanism. An example suggesting the lack of surrogacy of IOP to predict clinically relevant benefit has been provided by the recent Low-pressure Glaucoma Treatment Study.23 In the Low-pressure Glaucoma Treatment Study, the patients were randomized to timolol maleate 0.5% versus brimonidine tartrate 0.2% and followed over time with IOP measurements and monitoring of visual field status. Despite very similar mean treated IOP in both groups, patients using brimonidine 0.2% had a much lower incidence of visual field progression (9.1%) than those using timolol (39.2%). Clearly, IOP would not be a valid surrogate endpoint in this case, as it was not able to strongly predict the effect of the drugs on the clinically relevant endpoint. Despite this fact, both drugs have been approved for clinical use because of their IOP-lowering effects. Although the reasons for such lack of surrogacy are not certain at this point, the main conclusion is that a trial using solely IOP as the endpoint would erroneously conclude that both drugs would offer the same benefit in preventing clinically relevant outcomes in glaucoma.
Recently, the UKGTS24 investigated whether latanoprost was able to reduce visual field deterioration in glaucoma. In the UKGTS, 516 individuals were randomized to latanoprost versus placebo. At 24 months of follow-up, the mean IOP was significantly lower in the latanoprost group when compared with placebo (3.8 mm Hg vs. 0.9 mm Hg) and visual field preservation was significantly longer in the latanoprost group, with an adjusted hazard ratio (HR) of 0.44 (95% confidence interval [CI]: 0.28–0.69; P = 0.0003). Although these results clearly demonstrate beneficial effects of latanoprost on a clinically relevant endpoint (visual field loss), it is unclear whether IOP could be considered a valid surrogate endpoint in this case. That is, was all the effect of latanoprost in preserving visual function mediated by its effect in reducing IOP? With further analyses, the UKGTS investigators have an excellent opportunity to address this issue.
As IOP is an obviously inappropriate surrogate endpoint for clinical trials evaluating potential neuroprotective agents, a search has been conducted for other potential biomarkers that could serve as surrogate endpoints, such as imaging measurements.
Can Imaging Measurements Be Used as Surrogate Endpoints in Glaucoma Clinical Trials?
As with IOP, it is important to critically analyze the role and evidence for validation of imaging measurements as potential surrogate endpoints. Such an evaluation should be made in terms of biological plausibility, prognostic value, and whether treatment effects on the surrogate correspond to effects on the clinically relevant outcomes.
The biological plausibility is clear. The hallmark of glaucoma is progressive retinal ganglion cell loss, which results in loss of the retinal nerve fiber layer and characteristic changes in optic disc topography. This is supported by strong clinical, epidemiologic, and experimental data.25 In fact, the evidence linking structural damage of the optic nerve to visual field loss in the disease is actually stronger than that for intraocular pressure.26
In 2008 and subsequently in 2010, the National Eye Institute (NEI) and the FDA Center for Drug Evaluation and Research (FDA CDER) held the NEI/FDA CDER Glaucoma Clinical Trial Design and Endpoints Symposium to discuss the possible use of new structural and functional endpoints for evaluating glaucoma therapies in clinical trials.27 In the document summarizing the meeting, it is pointed out that for structural measurements to be considered as suitable endpoints, there is a need for demonstrating a strong correlation between these measurements and relevant functional endpoints in the disease. Evidence about the prognostic value of structural measurements has since been accumulated.28 Several studies have shown that imaging measurements of retinal nerve fiber layer thickness, optic disc topography, and the macular area are predictive of future development of visual field loss in glaucoma. However, are their predictive strengths good enough? The Table summarizes results for several studies investigating the predictive value of OCT measurements in predicting visual function outcomes in glaucoma. The studies have found statistically significant hazard ratios for baseline and longitudinal imaging measurements in predicting future visual field damage. However, although most studies have reported metrics such as hazard ratios, most of them lack a report of metrics that truly quantify the predictive strength of the associations. Hazard ratios can be made larger or smaller according to the units that are chosen to represent the scale of measurements, and statistical significance depends largely on sample size and is not a direct measure of clinical relevance.
Table.
Summary of Studies Investigating the Ability of Parameters From Optical Coherence Tomography in Predicting Visual Function Loss
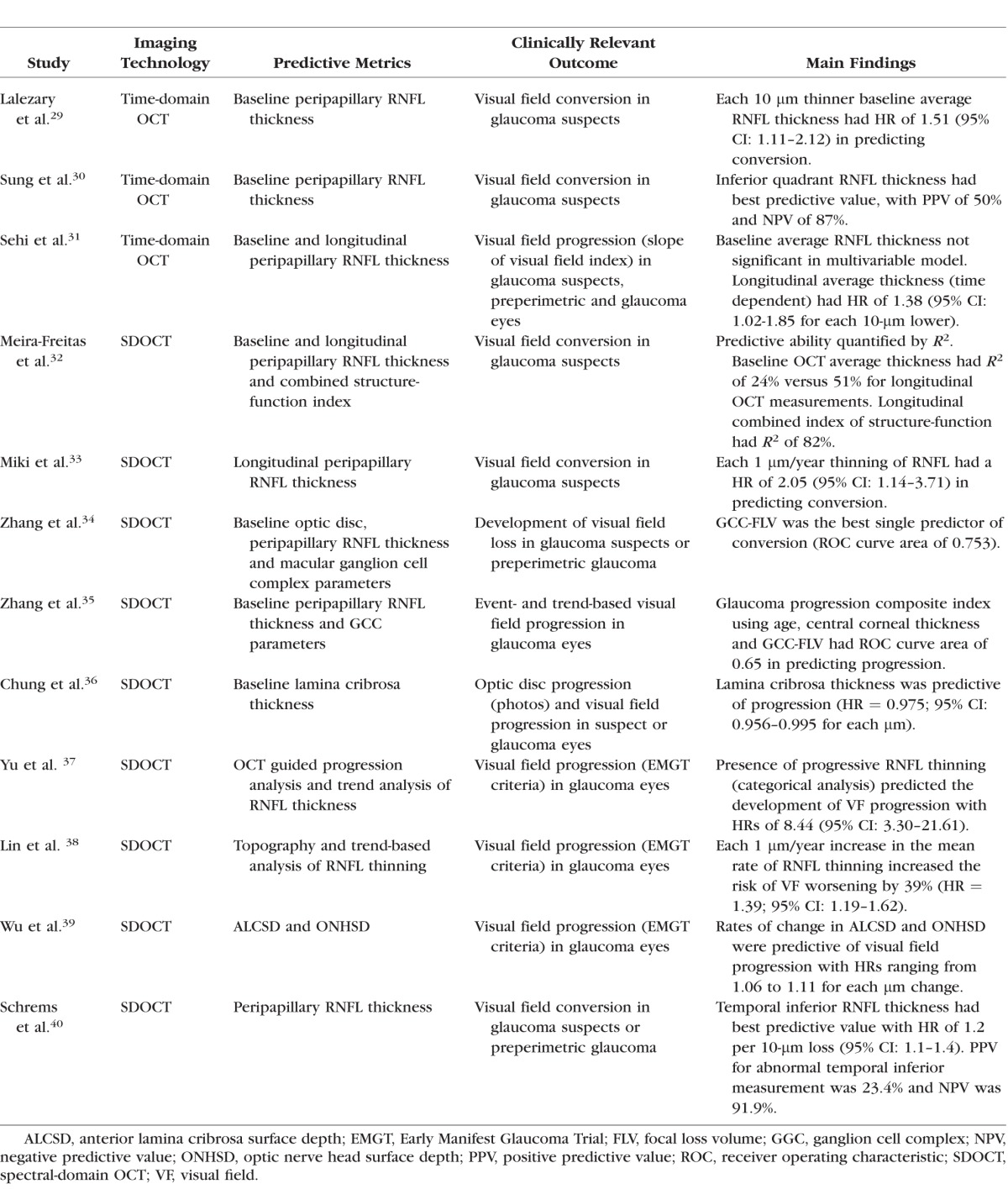
A critical analysis of the strength of baseline cross-sectional imaging measurements in predicting future development of visual field loss reveals that their predictive value is generally weak. For example, in the Confocal Scanning Laser Ophthalmoscopy Ancillary Study to the OHTS41, analysis of rim area with the Moorfields regression analysis parameter had a hazard ratio of 3.9 (95% CI: 2.09–7.28) in a multivariable model to predict development of glaucoma among ocular hypertensive eyes. At first look, this would appear as a strong predictive ability. However, an abnormal result on this parameter was associated with a positive predictive value of only 24%. That is, only 24% of those with an abnormal result actually converted to glaucoma during a median follow-up of over 10 years. Although the parameter had a very high negative predictive value of 0.95, such a value was to be generally expected for almost any parameter because of the very low rate of conversion in the study. For OCT, most studies reveal similar findings. A recent report from the Advanced Imaging for Glaucoma Group investigated the prognostic ability of baseline OCT RNFL and macular measurements for predicting visual field progression. Similarly, although most parameters had statistically significant hazard ratios, quantification of predictive strength by the area under the ROC curve revealed values below 0.65 for all parameters.35 It should be noted that nonrandomized studies attempting to quantify the strength of predictive factors may be subject to a fundamental limitation. If the treatment regimens can vary among patients per decisions of treating physicians, it is likely that those with worse values on the predictive factors at baseline (such as thinner RNFL thickness) will get more treatment. This will then subsequently decrease the impact of such factors in predicting progression, resulting in biased estimates of predictive strength. This has been a limitation inherent to almost all of the studies in this field.
Before dismissing the value of imaging measurements in predicting visual function loss, it is important of recognize that the main benefit of imaging comes from longitudinal monitoring of structural losses over time. Because of high inter-subject variability, it is hard to predict future outcomes based on a single, cross-sectional, baseline measurement. In fact, studies reporting on the ability of longitudinal imaging measurements in predicting visual field progression report much stronger predictive values. A recent study found a hazard ratio of 8.44 (95% CI: 3.30–21.61) for a trend-based analysis of OCT RNFL thinning in predicting visual field loss in a multivariable model.37 In another study, strengths of baseline and longitudinal OCT measures in predicting development of visual field loss were quantified by an R2 metric. Although baseline SDOCT RNFL thickness had an R2 of only 24% in predicting development of visual field loss, the R2 improved to 51% for longitudinal RNFL measurements.32 It should be noted that achieving higher R2 values is limited by the inherent variability of tests as well as by the fact that limited follow-up time leads to censoring of a significant number of patients.
Although not discussed in the previous NEI/FDA CDER documents,27 to show that structural measurements can be used as reliable surrogate endpoints, one has to also demonstrate that the effect of treatment on changes in structure is a reliable predictor of the effect of treatment on changes in function. A recent study attempted to address this issue in the context of IOP-lowering therapies. The study attempted to verify whether confocal scanning laser ophthalmoscopy neuroretinal rim area measurements could satisfy Prentice's criteria for surrogacy.42 The study demonstrated that, even though the effect of IOP lowering on rim area did not fully explain the effect of IOP lowering in preventing visual field loss, it explained a considerable part of it. Using a measure called proportion of treatment effect, the authors showed that rim area measurements were able to explain 65% of the effect of treatment on the risk of development of visual field loss. Although this effect can be considered only moderate, a proportion of the treatment effect of 100% has not been demonstrated for any surrogate endpoint in medicine. It should be noted, however, that this evaluation of surrogacy was not done in the context of a randomized trial investigating a single treatment but, rather, from an observational cohort study. Also, it is possible that stronger effects could be demonstrated for measurements derived from more recent imaging technologies, such as SDOCT. This remains to be investigated.
It is important to emphasize that the validation of surrogacy of structural measurements has not yet been made in the context of neuroprotective therapies. Extrapolation of surrogacy from studies evaluating IOP-lowering therapy is likely inappropriate. It is possible that a candidate neuroprotective drug could be beneficial on a structural surrogate while not showing net beneficial effects in the functional clinically relevant outcome. For example, a drug could preserve tissue anatomy without really preserving function. If only structural measurements are used as surrogate endpoints in this situation, they would tend to overestimate the benefit of the treatment. This highlights the importance of a comprehensive understanding of the mechanisms of action of the proposed therapy which should be gained from early experimental studies.
A caveat needs to be mentioned regarding the above studies on the predictive value of imaging measurements. These studies have only linked changes in structural measures to changes in automated perimetry. They have not directly shown a prognostic relationship between structural measurements and the endpoints directly representing measures of functional impairment or disability. However, a recent study has shown a significant relationship between longitudinal RNFL thickness measurements by SDOCT and quality of life as assessed by patient-reported outcomes.43
The use of structural measurements as sole endpoints in clinical trials is limited by the known relationship between disease severity and ability of these measurements to detect change.44 Although the odds of detecting progression by imaging are higher than visual fields for early disease, the situation is generally inverted for late stages, when the odds of detecting change by visual fields may be higher than by imaging.45 As changes in visual function may be seen in the absence of detectable structural losses in a significant number of cases, a trial using only imaging metrics as surrogate endpoints could potentially fail to detect clinically relevant effects of the proposed drug. However, this limitation could potentially be addressed by the use of composite endpoints,1 including structural measurements as well as functional endpoints.46 Another potential solution is the use of approaches that combine structure and function. Previous studies have shown that Bayesian models incorporating structural information may lead to better estimates of functional loss over time.46,47 Another proposed approach is to combine structure and function into a single metric for estimating neural losses in glaucoma.48,49
In addition to their potential use as endpoints in clinical trials, structural measurements could be used for risk stratification, identifying high-risk patients in whom certain interventions are most likely to be beneficial. For example, patients with rapidly progressing RNFL loss have been shown to be at high risk for development of field losses. Even under routine clinical care and under treatment with currently available therapies, a substantial proportion of glaucoma patients still present with significant rates of RNFL loss.50 These patients could then be identified using RNFL imaging techniques as a potential group that could benefit from candidate therapies.
In the United States, there are mechanisms available for accelerated drug approval based on surrogate endpoints to reduce the time to review an application for indications with no known effective therapy. Accelerated approval (also referred to as “conditional approval” or “subpart H”) refers to the acceleration of the overall development plan by allowing submission of an application and, if approved, marketing of a drug on the basis of surrogate endpoints while further studies demonstrating direct patient benefit are underway. However, accelerated approval is limited to severe diseases where no effective therapies exist. One could then claim that such pathway would not be applicable to glaucoma, as effective IOP-lowering therapies do exist. However, glaucoma is not a single disease. Although some patients respond well to currently available treatments to lower IOP, others fail to respond and continue to show progressive neural losses despite low pressures. For these patients, effective therapies currently do not exist and severe visual field loss or blindness may be the result of progressive disease.
Conclusions
Advances in molecular biology, genome sequencing techniques, and pharmacogenomics are dramatically reshaping the development of new drugs in several areas of medicine. It is likely that such advancements will quickly result in new proposed therapies to slow down or even reverse neural losses in glaucoma. The benefit of these new proposed therapies, however, will need to be clearly assessed by trials using suitable endpoints. Surrogate endpoints may be viable alternatives when obtaining the true endpoints would result in unfeasible studies. However, these surrogates need to be properly validated before widespread use in practice. Validation entails assessing biological plausibility and prognostic value and quantifying how much of the treatment effect on clinically relevant outcomes can be captured by the treatment's effect on the surrogate. Such validation studies have not yet been conducted for any of the potential currently available biomarkers in glaucoma, and efforts should be taken by the scientific community to properly design and conduct such studies.
Acknowledgments
Supported in part by National Institutes of Health Grants EY021818 and EY025056.
Disclosure: F.A. Medeiros, Carl-Zeiss Meditec (C, F), Heidelberg Engineering (C, F), Topcon (F), Reichert (C, F), Allergan (C, F), Alcon (C, F), Novartis (C)
References
- 1. Lesko LJ,, Atkinson AJ., Jr. Use of biomarkers and surrogate endpoints in drug development and regulatory decision making: criteria, validation, strategies. Annu Rev Pharmacol Toxicol. 2001; 41: 347–366. [DOI] [PubMed] [Google Scholar]
- 2. Biomarkers Definitions Working Group. Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther. 2001; 69: 89–95. [DOI] [PubMed] [Google Scholar]
- 3. Boissel JP,, Collet JP,, Moleur P,, Haugh M. Surrogate endpoints: a basis for a rational approach. Eur J Clin Pharmacol. 1992; 43: 235–244. [DOI] [PubMed] [Google Scholar]
- 4. Prentice RL. Surrogate endpoints in clinical trials: definition and operational criteria. Stat Med. 1989; 8: 431–440. [DOI] [PubMed] [Google Scholar]
- 5. Gobburu JV. Biomarkers in clinical drug development. Clin Pharmacol Ther. 2009; 86: 26–27. [DOI] [PubMed] [Google Scholar]
- 6. Lassere MN,, Johnson KR,, Boers M,, et al. Definitions and validation criteria for biomarkers and surrogate endpoints: development and testing of a quantitative hierarchical levels of evidence schema. J Rheumatol. 2007; 34: 607–615. [PubMed] [Google Scholar]
- 7. Desai M,, Stockbridge N,, Temple R. Blood pressure as an example of a biomarker that functions as a surrogate. AAPS J. 2006; 8: E146–E52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hughes MD,, Daniels MJ,, Fischl MA,, et al. CD4 cell count as a surrogate endpoint in HIV clinical trials: a meta-analysis of studies of the AIDS Clinical Trials Group. AIDS. 1998; 12: 1823–1832. [DOI] [PubMed] [Google Scholar]
- 9. Fleming TR,, DeMets DL. Surrogate end points in clinical trials: are we being misled? Ann Intern Med. 1996; 125: 605–613. [DOI] [PubMed] [Google Scholar]
- 10. Psaty BM,, Weiss NS,, Furberg CD,, et al. Surrogate end points, health outcomes, and the drug-approval process for the treatment of risk factors for cardiovascular disease. JAMA. 1999; 282: 786–790. [DOI] [PubMed] [Google Scholar]
- 11. Schatzkin A,, Gail M. The promise and peril of surrogate end points in cancer research. Nat Rev Cancer. 2002; 2: 19–27. [DOI] [PubMed] [Google Scholar]
- 12. Svensson S,, Menkes DB,, Lexchin J. Surrogate outcomes in clinical trials: a cautionary tale. JAMA Intern Med. 2013; 173: 611–612. [DOI] [PubMed] [Google Scholar]
- 13. Lassere MN. The Biomarker-Surrogacy Evaluation Schema: a review of the biomarker-surrogate literature and a proposal for a criterion-based, quantitative, multidimensional hierarchical levels of evidence schema for evaluating the status of biomarkers as surrogate endpoints. Stat Methods Med Res. 2008; 17: 303–340. [DOI] [PubMed] [Google Scholar]
- 14. Krumholz HM,, Lee TH. Redefining quality—implications of recent clinical trials. N Engl J Med. 2008; 358: 2537–2539. [DOI] [PubMed] [Google Scholar]
- 15. Lathia CD,, Amakye D,, Dai W,, et al. The value, qualification, and regulatory use of surrogate end points in drug development. Clin Pharmacol Ther. 2009; 86: 32–43. [DOI] [PubMed] [Google Scholar]
- 16. ICH Harmonised Tripartite Guideline. Statistical principles for clinical trials. International Conference on Harmonisation E9 Expert Working Group. Stat Med. 1999; 18: 1905–1942. [PubMed] [Google Scholar]
- 17. Halabi S,, Armstrong AJ,, Sartor O,, et al. Prostate-specific antigen changes as surrogate for overall survival in men with metastatic castration-resistant prostate cancer treated with second-line chemotherapy. J Clin Oncol. 2013; 31: 3944–3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. The Cardiac Arrhythmia Suppression Trial (CAST) Investigators. Preliminary report: effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. N Engl J Med. 1989; 321: 406–412. [DOI] [PubMed] [Google Scholar]
- 19. Buyse M,, Sargent DJ,, Grothey A,, et al. Biomarkers and surrogate end points—the challenge of statistical validation. Nat Rev Clin Oncol. 2010; 7: 309–317. [DOI] [PubMed] [Google Scholar]
- 20. Weinreb RN,, Leung CK,, Crowston JG,, et al. Primary open-angle glaucoma. Nat Rev Dis Primers. 2016; 2: 16067. [DOI] [PubMed] [Google Scholar]
- 21. Kass MA,, Heuer DK,, Higginbotham EJ,, et al. The Ocular Hypertension Treatment Study: a randomized trial determines that topical ocular hypotensive medication delays or prevents the onset of primary open-angle glaucoma. Arch Ophthalmol. 2002; 120: 701–713; discussion 829–830. [DOI] [PubMed] [Google Scholar]
- 22. Drance SM. The Collaborative Normal-Tension Glaucoma Study and some of its lessons. Can J Ophthalmol. 1999; 34: 1–6. [PubMed] [Google Scholar]
- 23. Krupin T,, Liebmann JM,, Greenfield DS,, et al. A randomized trial of brimonidine versus timolol in preserving visual function: results from the Low-Pressure Glaucoma Treatment Study. Am J Ophthalmol. 2011; 151: 671–681. [DOI] [PubMed] [Google Scholar]
- 24. Garway-Heath DF,, Lascaratos G,, Bunce C,, et al. The United Kingdom Glaucoma Treatment Study: a multicenter, randomized, placebo-controlled clinical trial: design and methodology. Ophthalmology. 2013; 120: 68–76. [DOI] [PubMed] [Google Scholar]
- 25. Weinreb RN,, Khaw PT. Primary open-angle glaucoma. Lancet. 2004; 363: 1711–1720. [DOI] [PubMed] [Google Scholar]
- 26. Francis BA,, Varma R,, Vigen C,, et al. Population and high-risk group screening for glaucoma: the Los Angeles Latino Eye Study. Invest Ophthalmol Vis Sci. 2011; 52: 6257–6264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Weinreb RN,, Kaufman PL. Glaucoma research community and FDA look to the future, II: NEI/FDA Glaucoma Clinical Trial Design and Endpoints Symposium: measures of structural change and visual function. Invest Ophthalmol Vis Sci. 2011; 52: 7842–7851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Medeiros FA,, Alencar LM,, Zangwill LM,, et al. Prediction of functional loss in glaucoma from progressive optic disc damage. Arch Ophthalmol. 2009; 127: 1250–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lalezary M,, Medeiros FA,, Weinreb RN,, et al. Baseline optical coherence tomography predicts the development of glaucomatous change in glaucoma suspects. Am J Ophthalmol. 2006; 142: 576–582. [DOI] [PubMed] [Google Scholar]
- 30. Sung KR,, Kim S,, Lee Y,, et al. Retinal nerve fiber layer normative classification by optical coherence tomography for prediction of future visual field loss. Invest Ophthalmol Vis Sci. 2011; 52: 2634–2639. [DOI] [PubMed] [Google Scholar]
- 31. Sehi M,, Zhang X,, Greenfield DS,, et al. Retinal nerve fiber layer atrophy is associated with visual field loss over time in glaucoma suspect and glaucomatous eyes. Am J Ophthalmol. 2013; 155: 73–82. e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Meira-Freitas D,, Lisboa R,, Tatham A,, et al. Predicting progression in glaucoma suspects with longitudinal estimates of retinal ganglion cell counts. Invest Ophthalmol Vis Sci. 2013; 54: 4174–4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Miki A,, Medeiros FA,, Weinreb RN,, et al. Rates of retinal nerve fiber layer thinning in glaucoma suspect eyes. Ophthalmology. 2014; 121: 1350–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang X,, Loewen N,, Tan O,, et al. Predicting development of glaucomatous visual field conversion using baseline fourier-domain optical coherence tomography. Am J Ophthalmol. 2016; 163: 29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang X,, Dastiridou A,, Francis BA,, et al. Baseline Fourier-domain optical coherence tomography structural risk factors for visual field progression in the Advanced Imaging for Glaucoma Study. Am J Ophthalmol. 2016; 172: 94–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chung HS,, Sung KR,, Lee JY,, Na JH. Lamina cribrosa-related parameters assessed by optical coherence tomography for prediction of future glaucoma progression. Curr Eye Res. 2016; 41: 806–813. [DOI] [PubMed] [Google Scholar]
- 37. Yu M,, Lin C,, Weinreb RN,, et al. Risk of visual field progression in glaucoma patients with progressive retinal nerve fiber layer thinning: a 5-year prospective study. Ophthalmology. 2016; 123: 1201–1210. [DOI] [PubMed] [Google Scholar]
- 38. Lin C,, Mak H,, Yu M,, Leung CK. Trend-based progression analysis for examination of the topography of rates of retinal nerve fiber layer thinning in glaucoma. JAMA Ophthalmol. 2017; 135: 189–195. [DOI] [PubMed] [Google Scholar]
- 39. Wu Z,, Lin C,, Crowther M,, et al. Impact of rates of change of lamina cribrosa and optic nerve head surface depths on visual field progression in glaucoma. Invest Ophthalmol Vis Sci. 2017; 58: 1825–1833. [DOI] [PubMed] [Google Scholar]
- 40. Schrems WA,, Schrems-Hoesl LM,, Mardin CY,, et al. Can glaucomatous visual field progression be predicted by structural and functional measures? J Glaucoma. 2017; 26: 373–382. [DOI] [PubMed] [Google Scholar]
- 41. Weinreb RN,, Zangwill LM,, Jain S,, et al. Predicting the onset of glaucoma: the confocal scanning laser ophthalmoscopy ancillary study to the Ocular Hypertension Treatment Study. Ophthalmology. 2010; 117: 1674–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Medeiros FA,, Lisboa R,, Zangwill LM,, et al. Evaluation of progressive neuroretinal rim loss as a surrogate end point for development of visual field loss in glaucoma. Ophthalmology. 2014; 121: 100–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gracitelli CP,, Abe RY,, Tatham AJ,, et al. Association between progressive retinal nerve fiber layer loss and longitudinal change in quality of life in glaucoma. JAMA Ophthalmol. 2015; 133: 384–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Medeiros FA,, Zangwill LM,, Bowd C,, et al. The structure and function relationship in glaucoma: implications for detection of progression and measurement of rates of change. Invest Ophthalmol Vis Sci. 2012; 53: 6939–6946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Abe RY,, Diniz-Filho A,, Zangwill LM,, et al. The relative odds of progressing by structural and functional tests in glaucoma. Invest Ophthalmol Vis Sci. 2016; 57: OCT421–OCT428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Medeiros FA,, Leite MT,, Zangwill LM,, Weinreb RN. Combining structural and functional measurements to improve detection of glaucoma progression using Bayesian hierarchical models. Invest Ophthalmol Vis Sci. 2011; 52: 5794–5803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Russell RA,, Malik R,, Chauhan BC,, et al. Improved estimates of visual field progression using bayesian linear regression to integrate structural information in patients with ocular hypertension. Invest Ophthalmol Vis Sci. 2012; 53: 2760–2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Medeiros FA,, Lisboa R,, Weinreb RN,, et al. A combined index of structure and function for staging glaucomatous damage. Arch Ophthalmol. 2012; 130: 1107–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Medeiros FA,, Zangwill LM,, Anderson DR,, et al. Estimating the rate of retinal ganglion cell loss in glaucoma. Am J Ophthalmol. 2012; 154: 814–824. e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Diniz-Filho A,, Abe RY,, Zangwill LM,, et al. Association between intraocular pressure and rates of retinal nerve fiber layer loss measured by optical coherence tomography. Ophthalmology. 2016; 123: 2058–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]