

Abstract
In endothelia, NO is synthesized by endothelial NO synthase (eNOS), which is negatively regulated by caveolin-1 (Cav-1), the primary coat protein of caveolae. We show that delivery of Cav-1 amino acids 82-101 (Cav) fused to an internalization sequence from Antennapedia (AP) blocks NO release in vitro and inflammation and tumor angiogenesis in vivo. To characterize the molecular mechanism by which the AP-Cav peptide and Cav-1 mediate eNOS inhibition, we subdivided the Cav portion of AP-Cav into three domains (Cav-A, -B, and -C), synthesized five overlapping peptides (AP-Cav-A, -AB, -B, -BC, and -C), and tested their effects on eNOS-dependent activities. Peptides containing the Cav-B domain (amino acids 89-95) induced time- and dose-dependent inhibition of eNOS-dependent NO release in cultured endothelial cells, NO-dependent inflammation in the ear, and hydraulic conductivity in isolated venules. Alanine scanning of AP-Cav-B revealed that Thr-90 and -91 (T90,91) and Phe-92 (F92) are crucial for AP-Cav-B- and AP-Cav-mediated inhibition of eNOS. Mutation of F92 to A92 in the Cav-1 cDNA caused the loss of eNOS inhibitory activity compared with wild-type Cav-1. These data highlight the importance of amino acids 89-95 and particularly F92 in mediating eNOS inhibition by AP-Cav and Cav-1.
Caveolae are cholesterol- and sphingomyelin-rich invaginations of the plasma membrane found throughout the cardiovascular system (1). Numerous lines of evidence suggest that these macromolecular organelles have the capacity to segregate or concentrate spatially distinct signaling molecules that may modulate many cellular responses. Electron microscopy studies have revealed the presence of caveolae in several cell types, and vascular endothelial cells are a major source of caveolae. Caveolin-1 (Cav-1), the main coat protein of caveolae in blood vessels, is believed to be instrumental in the caveolae-dependent compartmentalization and modulation of signaling systems. Emerging evidence also suggests that Cav-1 is in part targeted to the cell membrane, likely through palmitoylation, where it can bind to other molecules through its scaffolding domain (residues 82-101 in Cav-1) or carboxyl tail (2).
One well described molecular target of Cav-1 is endothelial nitric oxide (NO) synthase (eNOS), the NO synthase (NOS) isoform that produces low levels of NO in response to mechanical forces and diverse agonists in endothelial cells. eNOS in the plasma membrane is negatively regulated by its protein-protein interaction with Cav-1 (3, 4). This finding is supported by recent genetic data showing that Cav-1 knockout mice have increased endothelium-dependent relaxations and NOx levels in blood (5, 6). Additional evidence supporting a negative regulatory role for Cav-1 on eNOS functions stems from using a cell-permeable version of the inhibitory Cav-1 scaffolding domain that selectively blocks eNOS-mediated NO release, vasorelaxation, inflammation, and tumor angiogenesis in variety of settings (7, 8). Hence, this peptide, called Antennapedia (AP)-Cav or cavtratin, is a valuable tool for mimicking and studying Cav-1 molecular interactions with eNOS.
To gain insights into the molecular determinants of how Cav-1 and the peptide AP-Cav may mediate eNOS inhibition, we have designed several cell-permeable peptides containing truncated portions of Cav-1 and examined their effect on eNOS function in four distinct bioassays. Here, we show that truncated cell-permeable peptides that contain the hydrophobic sequence FTTFTVT (Cav-1 amino acids 89-95) can inhibit recombinant eNOS activity, NO release from endothelial cells, microvessel permeability in vitro, and inflammation in vivo similarly to AP-Cav (residues 82-101). By performing alanine scanning of this seven amino acid sequence, residues Thr-90 and -91 (T90,91) and Phe-92 (F92) are crucial for mediating eNOS inhibition by AP-Cav-derived peptides in vitro and in vivo. In a cell-based reconstitution system, expression of wild-type (WT) Cav-1 protein attenuates eNOS-dependent NO release, an effect diminished in cells expressing the F92 substitution to alanine (F92A) Cav-1 mutant. Taken together, these data identify the critical amino acids in Cav-1 that mediate eNOS inhibition and also validate the approach using cell-permeable peptides to characterize and mimic the molecular interactions between two client proteins.
Methods
Peptides and Reagents. Peptides, corresponding to the full-length (amino acids 82-101; DGIWKASFTTFTVTKYWFYR) or truncated scaffolding domain of Cav-1 were synthesized as a fusion peptide to the C terminus of the Antennapedia internalization sequence (RQIKIWFQNRRMKWKK) by standard fluorenylmethoxycarbonyl chemistry and analyzed by mass spectrometry to confirm purity by the W. M. Keck Foundation Biotechnology Resource Laboratory at the Yale University School of Medicine. Before each experiment, desiccated peptides were weighed, dissolved in DMSO (from J. T. Baker, Philipsburg, NJ) to 10 mM, and diluted to 1 mM with distilled water.
Cell Culture. Bovine aortic endothelial cells (BAEC) below passage 10, COS cells, and human embryonic kidney (HEK) cells were cultured in DMEM (Mediatech, Herndon, VA) supplemented with 10% FBS (HyClone) and 1× penicillin/streptomycin (Sigma) in a humidified incubator at 37°C with 7% CO2.
NO Release Experiments. BAEC cells were grown to confluence in six-well tissue culture plates (Becton Dickinson). Medium was removed, and serum-free DMEM containing various concentrations of peptides or vehicle (0.1% DMSO) was added and incubated for 6 h. After pretreatment, the medium was removed, and cells were stimulated with DMEM containing peptides and/or VEGF (1 nM; Genentech) for 30 min. For HEK cells, medium was removed after transfection, and serum-free DMEM was added and incubated for 24 h. After a 30-min (BAEC) or 24-h (HEK) incubation, the medium was collected, and cells were trypsinized and counted by using a Coulter counter. NO release from confluent BAEC or HEK was assessed in the medium by measuring nitrite levels using a NO-specific chemiluminescence analyzer (Sievers, Boulder, CO) as described in ref. 9. Data are reported as pmol of nitrite per 106 cells.
Inflammation Model. Plasma leakage in mouse skin was studied by using a modified Miles assay. Male Swiss mice (30-35 g; Charles River Laboratories) were pretreated with AP-Cav (2.5 mg/kg), truncated peptides (same molar dose as AP-Cav), or vehicle (1% DMSO in water) for 1 h. Mice were anesthetized with ketamine/xylazine, and a PD10 catheter (Becton Dickinson) was introduced in the left jugular vein for administration of Evans blue (30 mg/kg in PBS; Sigma). Phenylisothiocyanate, an analog of mustard oil (Pierce), was diluted to 5% (vol/vol) in mineral oil and applied 1 min after the administration of Evans blue onto the dorsal and ventral surfaces of the right ear with a cotton tip. The left ear was used as control and treated with mineral oil alone. After 30 min, the anesthetized animals were killed and perfused with saline, and ears were removed, dried, and weighed. Evans blue was extracted from the ears with 500 μl of formamide for 24 h at 55°C and quantified spectrophotometrically at 595 nm.
Plasmid Constructions and Cell Transfection. The human Cav-1 cDNA was amplified by RT-PCR from human umbilical vein endothelial cell mRNA. RNA isolation (5 μg) and cDNA synthesis was performed with the RNeasy (Qiagen; Valencia, CA) and SuperScript Reverse Transcriptase kits (Invitrogen). The Cav-1 cDNA was amplified by using Advantage-2 proofreading polymerase (Invitrogen) and the following primers: 5′-CCTCCTCACAGTTTTCATCCAGCC-3′ and 5′-GGAGTAGGCAGTTGAGGTTGTTGG-3′. The PCR product was ligated into pcDNA3.1(-) (Invitrogen) and sequenced. The single point mutation of the Cav-1 gene was performed by using the Pfu Turbo site-directed mutagenesis kit (Stratagene) and primers 5′-GGCCAGCTTCACCACCGCCACGGTGACCAAATACTGGTTTTACC-3′ and 5′-GGTAAAACCAGTATTTGGTCACCGTGGCGGTGGTGAAGCTGGCC-3′. Hemagglutinin-tagged (3′ end) versions of WT and F92A Cav-1 were obtained by PCR with the reverse primer 5′-TAATAAGCGGCCGCTTAAGCGTAGTCTGGGACGTCGTATGGGTATATTTCTTTCTGCAAGTTGATGCGGACATTG-3′. Positive clones were amplified and sequenced to confirm mutation. For cell transfection, semiconfluent HEK or COS cells were grown in six-well tissue culture plates and transfected for6hwith human eNOS (10) and/or WT or mutant human Cav-1 plasmids by using Lipofectamine 2000 in Opti-Mem (Invitrogen). A β-gal plasmid was used to normalize DNA quantities. Plasmid levels were adjusted to ensure equal expression levels of Cav-1 proteins. The supernatant was collected 24 h later; cells were trypsinized, counted, and lysed; and proteins were isolated.
Immunofluorescence. Immunofluorescence experiments were performed on control and transfected COS cells. COS cells were seeded on gelatin-coated coverslips and transfected as described above. Then, 36 h after transfection start, cells were washed, fixed in 3% paraformaldehyde for 15 min, and permeabilized in 0.1% Triton X-100 for 10 min. Nonspecific binding was blocked with 0.1% BSA plus 10% normal donkey serum (Invitrogen) for 1 h. Anti-eNOS (Transduction Laboratories, BD Biosciences, San Jose, CA) and anti-Cav-1 (Santa Cruz Biotechnologies) primary antibodies were added (1:200 dilutions) and incubated for 1 h, followed by anti-mouse and -rabbit secondary antibodies (1:1,000 dilution; Molecular Probes). The coverslips then were mounted on glass slides with Gelvatol/DAPI (Sigma) and analyzed with a Zeiss Axiovert epifluorescence microscope. Captured images were deconvoluted by using the openlab software (Improvision, Lexington, MA).
Coimmunoprecipitation and Western Blot Analyses. Cav-1 and eNOS coimmunoprecipitations and Western blotting were performed as described in ref. 10.
Hydraulic Conductivity. Hydraulic conductivity experiments were conducted as described in ref. 11. Briefly, freshly isolated rat mesenteric venules were cannulated with a glass micropipette, and measurements based on the modified Landis technique were performed. A charge-coupled device camera was connected to a microscope, and a video was recorded from a segment of the perfused microvessel throughout each experiment for hydraulic conductivity measurements.
Recombinant eNOS Activity. The conversion of l-[14C]arginine to l-[14C]citrulline was used to determine NOS activity (12). Briefly, recombinant purified bovine eNOS (50-100 ng) was incubated with Cav, Cav-AB, or Cav-A (10 μM) in a 100-μl reaction for 15 min at room temperature (13). The reaction was initiated by the addition of l-[14C]arginine and NOS cofactors for 10 min at 37°C and processed as described in ref. 10.
Statistical Analysis. Data are represented as mean ± SEM. Statistical comparisons were made by ANOVA followed by Dunnet's t test. Data were considered significantly different if values of P < 0.05 were observed.
Results
Truncated AP-Cav Peptides Block NO Release and Inflammation in Vivo. To determine which amino acids of the Cav-1 scaffolding domain (amino acids 82-101) mediate eNOS inhibition, we subdivided the domain into three subdomains: Cav-A (82-88), Cav-B (89-95), and Cav-C (96-101). We then synthesized five combinations of these regions as cell-permeable peptides using AP as the cellular internalization motif (Fig. 1A).
Fig. 1.

Truncated AP-Cav peptides attenuate endothelial cell NO release and Evans blue extravasation. (A) The caveolin scaffolding domain of AP-Cav (WT) was subdivided into three subdomains (Cav-A, -B, and -C), and five peptides containing combinations of these subdomains were synthesized (AP-Cav-AB, -BC, -A, -B, and -C). (B) Effect of truncated AP-Cav peptides on VEGF-induced NO release in BAEC. BAEC were pretreated with peptides (10 μM) for 6 h, and VEGF (1 nM)-induced NO release was accumulated for 30 min. Basal nitrite release (133 ± 21 pmol per 106 cells) was subtracted from all values. (C) Dose-dependent effect of AP-Cav-B on VEGF-induced NO release. BAEC were treated with AP-Cav-B (5-50 μM) as described above. Basal nitrite release (208 ± 22 pmol per 106 cells) was subtracted from all values. (D) Effect of truncated AP-Cav peptides on mustard oil-induced inflammation. Male CD-1 mice were treated for 1 h with AP-Cav (2.5 mg/kg i.p.) or truncated peptides (equivalent molar dose) and then were anesthetized (ketamine/xylazine) for administration of Evans blue (via the left jugular vein; 30 mg/kg) 1 min before painting right and left ears with 5% phenyl isothiocyanate and mineral oil (control), respectively. After 30 min, the animals were killed, and the content of Evans blue was determined spectrophotometrically at 595 nm. Respective basal Evans blue extravasation (left ear values for each group) was subtracted from stimulated (right ear) values. All graphs represent means of at least three experiments in triplicate ± SEM. *, P < 0.05 compared with vehicle-treated control (-) group.
Initially, we tested the inhibitory potential of the peptides on 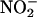 release from cultured BAEC, which was quantified by NO-specific chemiluminescence. Pretreatment of confluent BAEC with full-length AP-Cav (10 μM) reduced VEGF-induced
release from cultured BAEC, which was quantified by NO-specific chemiluminescence. Pretreatment of confluent BAEC with full-length AP-Cav (10 μM) reduced VEGF-induced 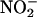 release by 50-60% compared with untreated or vehicle-treated cells (AP; Fig. 1B). Pretreatment with three truncated peptides containing the B subdomain of Cav (AP-Cav-AB, -BC, and -B; 10 μM) also blocked VEGF-induced
release by 50-60% compared with untreated or vehicle-treated cells (AP; Fig. 1B). Pretreatment with three truncated peptides containing the B subdomain of Cav (AP-Cav-AB, -BC, and -B; 10 μM) also blocked VEGF-induced 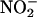 release to a similar extent, whereas AP-Cav-A or -C had no significant effect on
release to a similar extent, whereas AP-Cav-A or -C had no significant effect on 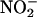 release. The inhibitory effect of AP-Cav-B was dose-dependent with maximal inhibition (≈80%) occurring at 50 μM (Fig. 1C).
release. The inhibitory effect of AP-Cav-B was dose-dependent with maximal inhibition (≈80%) occurring at 50 μM (Fig. 1C).
Next, we examined the effects of these permeable peptides on acute inflammation. Treatment of mice with cavtratin (also known as AP-Cav) blocked vascular leakage in the Miles assay to a similar extent as l-nitroarginine methyl ester (7). As expected, a 1-h pretreatment of mice with AP-Cav (2.5 mg/kg; i.p.) reduced mustard oil-induced Evans blue extravasation (Fig. 1D) compared with untreated or vehicle-treated mice (AP; equivalent molar dose). Similar pretreatment with AP-Cav-AB, -BC, and -B, but not AP-Cav-A or -C, reduced Evans blue leakage and interstitial edema in ear and surrounding tissues from 38% to 50%. These data suggest that key amino acids located within the Cav-B subdomain mediate eNOS inhibition and eNOS-dependent protein extravasation.
Peptides Containing the Cav-B Domain Attenuate Platelet-Activating Factor (PAF)-Induced Hydraulic Conductivity in Intact Vessels. Previous studies have revealed that eNOS-derived NO release also can promote increases in hydraulic conductivity (Lp) in intact microvessels, which is characterized by solute and water influx into the interstitium of isolated venules. As reported in ref. 11, AP-Cav blocks PAF-induced hydraulic conductivity of isolated microvessels; therefore, we examined the effects of two AP-Cav mutants on hydraulic conductivity in venules. First, a 2-h perfusion of microvessels with peptides (10 μM) did not significantly modulate basal permeability (Lp; Fig. 2A). Stimulation of vehicle-treated vessels (AP) with PAF (10 nM) induced a rapid and transient increase in mean Lp (Fig. 2A) where the mean peak increase in Lp was 5.9 ± 0.7 times the control value (Fig. 2B). In contrast, PAF-induced increases in Lp were reduced in vessels treated with either full-length AP-Cav or AP-Cav-AB, the truncated peptide that displayed the greater potency at blocking NO release and inflammation (Fig. 1). Vessels treated with AP-Cav-B showed a reduced, but significant, decrease in PAF-induced peak Lp (Fig. 2B).
Fig. 2.

Truncated AP-Cav peptides attenuate PAF-induced hydraulic conductivity in isolated venules. (A) Paired measurements of Lp in response to 10 nM PAF with and without peptide pretreatment. A 2-h peptide pretreatment had no effect on basal Lp (Left). The addition of PAF to control vessels (AP, 10 μM; ○) caused an increase in Lp, which was almost completely attenuated by pretreating vessels with AP-Cav (⋄) or AP-Cav-AB (•), whereas pretreatment with AP-Cav-B (▴) was not as profound. (B) PAF-induced mean peak increases in Lp with peptide pretreatment as described above. Basal Lp (0.92 ± 0.15) was subtracted from all groups. Values are means of five different experiments ± SEM. *, P < 0.05 compared with AP-treated control group.
Peptides Containing Cav Amino Acids 89-95 Block Recombinant eNOS Activity. Next, we compared the efficacy of Cav peptides to inhibit recombinant eNOS activity. WT bovine eNOS was expressed and purified from Escherichia coli, and activity was assayed by the conversion of l-[14C]arginine to l-[14C]citrulline. Incubation of purified eNOS with a conventional NOS inhibitor (l-nitroarginine methyl ester, 1 mM) completely blocked eNOS activity (Fig. 3A), and incubation with full-length Cav (82-101) had a similar dose-dependent effect on eNOS activity. Incubation with Cav-AB (1-25 μM), but not Cav-A, also inhibited eNOS activity by ≈80% (Fig. 3A). The Cav-B peptide lacking the internalization domain of AP was insoluble under these conditions and could not be assayed. These data suggest that Cav-B subdomain in the context of Cav-AB contains the crucial amino acids for direct eNOS inhibition.
Fig. 3.

Truncated AP-Cav peptides block recombinant eNOS activity and eNOS binding to Cav-1. (A) Purified eNOS was preincubated with either l-nitroarginine methyl ester (1 mM) or the peptides (Cav, Cav-AB, or Cav-A; 1-25 μM) for 15 min, and NOS activity was assessed as described in Methods. Data represent duplicate determinations from a single experiment that was repeated twice. (B) Confluent BAEC were treated for 6 h with AP, AP-Cav, AP-Cav-AB, AP-Cav-A, or AP-Cav-B (10 μM). Cells were lysed, Cav-1 was immunoprecipitated, and eNOS coassociation was detected by Western blot analysis. Experiments were performed in duplicate, and typical data are shown. IP, immunoprecipitation; WB, Western blot.
Cav-A and -B Subdomains Modulate eNOS and Endogenous Cav-1 Interactions. Previous reports have shown that eNOS and Cav-1 can be coimmunoprecipitated in a stable heteromeric complex and that the scaffolding domain of Cav-1 can interfere with the stability of this complex (14, 15). Because our data show that AP-Cav-AB and -B can attenuate eNOS-mediated NO release, inflammation, and hydraulic conductivity, we assayed the capacity of these truncated Cav-1 subdomains to compete with endogenous Cav-1 for interacting with eNOS. Confluent BAEC were treated for 6 h with peptides (10 μM) and lysed, and solubilized proteins were immunoprecipitated with a Cav-1 antibody. As shown in Fig. 3B, the immunoprecipitation and recovery of Cav-1 was identical under all conditions [Bottom, immunoprecipitation (IP) Cav-1, Western blot (WB) Cav-1]. eNOS was coimmunoprecipitated with Cav-1 in vehicle-treated cells (lane 1). Incubation of BAEC with full-length AP-Cav, and AP-Cav-AB, -A, and -B all caused a moderate attenuation of eNOS coimmunoprecipitation with endogenous Cav-1 (lanes 2-5) ranging from 34% to 78% of control (using semiquantitative densitometry). These data suggest that both Cav-A and -B domains can interact with eNOS, although Cav-A cannot block eNOS activity.
T90,91 and F92 Residues of Cav-1 Mediate eNOS Inhibition. Because our data show that the Cav-B subdomain (amino acids 89-95), as an AP-conjugated permeable peptide, mediates eNOS inhibition, we tested the individual importance of each of these seven residues by using an alanine scanning approach. Seven cell-permeable mutant peptides containing one alanine residue were synthesized and tested on VEGF-induced NO release in BAEC. Treatment of BAEC with F89A-, T93A-, Val-94 (V94)A- and T95A-AP-Cav-B mutants (10 μM) blocked VEGF-induced NO release in BAEC in a similar fashion to WT AP-Cav-B (10 μM; Fig. 4A). In contrast, alanine scans of residues 90-92 caused a significant (T90A- and T91A-AP-Cav-B) or complete (F92A-AP-Cav-B) loss of inhibitory activity compared with WT AP-Cav-B. These data reveal the importance of T90, T91, and F92 in mediating eNOS inhibition by AP-Cav-B.
Fig. 4.

F92 mutants fail to block eNOS activity in vitro and in vivo.(A) Seven mutant peptides were generated, and their inhibitory potential was tested on VEGF-induced NO release by BAEC. Basal nitrite release (199 ± 20 pmol per 106 cells) was subtracted for all groups. (B) BAEC were treated as described for 6 h with AP-Cav, F92A-AP-Cav, or T90/91/F92A-AP-Cav (10 μM of each) and stimulated with VEGF (1 nM) for 30 min. Basal nitrite release (180 ± 8 pmol per 106 cells) was subtracted for all groups. (C) Compared with AP-Cav, AP-T90/91/F92ACav does not block mustard oil-induced plasma protein leakage in vivo. Mice were pretreated as described for Fig. 1, and plasma leakage was stimulated with mustard oil. Basal Evans blue extravasation (left ear values for each group) was subtracted from the stimulated (right ear) values. All data are means of at least three experiments in triplicate ± SEM. *, P < 0.05 compared with vehicle-treated control group.
In a parallel series of experiments, the role of residues T90,91 and F92 in full-length AP-Cav were tested. Two cell-permeable AP-Cav peptides containing either (F92A) single or (T90/91/F92A) triple mutations were synthesized and tested on VEGF-stimulated NO release from BAEC. Our data showed that in contrast to WT AP-Cav, pretreatment with the F92A AP-Cav and the triple mutant T90/91/F92A AP-Cav did not cause a significant inhibition of VEGF-induced NO release (Fig. 4B). Moreover, in vivo pretreatment of mice with the triple mutant T90/91/F92A AP-Cav did not influence mustard oil-induced Evans blue extravasation (Fig. 4C). Taken together, these observations show that residues T90,91 and F92 in AP-Cav are important for the regulation of eNOS function.
Mutation of F92 in Cav-1 Protein Reduces the Inhibitory Effect of Cav-1 on eNOS Function. Because both Cav-1 protein and AP-Cav peptide inhibit eNOS, and F92 in the context of AP-Cav is critical for the inhibitory effects on eNOS, we mutated F92 to alanine in the human Cav-1 cDNA and examined the inhibitory effect of this construct on NO production in intact cells. As shown in Fig. 5A, expression of human eNOS cDNA in HEK cells caused a robust increase in 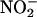 release compared with control transfected cells. Coexpression of WT human Cav-1 cDNA with eNOS markedly attenuated
release compared with control transfected cells. Coexpression of WT human Cav-1 cDNA with eNOS markedly attenuated 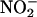 release. However, overexpression of human Cav-1 containing F92 substitution to alanine (F92A) impaired its inhibitory effect on eNOS. Western blotting showed that changes in
release. However, overexpression of human Cav-1 containing F92 substitution to alanine (F92A) impaired its inhibitory effect on eNOS. Western blotting showed that changes in 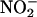 levels in HEK cells expressing eNOS and WT or (F92A) mutant Cav-1 were not due to variations in protein levels (see blots in Fig. 5A) or altered subcellular localization of mutant Cav-1 (Fig. 5B). β-gal (control) or Cav-1/eNOS-transfected COS cells incubated with control IgG antibodies or secondary alone, respectively, showed no staining (data not shown).
levels in HEK cells expressing eNOS and WT or (F92A) mutant Cav-1 were not due to variations in protein levels (see blots in Fig. 5A) or altered subcellular localization of mutant Cav-1 (Fig. 5B). β-gal (control) or Cav-1/eNOS-transfected COS cells incubated with control IgG antibodies or secondary alone, respectively, showed no staining (data not shown).
Fig. 5.

Mutant F92A Cav-1 does not block eNOS-mediated NO release despite colocalizing with eNOS. (A) HEK cells were transfected with plasmids encoding for a control protein (β-gal; lane 1), human eNOS (lane 2), human eNOS and WT Cav-1 (lane 3), and human eNOS and F92 Cav-1 (lane 4). Basal nitrite release (24-h accumulation) was assayed in the medium with a NO chemiluminescence analyzer. Cells then were lysed, and proteins were collected for Western blot analysis: eNOS (top blot) and Cav-1 (middle blot) and β-COP protein expression as a loading control (bottom blot). (B) Cav-1 F92A mutation does not affect its subcellular targeting and colocalization with eNOS. COS cells were transfected with plasmids coding for eNOS and hemagglutinin-tagged versions of WT (Upper) and F92A Cav-1 (Lower). Cells were fixed, incubated with antibodies against hemagglutinin (HA; red) and eNOS (green), and mounted on glass slides with medium containing nuclear dye DAPI (blue). (Right) Merged images with DAPI staining are shown.
Discussion
Our data demonstrate that eNOS inhibition by AP-Cav is mediated by hydrophobic amino acids located within the Cav-B subdomain (Cav-1 amino acids 89-95). This finding is supported by data showing that peptides containing this seven-amino-acid sequence reduce eNOS function in four distinct bioassays: VEGF-induced NO release from endothelial cells, irritant-induced inflammation in vivo, PAF-induced hydraulic conductivity in venules, and eNOS activity assays. In addition, the cell-permeable Cav-B subdomain competed with endogenous Cav-1 for eNOS binding. Alanine scanning of amino acids in the B domain show that F92 is largely responsible for mediating eNOS inhibition by AP-Cav-B and AP-Cav. Moreover, we showed that a F92A Cav-1 protein mutant failed to block eNOS-mediated NO release compared with WT Cav-1 in intact cells. Taken together, our results demonstrate the relative importance of the amino acids in the caveolin scaffolding domain that mediate eNOS inhibition and the utility of cell-permeable peptides as tools to dissect the structure/function relationships between two interacting proteins.
Another interesting aspect of our data is the role of the Cav-A domain on Cav-B-mediated eNOS inhibition. Although we observed no significant inhibitory activity from AP-Cav-A alone, AP-Cav-AB caused greater inhibition of several NO-mediated responses compared with AP-Cav-B. Part of the answer may reside in pharmacokinetic disparities between Cav-B and -AB. For instance, the addition to Cav-B of positively charged residues from Cav-A may greatly affect its bioavailability and distribution, especially in vivo. These latter properties were illustrated clearly in our recombinant eNOS assay where the free peptide, Cav-B, was totally insoluble and Cav-AB was soluble and blocked eNOS activity. Moreover, in vivo, AP-Cav-AB was more potent than AP-Cav-B. The amino acids in the A domain, although not inhibitory, per se (Fig. 1B), appear to bind to eNOS, because it can compete with Cav-1 for eNOS in coimmunoprecipitation experiments (Fig. 3B). Hence, AP-Cav-AB may functionally interact more efficiently than AP-Cav-B because it contains the two eNOS binding motifs (Cav-A and -B), although the amino acids responsible for eNOS inhibition are localized in the Cav-B subdomain. Further biophysical studies clearly are warranted to examine this idea.
As previously stated, the Cav-B subdomain is entirely composed of hydrophobic residues (FTTFTVT), which was clearly illustrated by the insolubility of the peptide lacking the polybasic amino acids of AP in eNOS activity assays. However, this finding raises the possibility that this domain may interact with a hydrophobic “pocket” on eNOS. Previous reports have suggested the presence of a Cav-1 consensus binding motif in the eNOS oxygenase domain, which also consists of a series of hydrophobic residues (FSAAPFSGW) (9, 16); however, based on the crystal structure of the eNOS oxygenase, this domain is buried in the protein, stabilizes the heme prosthetic group, and is unlikely to participate in protein-protein interactions (17). Based on biochemical studies, Cav-1 can interact with eNOS or neuronal NOS by means of binding to sites in both the oxygenase and reductase domains of the enzymes (9, 18-20). Mechanistically, Cav-1 binding to NOS inhibits the reductase function of the enzymes, thereby reducing NO synthesis (21). By using an alanine scanning approach, T90,91 and, most importantly, F92, were involved in eNOS inhibition by AP-Cav-B (FTTFTVT) and full-length AP-Cav. Interestingly, a scrambled version of Cav (Cav-X) that contains an intact Cav-B domain, with the exception of a F → S mutation in position 92 (FTTSTVT), was shown to have minimal eNOS inhibitory activity (14). Chemically, F residues are good candidates for disrupting electron flux because their phenyl rings contain aromatic side groups characterized by their bulk and ease to accept and stabilize electrons through resonance structures. However, because no crystal structure of Cav-1 bound to eNOS is currently available, the details of such inhibition remain speculative.
In solution, small peptides such as AP-Cav can take on numerous conformations. This property, attributable to their small size and absence of secondary stabilizing structures like α-helices and β-sheets, may cast doubt on the biological relevance of inhibitory mechanisms when compared with the inhibitory function of the oligomeric endogenous Cav-1. However, experiments defining the important role of F92 by using cell-permeable peptides were instrumental in guiding the rational mutagenesis strategy of the human Cav-1 cDNA. Indeed, mutation of F92 → A (F92A) in the Cav-1 cDNA markedly reduced the inhibitory effect of Cav-1 on NO release from cells. The F92A Cav-1 protein localized in a punctuate pattern in transfected cells similar to WT Cav-1, suggesting that the impaired function of F92A was not attributable to altered subcellular localization of the protein. Thus, our data demonstrate that F92 is crucial for eNOS inhibition by both AP-Cav and Cav-1. The broader importance of F92 in Cav-1 interactions with other potential proteins that may interact with Cav-1 (c-src, G-protein-coupled receptors, mitogen-activated protein kinases, etc.) remains to be determined (1).
Another salient feature of our study is the utility of cell-permeable peptides in dissecting protein-protein interactions in vivo. The use of synthetic peptides proved to be time-efficient and can be used to characterize the molecular interactions between any two client proteins, as documented with eNOS and Cav-1. Moreover, this approach allowed the identification of truncated peptides that show similar potency to full-length AP-Cav for inhibiting eNOS. These truncated mutants may prove useful for therapeutic application of eNOS inhibition, such vascular leakage associated with inflammation or tumor growth (7, 8).
In conclusion, our data reveal that eNOS inhibition by AP-Cav is mediated by amino acids 89-95 within the caveolin scaffolding domain and that F92 is crucial for such activity. In an intact cell system, we observed that Cav-1-mediated eNOS inhibition is also dependent on the presence of F92 in Cav-1. Because the data obtained with our cell-permeable peptides were transposed successfully into the context of the Cav-1 protein, this paper illustrates the feasibility of using cell-permeable peptides for identifying and describing at a molecular level the molecular determinants responsible for the interaction between two client proteins.
Acknowledgments
We thank Ms. Janet Crawford for her constant help with peptide synthesis. This work was funded by National Institutes of Health Grants R01 HL64793, R01 HL 61371, R01 HL 57665, and P01 HL 70295 and National Heart, Lung, and Blood Institute-Yale Proteomics Contract N01-HV-28186 (to W.C.S.). P.N.B. is supported by a fellowship from the Canadian Institutes of Health Research, and P.M.B is supported by National Institutes of Health Fellowship F32 HL072618.
Author contributions: P.N.B., J.S.P., and W.C.S. designed research; P.N.B., P.M.B., J.Y., J.S.P., and P.H. performed research; P.N.B., P.M.B., J.Y., J.S.P., P.H., and W.C.S. analyzed data; and P.N.B. and W.C.S. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: AP, Antennapedia; Cav-1, caveolin-1; NOS, NO synthase; eNOS, endothelial NOS; BAEC, bovine aortic endothelial cells; PAF, platelet-activating factor; HEK, human embryonic kidney.
References
- 1.Gratton, J. P., Bernatchez, P. N. & Sessa, W. C. (2004) Circ. Res. 94, 1408-1417. [DOI] [PubMed] [Google Scholar]
- 2.Smart, E. J., Graf, G. A., McNiven, M. A., Sessa, W. C., Engelman, J. A., Scherer, P. E., Okamoto, T. & Lisanti, M. P. (1999) Mol. Cell. Biol. 19, 7289-7304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Garcia-Cardena, G., Oh, P., Liu, J., Schnitzer, J. E. & Sessa, W. C. (1996) Proc. Natl. Acad. Sci. USA 93, 6448-6453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shaul, P. W., Smart, E. J., Robinson, L. J., German, Z., Yuhanna, I. S., Ying, Y., Anderson, R. G. & Michel, T. (1996) J. Biol. Chem. 271, 6518-6522. [DOI] [PubMed] [Google Scholar]
- 5.Drab, M., Verkade, P., Elger, M., Kasper, M., Lohn, M., Lauterbach, B., Menne, J., Lindschau, C., Mende, F., Luft, F. C., et al. (2001) Science 293, 2449-2452. [DOI] [PubMed] [Google Scholar]
- 6.Razani, B., Engelman, J. A., Wang, X. B., Schubert, W., Zhang, X. L., Marks, C. B., Macaluso, F., Russell, R. G., Li, M., Pestell, R. G., et al. (2001) J. Biol. Chem. 276, 38121-38138. [DOI] [PubMed] [Google Scholar]
- 7.Bucci, M., Gratton, J. P., Rudic, R. D., Acevedo, L., Roviezzo, F., Cirino, G. & Sessa, W. C. (2000) Nat. Med. 6, 1362-1367. [DOI] [PubMed] [Google Scholar]
- 8.Gratton, J. P., Lin, M. I., Yu, J., Weiss, E. D., Jiang, Z. L., Fairchild, T. A., Iwakiri, Y., Groszmann, R., Claffey, K. P., Cheng, Y. C. & Sessa, W. C. (2003) Cancer Cell 4, 31-39. [DOI] [PubMed] [Google Scholar]
- 9.Garcia-Cardena, G., Martasek, P., Masters, B. S., Skidd, P. M., Couet, J., Li, S., Lisanti, M. P. & Sessa, W. C. (1997) J. Biol. Chem. 272, 25437-25440. [DOI] [PubMed] [Google Scholar]
- 10.Garcia-Cardena, G., Fan, R., Stern, D. F., Liu, J. & Sessa, W. C. (1996) J. Biol. Chem. 271, 27237-27240. [DOI] [PubMed] [Google Scholar]
- 11.Zhu, L., Schwegler-Berry, D., Castranova, V. & He, P. (2003) Am. J. Physiol. 286, H195-H201. [DOI] [PubMed] [Google Scholar]
- 12.Bredt, D. S. & Snyder, S. H. (1990) Proc. Natl. Acad. Sci. USA 87, 682-685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martasek, P., Liu, Q., Liu, J., Roman, L. J., Gross, S. S., Sessa, W. C. & Masters, B. S. (1996) Biochem. Biophys. Res. Commun. 219, 359-365. [DOI] [PubMed] [Google Scholar]
- 14.Michel, J. B., Feron, O., Sacks, D. & Michel, T. (1997) J. Biol. Chem. 272, 15583-15586. [DOI] [PubMed] [Google Scholar]
- 15.Gratton, J. P., Fontana, J., O'Connor, D. S., Garcia-Cardena, G., McCabe, T. J. & Sessa, W. C. (2000) J. Biol. Chem. 275, 22268-22272. [DOI] [PubMed] [Google Scholar]
- 16.Couet, J., Li, S., Okamoto, T., Ikezu, T. & Lisanti, M. P. (1997) J. Biol. Chem. 272, 6525-6533. [DOI] [PubMed] [Google Scholar]
- 17.Raman, C. S., Li, H., Martasek, P., Kral, V., Masters, B. S. & Poulos, T. L. (1998) Cell 95, 939-950. [DOI] [PubMed] [Google Scholar]
- 18.Venema, V. J., Ju, H., Zou, R. & Venema, R. C. (1997) J. Biol. Chem. 272, 28187-28190. [DOI] [PubMed] [Google Scholar]
- 19.Ju, H., Zou, R., Venema, V. J. & Venema, R. C. (1997) J. Biol. Chem. 272, 18522-18525. [DOI] [PubMed] [Google Scholar]
- 20.Michel, J. B., Feron, O., Sase, K., Prabhakar, P. & Michel, T. (1997) J. Biol. Chem. 272, 25907-25912. [DOI] [PubMed] [Google Scholar]
- 21.Ghosh, S., Gachhui, R., Crooks, C., Wu, C., Lisanti, M. P. & Stuehr, D. J. (1998) J. Biol. Chem. 273, 22267-22271. [DOI] [PubMed] [Google Scholar]