

Abstract
Purpose
Hereditary factors play an important role in colorectal cancer (CRC) risk, yet the prevalence of germline cancer susceptibility gene mutations in patients with CRC unselected for high-risk features (eg, early age at diagnosis, personal/family history of cancer or polyps, tumor microsatellite instability [MSI], mismatch repair [MMR] deficiency) is unknown.
Patients and Methods
We recruited 1,058 participants who received CRC care in a clinic-based setting without preselection for age at diagnosis, personal/family history, or MSI/MMR results. All participants underwent germline testing for mutations in 25 genes associated with inherited cancer risk. Each gene was categorized as high penetrance or moderate penetrance on the basis of published estimates of the lifetime cancer risks conferred by pathogenic germline mutations in that gene.
Results
One hundred five (9.9%; 95% CI, 8.2% to 11.9%) of 1,058 participants carried one or more pathogenic mutations, including 33 (3.1%) with Lynch syndrome (LS). Twenty-eight (96.6%) of 29 available LS CRCs demonstrated abnormal MSI/MMR results. Seventy-four (7.0%) of 1,058 participants carried non-LS gene mutations, including 23 (2.2%) with mutations in high-penetrance genes (five APC, three biallelic MUTYH, 11 BRCA1/2, two PALB2, one CDKN2A, and one TP53), 15 of whom lacked clinical histories suggestive of their underlying mutation. Thirty-eight (3.6%) participants had moderate-penetrance CRC risk gene mutations (19 monoallelic MUTYH, 17 APC*I1307K, two CHEK2). Neither proband age at CRC diagnosis, family history of CRC, nor personal history of other cancers significantly predicted the presence of pathogenic mutations in non-LS genes.
Conclusion
Germline cancer susceptibility gene mutations are carried by 9.9% of patients with CRC. MSI/MMR testing reliably identifies LS probands, although 7.0% of patients with CRC carry non-LS mutations, including 1.0% with BRCA1/2 mutations.
INTRODUCTION
Hereditary factors play a key role in colorectal cancer (CRC) risk. Studies have estimated the prevalence of hereditary CRC syndromes by performing syndrome-specific genetic testing on patients with CRC with acknowledged high-risk features. For instance, studies that used microsatellite instability (MSI) or DNA mismatch repair (MMR) protein immunohistochemistry (IHC) tumor testing in preselected patients for germline MMR gene testing have identified Lynch syndrome (LS) in 2% to 4% of patients with CRC.1-5 Likewise, studies that demonstrated a < 1% prevalence of familial adenomatous polyposis (FAP) and MUTYH-associated polyposis (MAP) included germline APC testing only in patients with CRC with clinical features of FAP (eg, > 100 adenomas) and only sequenced for a limited panel of MUTYH mutations.6-10
Because these studies focused solely on high-risk patients with CRC identified by MSI/MMR status or clinical history, the true prevalence of inherited cancer susceptibility among unselected CRC populations is unknown. Furthermore, existing estimates do not account for the contribution of germline factors linked to modestly increased risks of CRC, including monoallelic MUTYH mutations and the Ashkenazi Jewish APC*I1307K founder allele.11,12 Finally, emerging data have suggested that some patients with CRC harbor mutations in breast and ovarian cancer susceptibility genes (eg, BRCA1/2), which raises the question about whether these mutations contribute to hereditary CRC risk.13,14 The primary aim of the current study was to assess the prevalence of cancer susceptibility gene mutations among patients with CRC but without preselection for high-risk features.
PATIENTS AND METHODS
All patients undergoing clinical care at the Dana-Farber Cancer Institute for GI cancer were eligible for enrollment in an institutional sample registry, which included collection of a one-time blood sample for clinical research. A cohort of 1,100 consecutively enrolled registry participants with CRC, from whom blood was collected between December 2008 to March 2014, were retrospectively identified for analysis in this study regardless of tumor MSI/MMR status, age at diagnosis, and personal/family history of cancer. Specimens were collected for research purposes only and, therefore, did not follow the Clinical Laboratory Improvement Amendments chain of custody regulations for clinical testing. Thus, genetic test results obtained in this study were considered research and were not returned to study participants or their clinicians (as was explained in the study consent form), which mirrored methods from a similar study in a cohort of women with breast cancer.15 The study was approved by the Dana-Farber/Harvard Cancer Center Institutional Review Board.
Structured medical record review and self-administered questionnaires were used to obtain information about participants’ personal/family history; CRC stage; anatomic location; and molecular features, including results of MSI, MMR IHC, and KRAS/NRAS and BRAF mutation analyses. Because somatic RAS and BRAF mutations are known to be mutually exclusive in CRC, tumors with KRAS/NRAS mutations were presumed to be BRAF wild type, and those with BRAF mutations were presumed to be KRAS/NRAS wild type. KRAS G12C mutations were specifically analyzed because prior studies found this mutation to be overrepresented in CRCs in patients with MAP.16,17
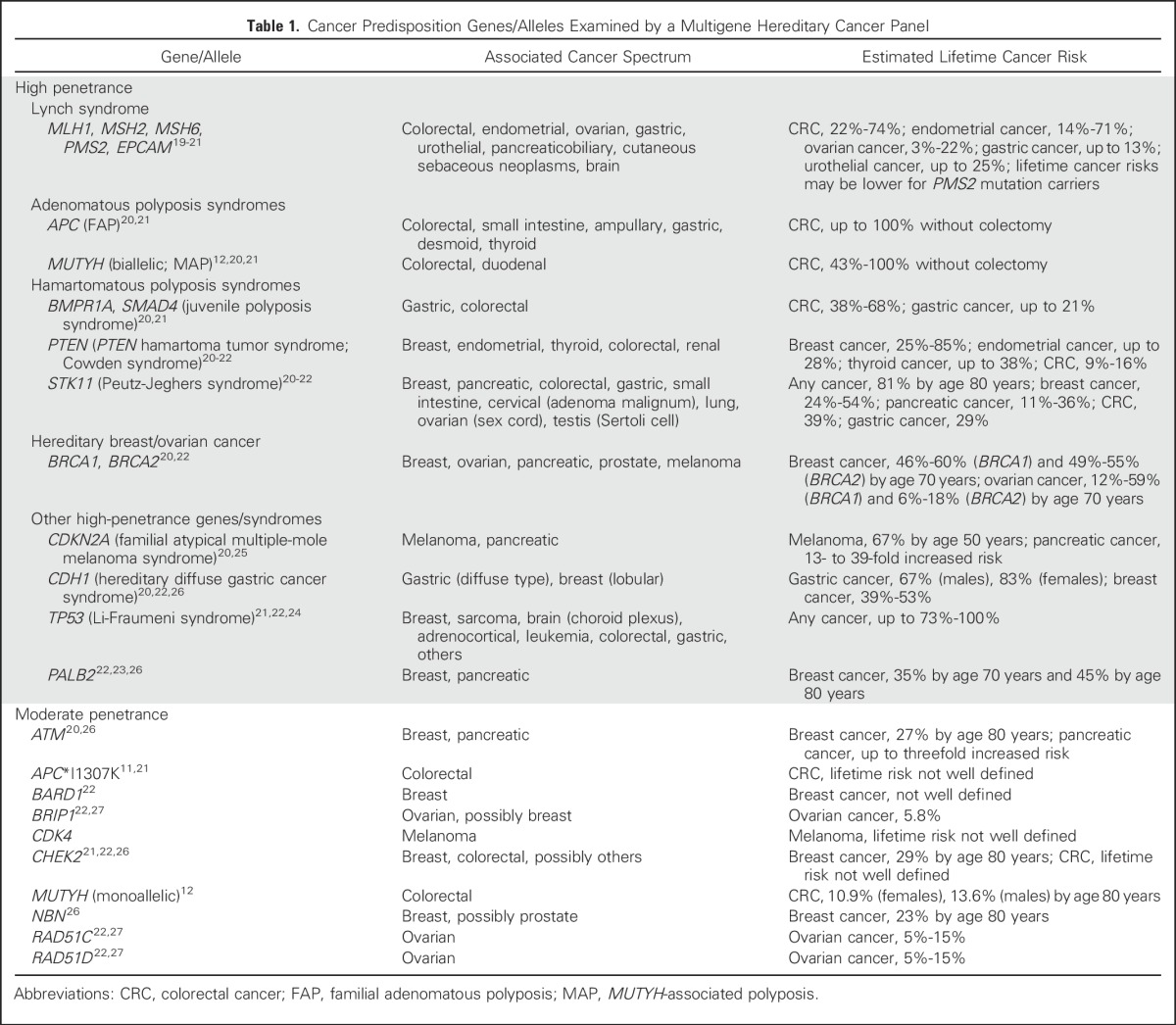
Germline DNA was extracted from frozen whole blood of each participant, polymerase chain reaction amplified with a custom amplicon library, and sequenced to detect sequence alterations and large rearrangements among 25 cancer susceptibility genes by following procedures developed in prior studies.13,18 Published calculations of the lifetime risks of cancer conferred by mutations in each gene were used to categorize genes as high penetrance or moderate penetrance (Table 1).11,12,19-27 All sequence variations and large rearrangements detected were classified for pathogenicity into the following categories, as previously described13: deleterious, suspected deleterious, variant of uncertain significance, favor polymorphism, and polymorphism. Variants were classified as being of uncertain significance if data were insufficient to support either a deleterious or a benign interpretation. Individuals with deleterious or suspected deleterious alterations were collectively defined as having pathogenic mutations. Individuals who carried pathogenic mutations were further assessed to determine whether they fulfilled clinical criteria for syndrome-specific germline analysis of the gene in which their mutation was found (Appendix, online only).
Table 1.
Cancer Predisposition Genes/Alleles Examined by a Multigene Hereditary Cancer Panel
The primary outcome of this study was the detection of pathogenic cancer susceptibility gene mutations. Age at CRC diagnosis was analyzed as a continuous variable, and all other characteristics were analyzed as categorical variables. Bivariate association of mutation status with clinical characteristics was assessed by Fisher’s exact test (categorical variables) and the two-sample t test (continuous variables). Clinically relevant characteristics, including those found to be significant on bivariate analysis, were examined in multivariable logistic regression models to assess for an association with the presence of a pathogenic non-LS mutation. Stata/SE version 14.0 (StataCorp, College Station, TX) and R version 3.2.2 (R Project) software were used in these analyses. The degree of association was expressed as the odds ratio (OR) and corresponding 95% CI. P values were two-tailed and considered statistically significant at < .05.
RESULTS
Forty-two registry participants were excluded as a result of failed germline sequencing (n = 34) or a non-CRC diagnosis determined by medical record review (n = 8), which resulted in an evaluable population of 1,058 patients with CRC. Four hundred seventy-one (44.5%) participants were female, 939 (88.8%) were non-Hispanic white, 29 (2.7%) had a history of more than one CRC diagnosis, and 160 (15.1%) had a history of a non-CRC diagnosis (Table 2). The median age at first CRC diagnosis was 55 years (range, 21 to 92 years), and 336 (31.8%) participants received a diagnosis of CRC before age 50 years. One hundred thirty-eight (13.0%) participants had one or more first-degree relatives with CRC, and 206 (19.5%) had one or more second-degree relatives with CRC.
Table 2.
Description of Cohort
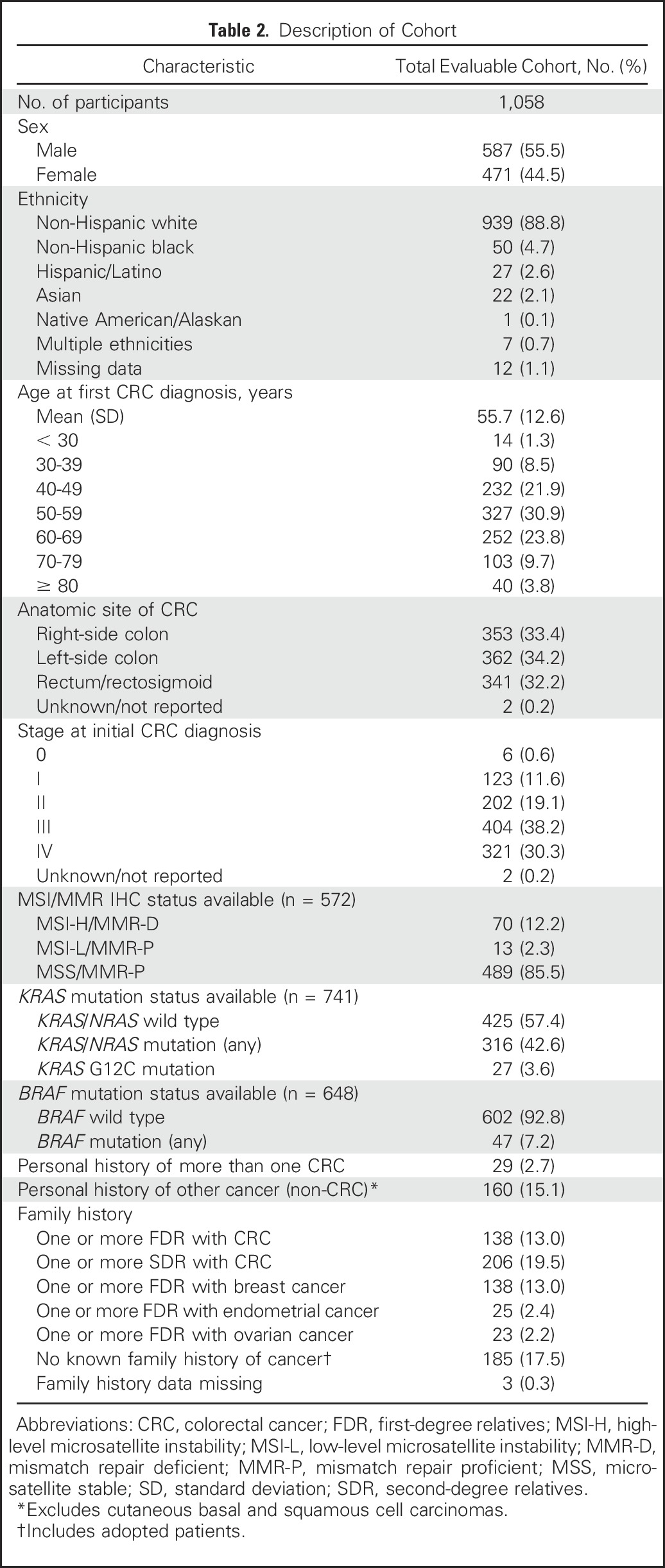
Five hundred seventy-two (54.1%) participants had tumors with MSI and/or MMR IHC data available, of which 70 (12.2%) were MSI-high (MSI-H) and/or MMR deficient (MMR-D). Seven hundred forty-one (70.0%) participants' tumors had available RAS testing results, of which 316 (42.6%) had KRAS/NRAS mutations, including 27 (3.6%) with KRAS G12C mutations. Six hundred forty-eight (61.2%) participants' tumors had BRAF test results available, of which 47 (7.2%) had BRAF mutations.
One hundred five (9.9%; 95% CI, 8.2% to 11.9%) of the 1,058 participants carried one or more pathogenic mutations, including 55 (5.2%; 95% CI, 4.0% to 6.8%) with high-penetrance mutations (four with concurrent moderate-penetrance mutations) and 50 (4.7%; 95% CI, 3.6% to 6.2%) with only moderate-penetrance mutations (Fig 1). Thirty-three (3.1%; 95% CI, 2.2% to 4.4%) of the 1,058 participants carried LS mutations (13 MLH1, seven MSH2, six MSH6, and seven PMS2; Appendix Table A1, online only) and 74 (7.0%; 95% CI, 5.6% to 8.7%) carried non-LS mutations (two both LS and non-LS mutations). Twenty-three (2.2%; 95% CI, 1.4% to 3.3%) participants carried high-penetrance non-LS mutations (five APC, three biallelic MUTYH, three BRCA1, eight BRCA2, two PALB2, one CDKN2A, and one TP53; Table 3). Thirty-eight (3.6%; 95% CI, 2.6% to 4.9%) participants had mutations in moderate-penetrance genes linked to CRC risk (19 monoallelic MUTYH, 17 APC*I1307K, and two CHEK2 mutations); 16 (1.5%; 95% CI, 0.9% to 2.5%) had mutations in other moderate-penetrance genes (Appendix Table A2, online only). Five participants carried more than one pathogenic mutation, including one with MLH1/BRCA2 mutations, one with BRCA2/monoallelic MUTYH mutations, one with MSH2/APC*1307K mutations, and two with BRCA1/APC*I1307K mutations.
Fig 1.

Pathogenic mutations identified with a multigene panel among 1,058 individuals with colorectal cancer. *Includes four probands with both a high- and a moderate-penetrance mutation. †One proband carried both an MLH1 and a BRCA2 mutation. ‡Does not include the four probands with concurrent high-penetrance mutations.
Table 3.
Clinical Characteristics of Probands With High-Penetrance Non-Lynch Syndrome Mutations

Twenty-eight (96.6%; 95% CI, 80.4% to 99.8%) of 29 LS probands had CRCs that demonstrated MSI-H and/or MMR-D (four had missing MSI/MMR data), and 32 (97.0%; 95% CI, 82.5% to 99.8%) of 33 met clinical criteria for LS testing. Eight (72.7%) of 11 BRCA1/2 carriers, two (40.0%) of five APC carriers, one (33.3%) of three biallelic MUTYH mutation carriers, and the TP53 carrier lacked personal/family histories that fulfilled criteria for germline testing for their respective syndrome. Although there are no guidelines for PALB2 or CDKN2A testing, neither PALB2 carrier had a personal/family history of breast or pancreatic cancer, and the CDKN2A carrier had no personal/family history of pancreatic cancer or melanoma. Thus, overall, 15 (65.2%; 95% CI, 42.8% to 82.8%) of 23 probands with high-penetrance non-LS mutations lacked clinical histories suggestive of their underlying mutation.
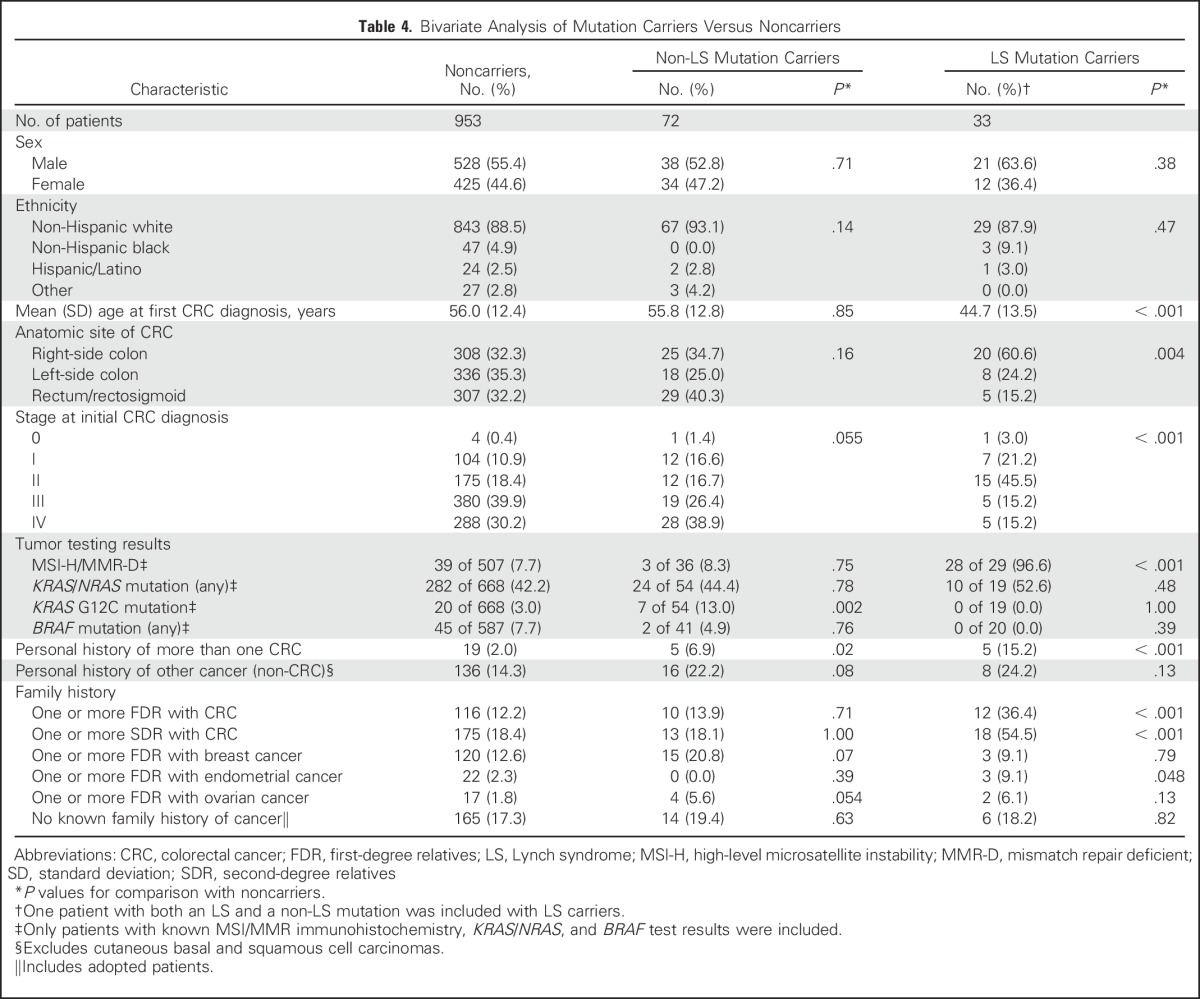
On bivariate analysis (Table 4), LS mutation carriers were significantly younger at CRC diagnosis and more likely to have an earlier stage of CRC, right-sided CRC, multiple CRC diagnoses, MSI-H/MMR-D CRC, a first- and/or second-degree relative with CRC, and a first-degree relative with endometrial cancer than noncarriers. On both bivariate and multivariable regression analyses (Appendix Table A3, online only), non-LS mutation carriers were significantly more likely to have had multiple CRC diagnoses (OR, 3.70; 95% CI, 1.31 to 10.49) and CRC with a somatic KRAS G12C mutation (OR, 4.58; 95% CI, 1.76 to 11.92) than noncarriers. Neither age at CRC diagnosis, anatomic CRC site, personal history of non-CRC, nor having a first-degree relative with CRC was significantly associated with the presence of a non-LS mutation. Forty (54%) of 74 non-LS mutation carriers were given a diagnosis of CRC at age ≥ 50 years and lacked a family history of CRC in a first-degree relative.
Table 4.
Bivariate Analysis of Mutation Carriers Versus Noncarriers
Four hundred eight germline variants of uncertain significance were detected among 330 (31.2%; 95% CI, 28.4% to 34.1%) of the 1,058 participants (Appendix Fig A1, online only; Appendix Table A4, online only). Six participants with MMR-D CRC carried variants of uncertain significance in MMR genes that corresponded to their tumor’s abnormal MMR protein staining, which suggests possible pathogenicity to these variants (Appendix Table A5, online only).
DISCUSSION
In this clinic-based cohort of 1,058 patients with CRC, 9.9% carried pathogenic germline mutations, most of which were in actionable, high-penetrance cancer susceptibility genes. Currently, patients with CRC are referred for germline testing based on the identification of high-risk phenotypic features (eg, age, family history), but beyond MSI/MMR tumor testing for LS, no systematic approach to hereditary risk assessment exists.20,21 Moreover, access to germline testing is inconsistent as a result of limited availability of genetic counseling and insurance coverage restrictions.28 The current results demonstrate that multigene germline testing without preselection on the basis of high-risk clinical features could significantly increase opportunities for primary and secondary cancer prevention because overall, 96.2% of mutation carriers (101 of 105) had mutations in genes for which guidelines recommend specific screening and/or risk-reduction practices.19-22
Consistent with prior studies that performed germline LS testing after preselection with MSI/MMR tumor testing,1-6 3.1% of participants in the current study had LS, virtually all of whom had abnormal MSI/MMR results and clinical phenotypes suggestive of LS. These data support the current practice of performing systematic MSI/MMR IHC for all CRCs to screen for LS4,5,19-21 and because MSI-H and MMR-D are important prognostic and predictive biomarkers in CRC. The large number of non-LS mutations identified in this cohort, however, demonstrates that tumor testing alone is insufficient for identifying inherited cancer risk among unselected patients with CRC.
Seven percent of patients with CRC carried mutations in non-LS genes, including 1.0% with BRCA1/2 mutations, and nearly two thirds of probands with high-penetrance non-LS mutations lacked clinical histories suggestive of their respective syndromes. The majority of non-LS mutation carriers in this study also lacked traditional phenotypic characteristics of hereditary CRC risk (age < 50 years, CRC in a first-degree relative). Although a personal history of more than one CRC diagnosis was significantly associated with presence of non-LS mutations, a minority of carriers had multiple CRCs demonstrating that such history has limited utility in identifying mutation carriers. Somatic KRAS G12C mutations were significantly associated with the presence of pathogenic non-LS mutations and were found predominantly in probands with biallelic or monoallelic germline MUTYH mutations, consistent with prior studies.16,17 These findings support the notion that individuals whose CRCs harbor somatic KRAS G12C mutations should undergo germline evaluation.16,17
One half of all FAP and MAP probands in this cohort lacked diffuse colorectal polyposis, which demonstrates that polyp number is an imperfect indicator of inherited risk.7 Beyond the classic hereditary CRC syndromes, mutations in other high-penetrance genes (BRCA1/2, PALB2, CDKN2A, and TP53) were identified in a notable number of CRC probands, most of whom lacked phenotypic features of these syndromes. BRCA1/2 mutations were more common than FAP and MAP combined in this cohort, and > 70% of BRCA1/2 probands lacked clinical features of hereditary breast and ovarian cancer, which suggests that these mutations are likely underdiagnosed among unselected patients with CRC. The finding of BRCA1/2 mutations in 1.0% of patients with CRC (approximately 1:96) is substantially higher than the estimated 0.25% general population prevalence (1:400) and raises questions about whether such mutations predispose to CRC.29,30 Previously, CRC has not been included in the BRCA1/2 tumor spectrum, and published guidelines do not recommend increased CRC screening for mutation carriers.22,31,32 However, some studies have suggested a possible modest link between CRC risk and mutations in BRCA1/2.14,33-35 Although the current data cannot establish a causal link between BRCA1/2 mutations and probands' CRC diagnoses, these findings further support the hypothesis that BRCA1/2 mutations predispose to a wider diversity of cancer than traditionally appreciated,13 potentially including CRC. Further studies with systematic somatic tumor testing (eg, BRCA1/2 loss of heterozygosity, homologous recombination repair deficiency analyses) will be key in understanding whether such germline mutations are truly causative of these cancers.
Although the absolute risk conferred by monoallelic MUTYH mutations and APC*I1307K is modest,36 these moderate-penetrance mutations account for a sizeable fraction of CRC risk. The current data suggest that the identification of patients likely to harbor these moderate-penetrance mutations on the basis of phenotype alone is difficult. Evidence continues to grow in support of the role of testing for moderate-penetrance mutations to identify individuals who stand to benefit from enhanced screening,12 and guidelines now recommend specialized colonoscopic surveillance for monoallelic MUTYH mutation and APC*I1307K carriers.21 Co-occurrence of these moderate-penetrance mutations with BRCA1/2 mutations in some participants raises the possibility that CRC risk is compounded by the presence of multiple germline mutations.
Given the high prevalence of mutations seen in this cohort and the inability to clinically identify many patients with CRC with actionable, high-penetrance mutations, these data raise the provocative question of whether all patients with CRC should undergo multigene germline testing for inherited cancer susceptibility. Although precedent exists for such a test everyone approach in ovarian cancer,22 the precise clinical significance of moderate-penetrance alterations28,36 and even unexpected high-penetrance mutations must be more fully characterized to understand the utility of performing multigene germline testing on all patients with CRC. From a care delivery perspective, the traditional practice of using high-risk phenotypic features (age, MSI/MMR status, family history) to guide referral for germline testing inherently limits the proportion of patients with CRC who undergo hereditary cancer risk assessment. Such phenotype-driven genetic testing strategies require consistent, systematic evaluation for clinical features of underlying germline risk, which is difficult to reliably execute in real-world clinical practice1,37,38 and, even if performed correctly, will still miss a large fraction of patients with CRC with germline risk according to the current data. As germline testing becomes less expensive and additional cancer predisposition genes are identified and their associated cancer risks better clarified, it is likely that there will be a threshold at which it becomes more efficient to systematically offer multigene germline testing to patients with CRC irrespective of phenotype.39
Although the identification of unexpected germline mutations in highly penetrant genes has obvious value to the healthy, at-risk family members of patients with CRC, who can pursue testing for the identified mutation and undergo appropriate risk-reducing interventions, emerging data also illustrate how treatment of probands' CRCs themselves might be influenced by the discovery of germline mutations. Immune checkpoint inhibitors, for example, have shown significant promise as treatment of patients with LS-associated CRCs and other MSI-H cancers,40 whereas PARP inhibitors show significant activity in the treatment of a wide array of cancers (eg, breast, ovarian, pancreatic, prostate) that develop not only in BRCA1/2 mutation carriers but also in individuals with ATM, CHEK2, and possibly PALB2 mutations.41,42 Whether PARP inhibitors have activity in individuals with CRC who have these mutations is unknown, but the current results demonstrate that a substantial fraction of patients with CRC could potentially benefit from therapies tailored to their germline status.
This study’s primary strength is its use of a large cohort of patients with CRC and detailed personal/family history and pathologic data. These data allowed for careful examination of each proband to determine the extent to which they demonstrated features of the proband’s particular syndrome. Furthermore, in contrast to prior studies that attempted to estimate the prevalence of hereditary CRC by performing syndrome-specific testing only in individuals with specific high-risk features, all patients with CRC in this cohort underwent multigene germline testing regardless of age at diagnosis, personal/family history, or tumor testing results.
The study’s limitations include the use of a clinic-based cohort recruited from a large academic center, which may limit generalizability to population-based patients with CRC. For instance, the median age at CRC diagnosis was 55 years, with 31.8% of participants receiving a diagnosis before age 50 years, whereas recent SEER data have shown that the median age at CRC diagnosis is 68 years, with only 15% of patients receiving a diagnosis before age 50 years.43,44 By comparison, prior studies of clinic-based cohorts of patients with CRC have found a median age at CRC diagnosis of 43 to 53 years, whereas published population-based CRC cohorts have had a median age at CRC diagnosis of 58 to 71 years.2-6 Because age at CRC diagnosis was not significantly associated with the presence of a non-LS mutation in the current study, the clinic-based setting may not have significantly affected the observed mutation prevalence. We acknowledge that any attempt to categorize cancer susceptibility genes as high penetrance versus moderate penetrance is somewhat arbitrary because cancer risk is a continuum rather than a categorical or binary phenomenon. The genes categorized as high penetrance in this study are well-established to predispose to a variety of malignancies, whereas the genes considered moderate penetrance generally have less-robust data to support their associated risks (Table 1). Furthermore, the prevalence of mutations identified by multigene panel testing inherently depends on the genes included in a given panel. Several genes newly linked to CRC risk, including POLD1, POLE, and NTHL1, were not examined in this study, and their penetrance and associated cancer spectrum are poorly understood. With additional testing for such genes, the prevalence of cancer susceptibility gene mutations among patients with CRC ultimately may be determined as even higher than the 9.9% rate observed in this study. Finally, we had limited data on participants who were of Ashkenazi Jewish ancestry, which precludes assessment of whether the founder BRCA1/2 and APC*I1307K mutations identified were attributable to their background population prevalence. Even with the exclusion of Ashkenazi founder mutations, the prevalence of nonfounder BRCA1/2 mutations (eight [0.8%] of 1,058 [approximately 1:132]; 95% CI, 0.4% to 1.6%) in this CRC cohort is higher than the 0.25% general population (1:400) prevalence,29 although such population-wide prevalence estimates remain a matter of debate.30
To date, clinical practice guidelines21,22,28 have been cautious about endorsing the use of multigene panel testing for hereditary cancer risk assessment because of a paucity of data about the clinical utility of such panels versus the established practice of phenotype-directed, syndrome-specific evaluation. Over the past decade, universal testing of all CRCs for MSI-H/MMR-D has increasingly become routine practice to systematize the recognition of LS, whereas patients with colorectal polyposis are recommended to undergo FAP and MAP testing.2,4,5,19-22 The current study findings, however, clearly illustrate that genetic factors that underlie CRC extend beyond these well-recognized familial CRC syndromes, are markedly more common than previously appreciated, frequently occur in patients with CRC who lack classic high-risk features, and are found predominantly in genes for which specialized risk-reducing interventions are recommended. These data demonstrate that comprehensive germline testing in unselected patients with CRC will identify substantially more opportunities for genetically driven cancer prevention than what is found in current practice, which suggests the need to reconsider contemporary approaches to hereditary cancer risk assessment.
Appendix
Methods
Patients who were found to carry pathogenic germline mutations in one or more cancer susceptibility genes underwent assessment of their personal and family cancer history to determine whether they fulfilled clinical criteria for undergoing syndrome-specific genetic testing of the gene(s) in which their mutation was found.
Lynch Syndrome
Participants with colorectal cancer (CRC) were considered to fulfill guidelines for Lynch syndrome (LS) testing19-21 (Stoffel EM, et al: J Clin Oncol 33:209-217, 2015) if they had any of the following: a known LS mutation in the family; CRC diagnosed before age 50 years; personal history of more than one CRC; personal history of any other LS-associated neoplasm (endometrial cancer; gastric cancer; ovarian cancer; pancreatic cancer; small intestine cancer; urothelial cancer of the bladder, ureter, or renal pelvis; biliary cancer; brain tumor; cutaneous sebaceous adenoma/carcinoma; or keratoacanthoma); CRC with high-level microsatellite instability (MSI-H) and/or abnormal DNA mismatch repair protein immunohistochemistry; Prediction Model for MLH1, MSH2, and MSH6 Gene Mutations score ≥ 5% (http://premm.dfci.harvard.edu; Kastrinos F, et al: Gastroenterology 140:73-81, 2011); one or more first-degree relatives (FDR) with LS-associated neoplasm diagnosed before age 50 years; two or more FDR and/or second-degree relatives (SDR) with LS-associated neoplasms.
BRCA1 and BRCA2
Participants with CRC were considered to fulfill guidelines for BRCA1/2 testing22 if they had any of the following: a known BRCA1/2 mutation in the family; a personal history of ovarian cancer; a personal history of male breast cancer; a personal history of breast or pancreatic cancer and Ashkenazi Jewish ancestry; a personal history of breast cancer diagnosed at age ≤ 45 years; a personal history of multiple breast cancers, with at least one diagnosed at age ≤ 50 years; a personal history of breast cancer diagnosed at age ≤ 50 years with one or more FDR, SDR, or third-degree relatives (TDR) diagnosed with breast, pancreatic, or Gleason ≥ 7 prostate cancer at any age; a personal history of triple-negative breast cancer diagnosed at age ≤ 60 years; a personal history of breast cancer (any age) with one or more FDR, SDR, or TDR with breast cancer diagnosed at age ≤ 50 years; a personal history of breast cancer (any age) with two or more FDR, SDR, or TDR with breast cancer (any age); a personal history of breast cancer (any age) with ≥ 2 FDR, SDR, or TDR with ovarian cancer (any age); a personal history of breast cancer (any age) with two or more FDR, SDR, or TDR with pancreatic and/or Gleason ≥ 7 prostate cancer (any age); a personal history of breast cancer (any age) with one or more FDR, SDR, or TDR with male breast cancer; a personal history of Gleason ≥ 7 prostate cancer (any age) with one or more FDR, SDR, or TDR with ovarian cancer (any age); a personal history of Gleason ≥ 7 prostate cancer (any age) with one or more FDR, SDR, or TDR with breast cancer diagnosed at age ≤ 50 years; a personal history of Gleason ≥ 7 prostate cancer (any age) with two or more FDR, SDR, TDR with breast, pancreatic, or Gleason ≥ 7 prostate cancer at any age; a personal history of pancreatic cancer (any age) with one or more FDR, SDR, or TDR with ovarian cancer (any age); a personal history of pancreatic cancer (any age) with one or more FDR, SDR, or TDR with breast cancer diagnosed at age ≤ 50 years; or a personal history of pancreatic cancer (any age) with two or more FDR, SDR, or TDR with breast, pancreatic, or Gleason ≥ 7 prostate cancer at any age. Participants with CRC were also considered to fulfill criteria for BRCA1/2 testing22 if they had a family history of an FDR or SDR who met any of these criteria or a TDR with breast and/or ovarian cancer who had two or more FDR, SDR, TDR with breast cancer (at least one who received a diagnosis at age ≤ 50 years) and/or ovarian cancer.
Adenomatous Polyposis Syndromes
Participants with CRC were considered to fulfill criteria for APC and MUTYH testing21 if they had any of the following: known APC and/or MUTYH mutations in the family or a personal history of ≥ 20 colorectal adenomas over their lifetime.
Li-Fraumeni Syndrome
Participants with CRC were considered to fulfill criteria for Li-Fraumeni syndrome (LFS) TP53 genetic testing22 (Tinat J, et al: J Clin Oncol 27:e108-e109, 2009) if they had a personal/family history of any of the following: a known TP53 mutation in the family; a personal history of breast cancer diagnosed at age < 31 years; a personal history of adrenocortical carcinoma, choroid plexus carcinoma, or rhabdomyosarcoma (any age); a personal history of any LFS-associated cancer (soft tissue sarcoma, osteosarcoma, CNS tumor, breast cancer, adrenocortical carcinoma) diagnosed at age < 46 years and one of more FDR, SDR, or TDR with any LFS-associated cancer diagnosed at age < 56 years (unless only breast cancer was present in proband/family); a personal history of any LFS-associated cancer and one or more FDR, SDR, or TDR with multiple LFS-associated cancers (any age); a personal history of two or more LFS-associated cancers (as long as at least one is a non–breast cancer) with one or more cancers diagnosed at age < 46 years; a personal history of sarcoma diagnosed at age < 45 years and an FDR with any cancer diagnosed at age < 45 years and another FDR or SDR with cancer diagnosed at age < 45 years; or a personal history of sarcoma diagnosed at age < 45 years and an FDR with any cancer diagnosed at age < 45 years and another FDR or SDR with sarcoma (any age).
Fig A1.

Number of germline variants of uncertain significance (VUS), per gene, detected with a 25-gene panel in 1,058 patients with colorectal cancer.
Table A1.
Clinical Characteristics of Patients With Lynch Syndrome Mutations

Table A2.
Clinical Characteristics of Patients With Moderate-Penetrance Germline Mutations

Table A3.
Factors That Predict the Presence of a Non–Lynch Syndrome Cancer Susceptibility Gene Mutation in Patients With CRC (versus patients with mutation-negative CRC)
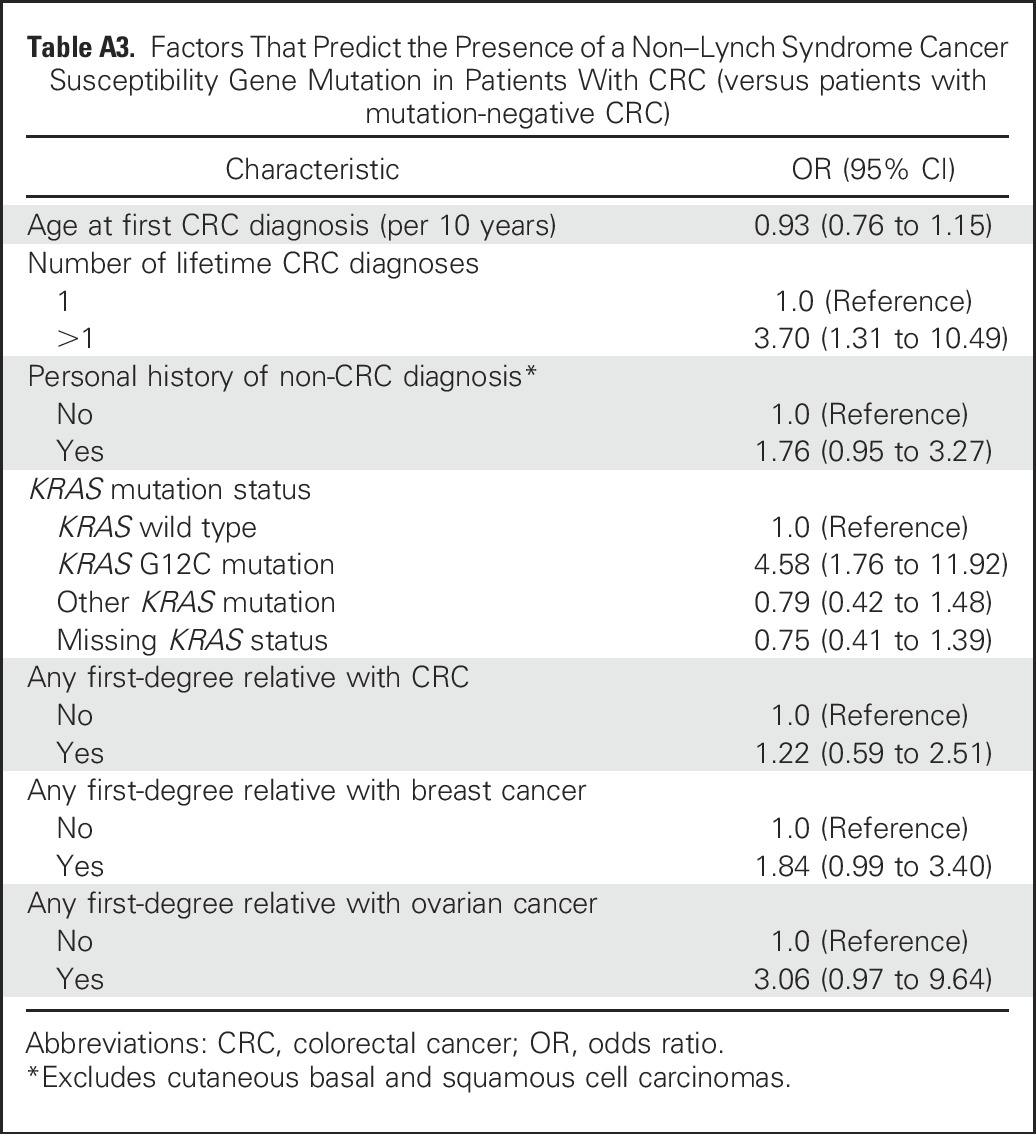
Table A4.
Germline Variants of Uncertain Significance Detected by a 25-Gene Panel Among 1,058 Participants With Colorectal Cancer

Table A5.
Clinical Characteristics of Patients With MMR-D CRC and Germline VUS in DNA MMR Genes

Footnotes
Listen to the podcast by Dr Ford at ascopubs.org/jco/podcasts
Supported by National Cancer Institute grants P50CA127003 (C.S.F.), K07CA148894 (K.N.), K24CA113433 (S.S.), and R01CA132829 (S.S.) and by Myriad Genetics.
Presented at the 52nd Annual Meeting of the American Society of Clinical Oncology, Chicago, IL, June 3-7, 2016, and the 20th Annual Meeting of the Collaborative Group of the Americas on Inherited Colorectal Cancer, Seattle, WA, October 2-3, 2016.
AUTHOR CONTRIBUTIONS
Conception and design: Matthew B. Yurgelun, Matthew H. Kulke, Charles S. Fuchs, Brian A. Allen, Robert J. Mayer, Deborah Schrag, Jeffrey A. Meyerhardt, Kimmie Ng, Anne-Renee Hartman, Richard J. Wenstrup, Sapna Syngal
Financial support: Matthew B. Yurgelun, Charles S. Fuchs, Kimmie Ng, Sapna Syngal
Administrative support: Matthew H. Kulke, Charles S. Fuchs, Philip G. McNamara, Richard J. Wenstrup, Sapna Syngal
Provision of study materials or patients: Matthew B. Yurgelun, Matthew H. Kulke, Charles S. Fuchs, Jason L. Hornick, Robert J. Mayer, Deborah Schrag, Jeffrey A. Meyerhardt, Kimmie Ng, Sapna Syngal
Collection and assembly of data: Matthew B. Yurgelun, Matthew H. Kulke, Brian A. Allen, Jason L. Hornick, Chinedu I. Ukaegbu, Lauren K. Brais, Philip G. McNamara, Anne-Renee Hartman, Richard J. Wenstrup, Sapna Syngal
Data analysis and interpretation: Matthew B. Yurgelun, Matthew H. Kulke, Charles S. Fuchs, Brian A. Allen, Hajime Uno, Jason L. Hornick, Chinedu I. Ukaegbu, Lauren K. Brais, Philip G. McNamara, Robert J. Mayer, Deborah Schrag, Jeffrey A. Meyerhardt, Kimmie Ng, John Kidd, Nanda Singh, Anne-Renee Hartman, Sapna Syngal
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Cancer Susceptibility Gene Mutations in Individuals With Colorectal Cancer
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/site/ifc.
Matthew B. Yurgelun
Research Funding: Myriad Genetics
Matthew H. Kulke
Consulting or Advisory Role: Lexicon Pharmaceuticals
Charles S. Fuchs
Consulting or Advisory Role: Genentech, Eli Lilly, Sanofi, Bayer AG, Celgene, Merck, Bristol-Meyers Squibb, Entrinsic Health Solutions, Five Prime Therapeutics, Agios
Brian A. Allen
Employment: Myriad Genetics
Stock or Other Ownership: Myriad Genetics
Hajime Uno
Consulting or Advisory Role: Ionis Pharmaceuticals
Jason L. Hornick
No relationship to disclose
Chinedu I. Ukaegbu
No relationship to disclose
Lauren K. Brais
No relationship to disclose
Philip G. McNamara
No relationship to disclose
Robert J. Mayer
Honoraria: Taiho Pharmaceutical
Consulting or Advisory Role: CASI Pharmaceuticals
Deborah Schrag
Consulting or Advisory Role: New Century Health, The Ohio State University
Other Relationship: Journal of the American Medical Association
Jeffrey A. Meyerhardt
Consulting or Advisory Role: Genentech
Kimmie Ng
Consulting or Advisory Role: Genentech, Eli Lilly, Tarrex Biopharma
Research Funding: Gilead Sciences, Trovagene, Celgene, Genentech (Inst), Pharmavite (Inst)
John Kidd
Employment: Myriad Genetics
Stock or Other Ownership: Myriad Genetics
Nanda Singh
Employment: Myriad Genetics
Stock or Other Ownership: Myriad Genetics
Anne-Renee Hartman
Employment: Myriad Genetics, Grail
Stock or Other Ownership: Myriad Genetics
Richard J. Wenstrup
Employment: Myriad Genetics
Leadership: Myriad Genetics
Stock or Other Ownership: Myriad Genetics
Sapna Syngal
Consulting or Advisory Role: Eisai
REFERENCES
- 1.Ward RL, Hicks S, Hawkins NJ. Population-based molecular screening for Lynch syndrome: Implications for personalized medicine. J Clin Oncol. 2013;31:2554–2562. doi: 10.1200/JCO.2012.46.8454. [DOI] [PubMed] [Google Scholar]
- 2.Moreira L, Balaguer F, Lindor N, et al. Identification of Lynch syndrome among patients with colorectal cancer. JAMA. 2012;308:1555–1565. doi: 10.1001/jama.2012.13088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kastrinos F, Ojha RP, Leenen C, et al. Comparison of prediction models for lynch syndrome among individuals with colorectal cancer J Natl Cancer Inst 108djv308, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hampel H, Frankel WL, Martin E, et al. Feasibility of screening for Lynch syndrome among patients with colorectal cancer. J Clin Oncol. 2008;26:5783–5788. doi: 10.1200/JCO.2008.17.5950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hampel H, Frankel WL, Martin E, et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer) N Engl J Med. 2005;352:1851–1860. doi: 10.1056/NEJMoa043146. [DOI] [PubMed] [Google Scholar]
- 6.Woods MO, Younghusband HB, Parfrey PS, et al. The genetic basis of colorectal cancer in a population-based incident cohort with a high rate of familial disease. Gut. 2010;59:1369–1377. doi: 10.1136/gut.2010.208462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grover S, Kastrinos F, Steyerberg EW, et al. Prevalence and phenotypes of APC and MUTYH mutations in patients with multiple colorectal adenomas. JAMA. 2012;308:485–492. doi: 10.1001/jama.2012.8780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lubbe SJ, Di Bernardo MC, Chandler IP, et al. Clinical implications of the colorectal cancer risk associated with MUTYH mutation. J Clin Oncol. 2009;27:3975–3980. doi: 10.1200/JCO.2008.21.6853. [DOI] [PubMed] [Google Scholar]
- 9.Bertario L, Russo A, Sala P, et al. Multiple approach to the exploration of genotype-phenotype correlations in familial adenomatous polyposis. J Clin Oncol. 2003;21:1698–1707. doi: 10.1200/JCO.2003.09.118. [DOI] [PubMed] [Google Scholar]
- 10.Cleary SP, Cotterchio M, Jenkins MA, et al. Germline MutY human homologue mutations and colorectal cancer: A multisite case-control study. Gastroenterology. 2009;136:1251–1260. doi: 10.1053/j.gastro.2008.12.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boursi B, Sella T, Liberman E, et al. The APC p.I1307K polymorphism is a significant risk factor for CRC in average risk Ashkenazi Jews. Eur J Cancer. 2013;49:3680–3685. doi: 10.1016/j.ejca.2013.06.040. [DOI] [PubMed] [Google Scholar]
- 12.Win AK, Dowty JG, Cleary SP, et al: Risk of colorectal cancer for carriers of mutations in MUTYH, with and without a family history of cancer. Gastroenterology 146:1208-1211.e5, 2014 [DOI] [PMC free article] [PubMed]
- 13.Yurgelun MB, Allen B, Kaldate RR, et al: Identification of a variety of mutations in cancer predisposition genes in patients with suspected Lynch syndrome. Gastroenterology 149:604-613.e20, 2015 [DOI] [PMC free article] [PubMed]
- 14.Phelan CM, Iqbal J, Lynch HT, et al. Incidence of colorectal cancer in BRCA1 and BRCA2 mutation carriers: Results from a follow-up study. Br J Cancer. 2014;110:530–534. doi: 10.1038/bjc.2013.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tung N, Lin NU, Kidd J, et al. Frequency of germline mutations in 25 cancer susceptibility genes in a sequential series of patients with breast cancer. J Clin Oncol. 2016;34:1460–1468. doi: 10.1200/JCO.2015.65.0747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guarinos C, Juárez M, Egoavil C, et al. Prevalence and characteristics of MUTYH-associated polyposis in patients with multiple adenomatous and serrated polyps. Clin Cancer Res. 2014;20:1158–1168. doi: 10.1158/1078-0432.CCR-13-1490. [DOI] [PubMed] [Google Scholar]
- 17.Aimé A, Coulet F, Lefevre JH, et al. Somatic c.34G>T KRAS mutation: A new prescreening test for MUTYH-associated polyposis? Cancer Genet. 2015;208:390–395. doi: 10.1016/j.cancergen.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 18.Judkins T, Leclair B, Bowles K, et al. Development and analytical validation of a 25-gene next generation sequencing panel that includes the BRCA1 and BRCA2 genes to assess hereditary cancer risk BMC Cancer 15215, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Giardiello FM, Allen JI, Axilbund JE, et al. Guidelines on genetic evaluation and management of Lynch syndrome: A consensus statement by the US Multi-Society Task Force on Colorectal Cancer. Gastroenterology. 2014;147:502–526. doi: 10.1053/j.gastro.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 20.Syngal S, Brand RE, Church JM, et al. ACG clinical guideline: Genetic testing and management of hereditary gastrointestinal cancer syndromes Am J Gastroenterol 110223–262.2015; quiz 263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.NCCN Clinical Practice Guidelines in Oncology: Genetic/familial high-risk assessment: Colorectal. Version 2, 2016. http://www.nccn.org/professionals/physician_gls/pdf/genetics_colon.pdf
- 22.NCCN Clinical Practice Guidelines in Oncology: Genetic/familial high-risk assessment: Breast and ovarian. Version 1, 2017. http://www.nccn.org/professionals/physician_gls/pdf/genetics_screening.pdf
- 23.Antoniou AC, Casadei S, Heikkinen T, et al. Breast-cancer risk in families with mutations in PALB2. N Engl J Med. 2014;371:497–506. doi: 10.1056/NEJMoa1400382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Malkin D. Li-Fraumeni syndrome. Genes Cancer. 2011;2:475–484. doi: 10.1177/1947601911413466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bishop DT, Demenais F, Goldstein AM, et al. Geographical variation in the penetrance of CDKN2A mutations for melanoma. J Natl Cancer Inst. 2002;94:894–903. doi: 10.1093/jnci/94.12.894. [DOI] [PubMed] [Google Scholar]
- 26.Easton DF, Pharoah PD, Antoniou AC, et al. Gene-panel sequencing and the prediction of breast-cancer risk. N Engl J Med. 2015;372:2243–2257. doi: 10.1056/NEJMsr1501341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ramus SJ, Song H, Dicks E, et al. Germline mutations in the BRIP1, BARD1, PALB2, and NBN genes in women with ovarian cancer J Natl Cancer Inst 107djv214, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Robson ME, Bradbury AR, Arun B, et al. American Society of Clinical Oncology policy statement update: Genetic and genomic testing for cancer susceptibility. J Clin Oncol. 2015;33:3660–3667. doi: 10.1200/JCO.2015.63.0996. [DOI] [PubMed] [Google Scholar]
- 29.McClain MR, Palomaki GE, Nathanson KL, et al. Adjusting the estimated proportion of breast cancer cases associated with BRCA1 and BRCA2 mutations: Public health implications. Genet Med. 2005;7:28–33. doi: 10.1097/01.gim.0000151155.36470.ff. [DOI] [PubMed] [Google Scholar]
- 30.Maxwell KN, Domchek SM, Nathanson KL, et al. Population frequency of germline BRCA1/2 mutations. J Clin Oncol. 2016;34:4183–4185. doi: 10.1200/JCO.2016.67.0554. [DOI] [PubMed] [Google Scholar]
- 31.Niell BL, Rennert G, Bonner JD, et al. BRCA1 and BRCA2 founder mutations and the risk of colorectal cancer. J Natl Cancer Inst. 2004;96:15–21. doi: 10.1093/jnci/djh008. [DOI] [PubMed] [Google Scholar]
- 32.Kirchhoff T, Satagopan JM, Kauff ND, et al. Frequency of BRCA1 and BRCA2 mutations in unselected Ashkenazi Jewish patients with colorectal cancer. J Natl Cancer Inst. 2004;96:68–70. doi: 10.1093/jnci/djh006. [DOI] [PubMed] [Google Scholar]
- 33.Kadouri L, Hubert A, Rotenberg Y, et al. Cancer risks in carriers of the BRCA1/2 Ashkenazi founder mutations. J Med Genet. 2007;44:467–471. doi: 10.1136/jmg.2006.048173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Suchy J, Cybulski C, Górski B, et al. BRCA1 mutations and colorectal cancer in Poland. Fam Cancer. 2010;9:541–544. doi: 10.1007/s10689-010-9378-x. [DOI] [PubMed] [Google Scholar]
- 35.Garre P, Martín L, Sanz J, et al. BRCA2 gene: A candidate for clinical testing in familial colorectal cancer type X. Clin Genet. 2015;87:582–587. doi: 10.1111/cge.12427. [DOI] [PubMed] [Google Scholar]
- 36.Tung N, Domchek SM, Stadler Z, et al. Counselling framework for moderate-penetrance cancer-susceptibility mutations. Nat Rev Clin Oncol. 2016;13:581–588. doi: 10.1038/nrclinonc.2016.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wood ME, Kadlubek P, Pham TH, et al. Quality of cancer family history and referral for genetic counseling and testing among oncology practices: A pilot test of quality measures as part of the American Society of Clinical Oncology Quality Oncology Practice Initiative. J Clin Oncol. 2014;32:824–829. doi: 10.1200/JCO.2013.51.4661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grover S, Stoffel EM, Bussone L, et al. Physician assessment of family cancer history and referral for genetic evaluation in colorectal cancer patients. Clin Gastroenterol Hepatol. 2004;2:813–819. doi: 10.1016/s1542-3565(04)00352-0. [DOI] [PubMed] [Google Scholar]
- 39.Gallego CJ, Shirts BH, Bennette CS, et al. Next-generation sequencing panels for the diagnosis of colorectal cancer and polyposis syndromes: A cost-effectiveness analysis. J Clin Oncol. 2015;33:2084–2091. doi: 10.1200/JCO.2014.59.3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Le DT, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372:2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mateo J, Carreira S, Sandhu S, et al. DNA-repair defects and olaparib in metastatic prostate cancer. N Engl J Med. 2015;373:1697–1708. doi: 10.1056/NEJMoa1506859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaufman B, Shapira-Frommer R, Schmutzler RK, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol. 2015;33:244–250. doi: 10.1200/JCO.2014.56.2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Abdelsattar ZM, Wong SL, Regenbogen SE, et al. Reply to percentage of colorectal cancer diagnoses in adults aged younger than 50 years. Cancer. 2016;122:1463–1464. doi: 10.1002/cncr.29977. [DOI] [PubMed] [Google Scholar]
- 44.Bibbins-Domingo K, Grossman DC, Curry SJ, et al. Screening for colorectal cancer: US Preventive Services Task Force recommendation statement. JAMA. 2016;315:2564–2575. doi: 10.1001/jama.2016.5989. [DOI] [PubMed] [Google Scholar]