

Abstract
The thiamin diphosphate (ThDP)-dependent enzyme acetohydroxyacid synthase (AHAS) catalyzes the first common step in branched-chain amino acid biosynthesis. By specific ligation of pyruvate with the alternative acceptor substrates 2-ketobutyrate and pyruvate, AHAS controls the flux through this branch point and determines the relative rates of synthesis of isoleucine, valine, and leucine, respectively. We used detailed NMR analysis to determine microscopic rate constants for elementary steps in the reactions of AHAS II and mutants altered at conserved residues Arg-276, Trp-464, and Met-250. In Arg276Lys, both the condensation of the enzyme-bound hydroxyethyl-ThDP carbanion/enamine (HEThDP) with the acceptor substrates and acetohydroxyacid release are slowed several orders of magnitude relative to the wild-type enzyme. We propose that the interaction of the guanidinium moiety of Arg-264 with the carboxylate of the acceptor ketoacid provides an optimal alignment of substrate and HEThDP orbitals in the reaction trajectory for acceptor ligation, whereas its interaction with the carboxylate of the covalent HEThDP-acceptor adduct plays a similar role in product release. Both Trp-464 and Met-250 affect the acceptor specificity. The high preference for ketobutyrate in the wild-type enzyme is lost in Trp464Leu as a consequence of similar forward rate constants of carboligation and product release for the alternative acceptors. In Met250Ala, the turnover rate is determined by the condensation of HEThDP with pyruvate and release of the acetolactate product, whereas the parallel steps with 2-ketobutyrate are considerably faster. We speculate that the specificity of carboligation and product liberation may be cumulative if the former is not completely committed.
Keywords: mechanism, thiamin diphosphate, amino acid biosynthesis
Acetohydroxyacid (AHA) synthase (AHAS), which catalyzes the first common step in the biosynthesis of branched-chain amino acids, belongs to a homologous family of thiamin diphosphate (ThDP)-dependent enzymes whose initial step is decarboxylation of pyruvate or another 2-ketoacid (1–3). In the process catalyzed by AHAS, the decarboxylation of pyruvate to form the hydroxyethyl-ThDP- anion/enamine (HEThDP-) is followed by the specific condensation of the intermediate with a second aliphatic ketoacid to form an AHA (Fig. 1). Whereas the role of the enzyme in the first steps in AHAS catalysis, i.e., activation of ThDP, decarboxylation of pyruvate, and formation of HEThDP (3), is comparable with the function of other members of its homologous family (4), the function of the protein in controlling the fate of HEThDP is poorly understood.
Fig. 1.

The alternative physiological reactions catalyzed by AHAS and the metabolic roles of the products.
The competition between the alternative “second substrates” 2-ketobutyrate and pyruvate, leading to partition of the flux through AHAS between acetohydroxybutyrate (AHB) and acetolactate (AL), determines the relative rates of formation of isoleucine and of valine, leucine, and pantothenate, respectively (Fig. 1) (5). Most AHASs show a strong preference for 2-ketobutyrate as the second substrate (6), which allows the pathway to function with an intracellular concentration of 2-ketobutyrate almost 2 orders of magnitude lower than that of pyruvate (7). The magnitude of the preference for ketobutyrate, “R,” is characteristic of a given AHAS:
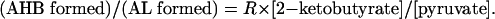 |
[1] |
AHAS isozyme II from Escherichia coli shows one of the highest preferences for 2-ketobutyrate that we have observed (6), R = 60. It is significant that for AHAS II and at least four other AHASs we have examined, the competition between pyruvate and 2-ketobutyrate does not affect the total rate of formation of AHAs. As the concentration of 2-ketobutyrate is increased in the presence of a fixed pyruvate concentration, AHB replaces AL as the product, but the sum of the rates of formation of the two remains essentially constant (6, 8). Such behavior requires that the rate-determining step for enzyme turnover be different from the product-determining step (8). Unless the rate constants of parallel steps with pyruvate and 2-ketobutyrate as the second substrate are coincidentally identical, it further implies that the rate-determining step occurs before binding of the second substrate. Tittmann et al. (9) recently used an acid-quench/1H NMR technique to assess the distribution of ThDP-containing intermediates in wild-type (WT) AHAS II at the steady state and showed that the slowest step(s) by far in the reaction is the formation of the first covalent adduct, lactyl-ThDP. It thus is not surprising that the turnover rate for WT AHAS II is unaffected by the competition between second substrates.
We have been interested for some time in the mechanistic origin of the high specificity of the biosynthetic AHASs for 2-ketobutyrate as the second substrate. Mutagenesis experiments have identified a critical Trp residue (Trp-464 in AHAS II), whose replacement with any other amino acid leads to at least a 10-fold reduction in R (10). However, the kinetic behavior of these and many other mutants of the enzyme is like that of the WT enzyme, in that the concentration of 2-ketobutyrate does not affect the total rate of formation of AHA products (10). Conversely, we recently found several residues in AHAS II whose mutagenesis causes a different sort of change in the kinetic behavior of the enzyme: In mutants at Arg-276 and its neighbors in the active site, Met-250 and Phe-109, the total rate of product formation increases upon addition of 2-ketobutyrate (11). Arg-276 seems to have a crucial role in the specific carboligation of a second ketoacid with bound HEThDP-. We have now determined individual rate constants for the reactions of AHAS II mutants Arg276Lys (Arg-276 → Lys), Met250Ala, and Trp464Leu in the presence of pyruvate as sole substrate and of pyruvate plus 2-ketobutyrate, by analysis of the distribution of ThDP-containing intermediates with the quench NMR technique. These experiments confirm that, in the mutants Arg276Lys and Met250Ala, steps involving the second substrate become rate determining for turnover. The comparison of mutant Trp464Leu with the WT enzyme suggests that the specificity of AHAS II for reaction with 2-ketobutyrate as second substrate is a cumulative result of higher rates for both the carboligation and product-release steps with 2-ketobutyrate. Trp-464 is required for this rate differential. The implications of this behavior for the mechanism of specificity in AHAS are discussed in detail below.
Experimental Procedures
Reagents. Sodium pyruvate, 2-ketobutyrate sodium salt, FAD, ThDP, and creatine were obtained from Sigma, and 1-naphthol was obtained from Carlo Erba Reagenti (Milan). All other chemicals and reagents were of analytical grade and purchased from commercial sources. Restriction and modification enzymes were purchased from New England Biolabs and MBI Fermentas (Amherst, NY).
Bacterial Strains and Plasmids. E. coli BL21(DE3) (Novagen) XL1-Blue MRF′ strains were used for isopropyl β-d-thiogalactoside-induced overexpression of the enzymes. The construction of plasmids pRSET-GM and pQEV-GM, both of which express the same protein with a sequence identical to the original AHAS II enzyme of E. coli ilvG2096 fused at its N terminus to the pRSET hexahistidine leader, is described in ref. 11. The preparation of all of the site-directed mutants discussed here is as described in ref. 11.
Production and Purification of Enzymes. Bacterial cells were transformed and grown as described in ref. 4, and expression was induced by the addition of 0.5 mM isopropyl β-d-thiogalactoside to cultures at OD660 0.5–0.6. The N-terminal hexahistidine-fused constructs expressed from pRSET-GM or pQEV-GM were purified by affinity chromatography on Ni-NTA agarose columns (Qiagen, Hilden, Germany). Bacterial cells were disrupted by sonication in a buffer (buffer A) containing 10 mM imidazole, 0.5 M KCl, 50 mM Tris·HCl (pH 8.0), and 20 μM FAD. After 1-h centrifugation at 27,000 × g, the supernatant was loaded on a 1 × 5.5-cm column of Qiagen Ni-NTA agarose previously washed with buffer A. The column then was washed with ≈50 ml of buffer A (until no more protein eluted) and the His′-GM protein eluted with buffer A plus 0.4 M imidazole. The fractions then were dialyzed against 50 mM KPi (pH 7.6) containing 20 μM FAD and 1 mM DTT.
Enzyme assays were carried out by using the methods described in refs. 4 and 11 in a 0.1 M KPi buffer (pH 7.6) containing 10 mM MgCl2, 0.1 mM ThDP, and 75 μM FAD, with 100 mM pyruvate as substrate, except where otherwise indicated.
Rapid mixing and NMR analysis experiments were carried out and analyzed by the method of Tittmann et al. (9). A hexahistidine-fused enzyme (15 mg/ml enzyme) was reconstituted with 250 μM FAD, 10 mM Mg2+, and an equimolar (active site concentration) amount of ThDP, in 0.1 M KPi buffer (pH 7.6). It then was mixed with 100 mM pyruvate or 50 mM pyruvate plus 50 mM 2-ketobutyrate in a RFQ 3 chemical quenched-flow machine (Kintek, Austin, TX), and the reaction was stopped by the addition of an equal volume of 12.5% (wt/vol) trichloroacetic acid/1 M DCl in D2O. Note that after quench under these conditions, the C2-H of ThDP does not exchange with solvent (12). The reaction was quenched at times between 0.1 and 10 sec, to make certain that the intermediate distributions were determined under true steady-state conditions. For Arg276Lys, the intermediate distribution also was analyzed for different pyruvate and ketobutyrate concentrations (5–50 mM).
In some cases, the reaction was carried out under anaerobic conditions with reduced FAD. AHAS II mutant Arg276Lys was reduced with pyruvate under anaerobic conditions (13). Subsequently, products, substrates, and ThDP/intermediates were removed by gel filtration under anaerobic conditions. The apo-FADH2 enzyme then was reconstituted with 10 mM Mg2+ and equimolar amounts of ThDP and reacted with Pyr/2KB. When Arg276Lys was reacted with its substrates under aerobic conditions, a large fraction of AcTHDP could be isolated as a consequence of the redox side reaction and the slow hydrolysis of AcThDP (14).
After separation of the denatured protein by centrifugation, the distribution of ThDP intermediates in the protein-free solution was analyzed by 1H NMR spectroscopy (9). The integrated areas of the C6′-H signals were used to determine the relative amounts of the covalent intermediates in the solution, whereas the amount of ThDP itself was determined from the area of the C2-H peak because its C6′-H signal was obscured by signals because of FAD (see, for example, Fig. 3).
Fig. 3.

Detection and quantitation of covalent intermediates in the reactions of AHAS II mutant Arg276Lys. Steady-state intermediate distribution of AHAS II mutant Arg276Lys in the presence of 100 mM pyruvate (A) or 50 mM pyruvate and 50 mM 2-ketobutyrate (B) in 0.1 M KPi at 37°C. Reactions were quenched after 300 msec, and the protein-free solutions obtained after centrifugation were analyzed by 1H NMR spectroscopy. Note that the fingerprint region (7.0–8.0 ppm) is zoomed 3- and 2-fold in A and B, respectively, relative to the downfield region where the ThDP C2-H appears.
The major source of error in the analysis of the distribution of intermediates is the error in the integration of the individual signals of the intermediate species, and it is therefore dependent on the signal-to-noise ratio of the respective signals. Signals with a fair signal-to-noise ratio have moderate errors, whereas poorly resolved signals lead to larger errors. When the NMR analysis was repeated with samples of independent preparations of the mutant proteins, no differences greater than the integration error were found in the steady-state intermediate distribution. For all rate constants given in this paper, the indicated errors were derived from the integration error of the individual intermediate signals. Noise as false-positive signals was excluded on the basis of the NMR spectra of quenched protein in the absence of the substrates and of the substrates/products themselves.
The fraction of isolated cofactor as unsubstituted ThDP was corrected for nonsaturation of the enzyme with pyruvate according to the Michaelis–Menten equation.
When the peak corresponding to a given intermediate could not be resolved, e.g., for the product adduct AHBThDP in some cases, lower limits for the microscopic rate constant for its breakdown were calculated from the upper limit of the peak area, defined as a hypothetical peak height of 3 times the signal-to-noise ratio.
Rate constants were derived from the NMR data as follows: According to Cleland's transit times model (15), if substrate is saturating, Eq. 2 relates kcat to the forward (net) rate constants of the catalytic cycle of AHAS (Fig. 2) as follows:
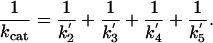 |
[2] |
At steady state and substrate saturation, the rates of formation and decomposition of enzymatic intermediates are balanced.
Fig. 2.

Minimal catalytic cycle for the reactions of AHAS II studied in this work, with the net rate constants identified.
Therefore, the ratio of the relative concentrations of the intermediates in AHAS catalysis can be directly correlated with the forward net rate constants for their interconversion as follows:
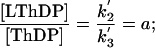 |
[3] |
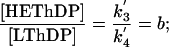 |
[4] |
and
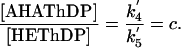 |
[5] |
The forward (net) rate constants of elementary catalytic steps of AHAS then can be calculated from kcat and the experimentally determined intermediate distribution according to Eqs. 6, 7, 8, 9, which are derived from Eqs. 2, 3, 4, 5, as follows:
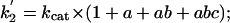 |
[6] |
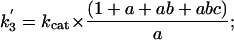 |
[7] |
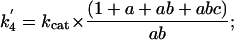 |
[8] |
and
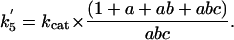 |
[9] |
Results
Duggleby and coworkers (16) demonstrated that AHAS isozyme II could be expressed with an N-terminal hexahistidine fusion to the large, catalytic subunit and readily purified as the holoenzyme by using a Ni-chelate column. We have used a slightly different construct whose hexahistidine fusion is derived from the pRSET plasmid (Invitrogen, Leek, The Netherlands) and expressed the protein from this plasmid or from plasmids based on pQE60 (Qiagen). The kinetic properties of the fusion protein obtained after affinity chromatography on a Ni-chelate column are very similar to those of the enzyme expressed without a fusion and purified in several steps by the protocol we previously developed (4). The steady-state kinetic properties of the WT protein with and without the fusion are shown in Table 1. Constructs with mutations in or around the putative active site also show kinetic properties that are affected only slightly by the presence or absence of the N-terminal fusion and the difference in purification protocols (data not shown). The pH dependence of the activity of these mutants is not significantly different from that of the WT enzyme, and has a maximum pH near 7.5 (data not shown).
Table 1. Steady-state kinetic properties of E. coli AHAS isozyme II and some derivatives.
| Specific act,† | Km for pyruvate,† | kcat/Km,† | K0.5 for ThDP,† | ||||
|---|---|---|---|---|---|---|---|
| Construct* | units/mg | mM | kcat,† s-1 | M-1·s-1 | R‡ | Vb/Va§ | μM |
| WT | 20 | 5.2 | 23.3 | 4,490 | 56 ± 7 | 1.0 | 0.63 |
| His′-WT | 17 | 6 | 19.8 | 3,305 | 59 ± 3 | 1.0 | 1.2 |
| His′-Met250Ala | 2.6 | 36 | 3.03 | 84 | 56 | 8.0 | 1.2 |
| His′-Trp464Leu | 11.9 | 11.2 | 13.9 | 1,240 | 3.0 ± 0.2 | 1.0 | 1.7 |
| His′-Arg276Lys | 2.8 | 38 | 3.27 | 85 | 28 | 4.0 | 2 |
The WT AHAS and the mutated enzymes were expressed and purified either as the natural holoenzyme (4) or as a protein with an N-terminal hexahistidine fusion (His′-) (11). Some of this data has appeared in a previous publication (11)
These kinetic parameters were determined in steady-state experiments using the colorimetric assay for acetoin (17), with 100 mM pyruvate as the sole substrate, in the presence of 0.1 M KPi buffer (pH 7.6) containing 10 mM MgCl2, 0.1 mM ThDP, and 75 μM FAD, except for variation of the relevant reactant. kcat is defined as the turnover rate per site, assuming two sites in the 138-kDa heterotetramer. The parameters have experimental uncertainties of 5–10%
R is the substrate specificity for 2-ketobutyrate as second substrate, (AHB formed)/(AL formed) = R × [2-ketobutyrate]/[pyruvate], and was determined by measuring AHB and AL formation simultaneously (8) in a competition experiment with varying 2-ketobutyrate and 50 mM pyruvate, in 0.1 M KPi buffer (pH 7.6) containing 10 mM MgCl2, 0.1 mM ThDP, and 75 μM FAD (11)
Vb/Va is the ratio between the rate of AL formation with 50 mM pyruvate as sole substrate and the extrapolated rate of AHB formation in the presence of 50 mM pyruvate at saturation with 2-ketobutyrate. The experimental uncertainty in Vb/Va is 10–15%
Kinetics of Differential Product Formation in the Steady State. As we have shown in ref. 10, mutation of Trp-464 leads to a very significant decrease in R, the specificity factor for reaction with 2-ketobutyrate as opposed to pyruvate. For mutant Trp464Leu, R is 25-fold lower than for WT AHAS II (Table 1). Its sensitivity to the sulfonylurea herbicide sulfometuron methyl also is decreased by nearly 2 orders of magnitude (data not shown). However, this mutation has only a limited effect on the specific activity of the enzyme or on its affinity for pyruvate as substrate, and most of the other properties of the mutant are very similar to those of the WT (Table 1), with or without N-terminal fusion.
The mutation of Met-250, Arg-276, or Phe-109 has a very different effect. The specific activity for formation of AL with pyruvate as sole substrate is decreased by 10- to 100-fold in some mutants at these positions (11). In mutants Met250Ala and Arg276Lys, kcat/KM is reduced some 40-fold, but the apparent specificity for reaction with 2-ketobutyrate is changed slightly or not at all (Table 1). What is notable for these mutants is that the overall reaction rate depends on the concentration of 2-ketobutyrate (11). At 50 mM pyruvate, the rate of formation of AHB by Met250Ala, extrapolated to saturation with 2-ketobutyrate, is 8-fold higher than the rate of formation of AL in the absence of 2-ketobutyrate (Table 1, Vb/Va). In the presence of a sufficiently high 2-ketobutyrate concentration, the activity of the Met250Ala mutant approaches that of the WT. These results have an important implication: 2-ketobutyrate must be involved in one (or more) steps that are partially rate determining for this mutant.
Many other mutants of AHAS II, altered at Phe-109, Met-250, or Arg-276, exhibit similar behavior: low activity in the formation of AL and a total rate of AHA formation that increases with increasing 2-ketobutyrate (11). The rate at which these mutants catalyze a nonnatural reaction, formation of the uncharged product R-phenylacetylcarbinol from pyruvate and benzaldehyde, is far less seriously compromised. This observation led us to propose that Arg-276 might be directly involved in the specific recognition of the ketoacid carboxylate group in the second phase of the AHAS II reaction (11). We further postulated that Met-250 and Phe-109, which buttress Arg-276, influence the reaction through their effects on the conformation of Arg-276.
Microscopic Rate Constants Determined from Intermediate Distributions. The detailed kinetics of the three mutants studied here shed light on the mechanism of the carboligation reaction catalyzed by AHAS II and on the roles of some key residues in its active site. For each of these mutant proteins, the distribution of the intermediates bound to ThDP during turnover was determined by the rapid mixing/quench NMR method and compared with the behavior of the WT enzyme described in ref. 9. Reactions were carried out in the presence of 100 mM pyruvate, or a mixture of 50 mM pyruvate and 50 mM 2-ketobutyrate, and the enzymatic reaction mixture was quenched with trichloroacetic acid/DCl in D2O. Fig. 3 shows the low field region in the 1H-NMR spectra of the protein-free solutions obtained from reactions with the Arg276Lys mutant. In contrast to the results with the WT enzyme (9), a large fraction of the coenzyme could be isolated as HEThDP and ALThDP when pyruvate was the sole substrate (Fig. 3A). Because this mutant, like the WT enzyme, has a high specificity for reaction with 2-ketobutyrate, when pyruvate and 2-ketobutyrate are used in equimolar amounts almost all of the product of the reaction is AHB. Under these conditions, a large quantity of a new ThDP-derived intermediate could be isolated (Fig. 3B). Because this characteristic signal decreases at lower 2-ketobutyrate concentrations (data not shown), it is reasonable to conclude that it refers to AHBThDP.
The spectra were analyzed in terms of the minimal mechanistic scheme of Fig. 2. The microscopic rate constants derived from the relative peak areas and the turnover numbers for the enzyme at 37°C, by using Eqs. 6, 7, 8, 9 (see Experimental Procedures), are summarized in Table 2. The corresponding rate constants for the WT enzyme, determined previously (9), also are given in Table 2. Both the C–C bond-forming step (k4′) and product-release step (k5′) are affected significantly by the replacement of Arg-276. These results support the proposition that Arg-276 is a key residue in both the ligation of the second ketoacid with the HEThDP enamine and the elimination of ThDP from the product complex.
Table 2. Microscopic rate constants for elementary steps in the reactions of WT AHAS II and three mutants.
| Enzyme | Reaction* | kcat, s-1 | k′2, s-1 | k′3, s-1 | k′4, s-1 | k′5, s-1 |
|---|---|---|---|---|---|---|
| WT | Pyr + Pyr | 20 ± 0.2 | 24 ± 4 | 530 ± 70 | 1,060 ± 175 | 176 ± 36 |
| Pyr + 2KB | 20 ± 0.2 | 21 ± 4 | 399 ± 54 | >2,000 | >2,000 | |
| Met250Ala | Pyr + Pyr | 3 ± 0.1 | 28 ± 7 | 35 ± 7 | 11.1 ± 1.3 | 5.6 ± 0.3 |
| Pyr + 2KB | 25 ± 0.2 | 42 ± 3 | 87 ± 16 | 262 ± 81 | >1,050 | |
| Arg276Lys | Pyr + Pyr | 4.6 ± 0.1 | 9.3 ± 1.4 | >75 | 15 ± 2 | 35 ± 3 |
| Pyr + 2KB | 9.1 ± 0.2 | 14.1 ± 1.5 | >288 | 57 ± 8 | 48 ± 7 | |
| Trp464Leu | Pyr + Pyr | 13 ± 0.1 | 16.4 ± 1.1 | 234 ± 68 | 172 ± 44 | 140 ± 37 |
| (Pyr + 2KB) ⇒ AHA† | 13 ± 0.1 | 16.2 ± 1.2 | 208 ± 49 | 310 ± 81 | 180 ± 47 | |
| (Pyr + 2KB) ⇒ AHB† | ≈360 | ≈210 | ||||
| (Pyr + Pyr) ⇒ AL† | ≈120 | ≈140 |
Net rate constants (see (Fig. 2) for formation of lactyl-ThDP from bound pyruvate (k′2), decarboxylation of lactyl-ThDP (k′3), covalent ligation of the second ketoacid (k′4), and release of AHA product (k′5) were calculated by using Eqs. 6, 7, 8, 9 from kcat and the distribution of ThDP-bound intermediates, determined by the rapid mixing-quench/NMR method at 37°C in 0.1 M KPi (pH 7.6). The constants for the WT enzyme are described in ref. 9.
Pyr + Pyr is the formation of AL from two molecules of pyruvate, in the presence of 100 mM pyruvate. Pyr + 2KB is the formation of AHB from pyruvate and 2-ketobutyrate, in the presence of 50 mM of each. Under these conditions, <2% of the reaction catalyzed by WT, Met250Ala, or Arg276Lys enzymes goes to AL
Pyr + 2KB ⇒ AHA is the formation of the total of acetohydroxyacids [Σ(AL + AHB)] in the presence of equimolar amounts of pyruvate and 2-ketobutyrate (50 mM each). The separate rates for formation of AHB and AL under these conditions are estimated on the following two rows of the table
The detailed kinetics of Met250Ala were examined by the same method (data not shown). The covalently bound product, ALThDP, is the most populated intermediate when pyruvate is the sole substrate, and its release must be partially rate determining. In contrast, free ThDP, lactyl-ThDP, and a small amount of HEThDP can be observed in the steady state in the presence of a mixture of pyruvate and ketobutyrate. The microscopic rate constants derived from the relative peak areas and the turnover numbers for the enzyme at 37°C also are summarized in Table 2. These data confirm that the rate constants for the steps involving the second substrate are also slower in the Met250Ala mutant than in the WT enzyme.
In the presence of pyruvate as sole substrate, a somewhat larger fraction of HEThDP is observed with mutant Trp464Leu than with the WT. Analysis of the NMR spectrum shows that k4′ is 5 times smaller than in the WT (Table 2). Because the specificity of this mutant toward 2-ketobutyrate is much lower than that of the other enzymes studied here, both AHB and AL are formed in the presence of equimolar amounts of pyruvate and 2-ketobutyrate (Fig. 4). The calculation of individual microscopic rate constants is somewhat more complex in this case, but it is possible to extract the separate rates of carboligation and product release (k4′ and k5′) for each substrate (Table 2). With Trp464Leu, in contrast to the WT and to the two other mutants studied here, each of these steps is only slightly faster for 2-ketobutyrate than for pyruvate.
Fig. 4.

Detection and quantitation of covalent intermediates in the reactions of AHAS II mutant Trp464Leu. Steady-state intermediate distribution of AHAS II mutant Trp464Leu in the presence of 100 mM pyruvate (A) or 50 mM pyruvate and 50 mM 2-ketobutyrate (B) in 0.1 M KPi at 37°C. Reactions were quenched and analyzed as in Fig. 3, except that the fingerprint region is zoomed 5- or 8-fold.
Discussion
Our previous examination of the microscopic rate constants for AHAS II, by using NMR analysis of intermediates covalently bound to ThDP, showed that the slowest step by far in the reactions catalyzed by the WT enzyme is the formation of the first covalent adduct, lactyl-ThDP (9, 18). This result explained why the turnover rate for WT AHAS II is unaffected by the competition between the two alternative acceptor substrates, and confirmed observations, made nearly 20 years ago, that HEThDP does not accumulate in steady state with this enzyme (19). Conversely, this first step leading to a covalent change is so overwhelmingly rate determining that it was difficult to resolve the rates of the other steps for the WT enzyme, particularly the product-determining steps in the process (Table 2). These experiments did indicate, however, that elimination of the covalently bound product AHA (k5′) is at least 10 times faster for AHB than for AL. This intriguing observation suggested the possibility that the elimination step contributes to the specificity of AHAS II.
We have identified residues in the active site of AHAS II that appear to affect the rates of the product-determining steps in the reaction (k4′ and/or k5′). Two different kinds of effects are observed: Mutation of Trp-464 leads to a loss of preference for 2-ketobutyrate (10), whereas mutation of Arg-276, Met-250, or Phe-109 appears to lead to specific decreases in the rates of the product-determining step(s) so that the reaction of the second substrate becomes rate determining (11). We suggested a hypothetical structure for the complex of AHAS II-HEThDP- with a molecule of 2-ketobutyrate in position to form a new C–C bond (based on the crystal structure of a complex between Saccharomyces cerevisiae AHAS and the herbicide chlorimuron ethyl (20) and on the stereochemistry of the product S-AHAs), which might explain these effects (11). In this hypothetical structure, Trp-464 interacts with the methyl group (C4) of 2-ketobutyrate, whereas Arg-276 is involved in a charge–charge interaction with its carboxylate (Fig. 5). The detailed microscopic rate constants for the three mutants studied here support this proposal. The rate constants for reactions of the Arg276Lys mutant (Table 2) confirm that both the addition of the HEThDP enamine to the carbonyl of the second substrate (k4′) and the release of the AHA product (k5′) are significantly slower for this mutant than for the WT enzyme (Table 2), so that significant amounts of the covalent intermediates are persistent in the steady-state. The rate of release of AHB (48 s-1) is only slightly faster than release of AL (35 s-1). The detailed microscopic rate constants for the Met250Ala mutant show similar changes relative to the WT, except that in this case k5′ for AHB is >30-fold faster than for AL. In the Trp464Leu mutant, the microscopic rate constants k4′ and k5′ are not decreased sufficiently for the reaction with the acceptor substrate to become rate-determining. However, for this mutant k4′ and k5′ are similar for the two alternative ketoacid second substrates rather than being faster for the reactions of 2-ketobutyrate.
Fig. 5.

The active site of AHAS II, based on the structure of the yeast AHAS–CIE complex (Protein Data Bank ID code 1N0H) (20). Trp-464 is shown in configuration A of Trp-586 in 1N0H. The residues are labeled with the numbering of AHAS II, and residues belonging to the second catalytic polypeptide are marked with ′. FAD is shown with thin gray bonds, and the enzyme is shown in the E-HEThDP·KB form. The software molscript (21) was used to make this figure.
It is not simple to provide structural-mechanistic rationalizations of the effects on the rate constants seen in Table 2. These measured rate constants are based on the distribution of covalent derivatives of ThDP after an acid quench, but each species detected may represent a mixture of the covalent intermediate on the enzyme with and without noncovalently associated substrates or products. In addition, rate constants derived from the distribution of intermediates are net rate constants, which reflect the true forward rate constant multiplied by the fraction of the resulting enzyme form that partitions onto products as opposed to undergoing reversal (15). The decarboxylation step (k3′) is presumably irreversible, but we know that steps 4 and 5 are reversible, because chemically synthesized AL can be converted to, e.g., peracetate (22) or phenylacetyl carbinol (ref. 23 and D.M.C., unpublished results) by AHAS II. On the basis of Schloss and coworkers' observations on the peracetate-forming oxygenase reaction (22, 24), one also can assume that there is reversible binding of the acceptor ketoacid to the enzyme–HEThDP complex before its reaction (as opposed to Theorell–Chance kinetics for step 4): Both pyruvate and 2-ketobutyrate can compete with oxygen for enzyme-bound HEThDP, but the competition saturates at <100% inhibition of the oxygenase reaction. Despite the difference in microscopic rate constants for ligation of ketobutyrate and pyruvate to bound HEThDP-, the slow reduction of FAD observed during substrate turnover by AHAS II is also only slightly dependent on the second substrate (14). Thus, steps k4′ and k5′ might be described in more detail as follows, where KA is a ketoacid:
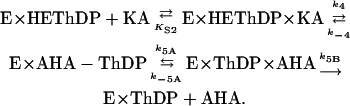 |
[10] |
The large observed decreases in the net rate k4′ in mutants Arg276Lys and Met250Ala thus might be entirely due to a direct effect on the bond-making step k4, or it might alternatively be due in part to the decrease in k5′. The decline in rate constant k5′ could, further, be due to changes in the bond-breaking step k5A or in the diffusion of the product AHA out of the active site (k5B).
Despite this ambiguity, the significant decreases in k4′ and k5′ observed in the mutant Arg276Lys clearly support a role for the interaction of Arg-276 with the second substrate/product in the transition states for both the carboligation and the product release steps. We propose that the interaction of the guanidinium moiety of Arg-264 and the carboxylate of the acceptor ketoacid/covalent HEThDP-acceptor adduct provides an optimal alignment of substrate and intermediate orbitals in the reaction trajectory of acceptor ligation and product cleavage (Fig. 6).
Fig. 6.

Proposed interactions in the transition states for steps 4 and 5 in the reaction of WT AHAS II with 2-ketobutyrate as acceptor substrate.
The possible proximity of Met-250 to Trp-464, the only residue we have identified which has a critical effect on the substrate specificity of the enzyme (10), provided the original impetus for making mutants at this position. Its replacement by alanine leads to decreases in the decarboxylation rate constant k′3, as well as in k′4 and k′5. Somewhat surprisingly, k′5, the rate of release of the product, is >2 orders of magnitude higher for AHB than for AL. The elimination of the side chain of Met-250 presumably can affect the local conformation of its neighbors in the active site, including Arg-276 and Trp-464, and thus affect interactions of the incoming second substrate and the departing product with the enzyme in unexpected ways.
The rate of product release, k5′, is similar in magnitude to that of the carboligation step, k4′, in all of the mutants of AHAS II studied here. This finding has important implications for understanding the overall specificity of the reaction catalyzed by AHAS II. The identification of Trp-464 as a residue critical for the preference of the enzyme for 2-ketobutyrate does not fully explain the substrate preference, R, near 60. If this preference were due to only a single step of the reaction, it would be equivalent to a difference in the free energies of activation, ΔΔG°, between reactions leading to AHB and AL, of 2.5 kcal/mol. This amount would be an unusually large contribution for the contacts between a single methyl group and a protein site. However, we have shown here (Table 2) that both the carboligation step (step 4, Fig. 2) and the product-release step (step 5) are significantly faster for the reaction with 2-ketobutyrate in each enzyme with a strong preference for reaction with 2-ketobutyrate. We thus can speculate that the specificities of steps 4 and 5 might be cumulative: If the formation of ALThDP in step 4 has a lower forward commitment factor than formation of AHBThDP, the reversal of ALThDP formation and release of pyruvate would allow 2-ketobutyrate another chance to react. The slower release of product from ALThDP could in effect allow for “kinetic proofreading” (25) and thus lead to the observed high preference for formation of AHB.
Acknowledgments
This work was supported in part by Israel Science Foundation Grant 660/01 (to Z.B.), by a seed grant to D.M.C. from the Vice-President for Research and Development at Ben-Gurion University, and by the Deutsche Forschungsgemeinschaft and the Fonds der chemischen Industrie (Halle). D.M.C. is an Incumbent of the Lily and Sidney Oelbaum Chair in Applied Biochemistry at Ben-Gurion University.
Author contributions: K.T., G.H., Z.B., and D.M.C. designed research; K.T. and M.V. performed research; K.T. and M.V. analyzed data; and D.M.C. wrote the paper.
Abbreviations: AHA, acetohydroxyacid; AHAS, AHA synthase; AHB, acetohydroxybutyrate; AL, acetolactate; ThDP, thiamin diphosphate; HEThDP, hydroxyethyl-ThDP anion/enamine.
References
- 1.Green, J. B. (1989) FEBS Lett. 246, 1-5. [DOI] [PubMed] [Google Scholar]
- 2.Bowen, T. L., Union, J., Tumbula, D. L. & Whitman, W. B. (1997) Gene 188, 77-84. [DOI] [PubMed] [Google Scholar]
- 3.Schowen, R. L. (1998) in Comprehensive Biological Catalysis, ed. Sinnot, M. (Academic, London), Vol. 2, pp. 217-266. [Google Scholar]
- 4.Bar-Ilan, A., Balan, V., Tittmann, K., Golbik, R., Vyazmensky, M., Hubner, G., Barak, Z. & Chipman, D. M. (2001) Biochemistry 40, 11946-11954. [DOI] [PubMed] [Google Scholar]
- 5.Barak, Z., Chipman, D. M. & Gollop, N. (1987) J. Bacteriol. 169, 3750-3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gollop, N., Damri, B., Chipman, D. M. & Barak, Z. (1990) J. Bacteriol. 172, 3444-3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Epelbaum, S., LaRossa, R. A., VanDyk, T. K., Elkayam, T., Chipman, D. M. & Barak, Z. (1998) J. Bacteriol. 180, 4056-4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gollop, N., Damri, B., Barak, Z. & Chipman, D. M. (1989) Biochemistry 28, 6310-6317. [DOI] [PubMed] [Google Scholar]
- 9.Tittmann, K., Golbik, R., Uhlemann, K., Khailova, L., Schneider, G., Patel, M., Jordan, F., Chipman, D. M., Duggleby, R. G. & Hubner, G. (2003) Biochemistry 42, 7885-7891. [DOI] [PubMed] [Google Scholar]
- 10.Ibdah, M., Bar-Ilan, A., Livnah, O., Schloss, J. V., Barak, Z. & Chipman, D. M. (1996) Biochemistry 35, 16282-16291. [DOI] [PubMed] [Google Scholar]
- 11.Engel, S., Vyazmensky, M., Vinogradov, M., Berkovich, D., Bar-Ilan, A., Qimron, U., Rosiansky, Y., Barak, Z. & Chipman, D. M. (2004) J. Biol. Chem. 279, 24803-24812. [DOI] [PubMed] [Google Scholar]
- 12.Kern, D., Kern, G., Neef, H., Tittmann, K., Killenberg-Jabs, M., Wikner, C., Schneider, G. & Huebner, G. (1997) Science 275, 67-70. [DOI] [PubMed] [Google Scholar]
- 13.Tittmann, K., Golbik, R., Ghisla, S. & Huebner, G. (2000) Biochemistry 39, 10747-10754. [DOI] [PubMed] [Google Scholar]
- 14.Tittmann, K., Schroeder, K., Golbik, R., McCourt, J., Kaplun, A., Duggleby, R. G., Barak, Z., Chipman, D. M. & Huebner, G. (2004) Biochemistry 43, 8652-8661. [DOI] [PubMed] [Google Scholar]
- 15.Cleland, W. W. (1975) Biochemistry 14, 3220-3224. [DOI] [PubMed] [Google Scholar]
- 16.Hill, C. M., Pang, S. S. & Duggleby, R. G. (1997) Biochem. J. 327, 891-898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Epelbaum, S., Chipman, D. M. & Barak, Z. (1990) Anal. Biochem. 191, 96-99. [DOI] [PubMed] [Google Scholar]
- 18.Chipman, D. M., Barak, Z., Engel, S., Mendel, S. & Vyazmensky, M. (2003) in Thiamine: Catalytic Mechanisms in Normal and Disease States, eds. Jordan, F. & Patel, M. S. (Marcel Dekker, New York), pp. 233-250.
- 19.Ciskanik, L. M. & Schloss, J. V. (1985) Biochemistry 24, 3357 (lett.). [Google Scholar]
- 20.Pang, S. S., Guddat, L. W. & Duggleby, R. G. (2003) J. Biol. Chem. 278, 7639-7644. [DOI] [PubMed] [Google Scholar]
- 21.Kraulis, P. J. (1991) J. Appl. Crystallogr. 24, 946-950. [Google Scholar]
- 22.Abell, L. M. & Schloss, J. V. (1991) Biochemistry 30, 7883-7887. [DOI] [PubMed] [Google Scholar]
- 23.Engel, S., Vyazmensky, M., Geresh, S., Barak, Z. & Chipman, D. M. (2003) Biotechnol. Bioeng. 83, 833-840. [DOI] [PubMed] [Google Scholar]
- 24.Tse, J. M. T. & Schloss, J. V. (1993) Biochemistry 32, 10398-10403. [DOI] [PubMed] [Google Scholar]
- 25.Hopfield, J. J. (1974) Proc. Natl. Acad. Sci. USA 71, 4135-4139. [DOI] [PMC free article] [PubMed] [Google Scholar]