

Abstract
The two-step energy-transfer process in a self-assembled complex comprising a cationic conjugated polymer (CCP) and a dsDNA is investigated by using pump-dump-emission spectroscopy and time-correlated single-photon counting; energy is transferred from the CCP to an ethidium bromide (EB) molecule intercalated into the dsDNA through a fluorescein molecule linked to one terminus of the DNA. Time-dependent anisotropy measurements indicate that the inefficient direct energy transfer from the CCP to the intercalated EB results from the near orthogonality of their transition moments. These measurements also show that the transition moment of the fluorescein spans a range of angular distributions and lies between that of the CCP and EB. Consequently, the fluorescein acts as a fluorescence resonance energy-transfer gate to relay the excitation energy from the CCP to the EB.
Keywords: conjugated polymers, DNA sensors, time-resolved anisotropy, ultrafast spectroscopy
Cationic conjugated polymers (CCPs) have been demonstrated to have utility in DNA sequence detection (1–3). In the approach initially proposed by Gaylord et al. (1), the luminescent conjugated polymer serves as the light-harvesting entity with subsequent energy transfer to a fluorescein (FL) molecule attached to a peptide nucleic acid (PNA) oligomer comprised of a specific sequence of bases that defines the target. Fluorescence resonance energy transfer (FRET) from the CCP to the PNA-FL occurs by self-assembly of the CCP and the PNA in a double helix with a complementary single-stranded DNA target. In this approach, the electrostatic interaction between the CCP and the negatively charged PNA-FL/DNA duplex brings the donor (CCP) and the acceptor (FL) in sufficiently close proximity to enable efficient FRET. When the base sequence on the DNA is not complementary to the base sequence on the FL-labeled PNA, the PNA-FL remains as a charge-neutral single-strand oligomer. At the dilute concentrations used, the typical distance between a neutral PNA-FL and a CCP is too large for efficient FRET. By using this FRET-based biosensor, specific targeted sequences of DNA were detected at concentrations as low as 10 pM (1).
Wang et al. (2) reported an alternative DNA sequence-detection scheme (Scheme 1) based on intercalation of ethidium bromide (EB) within a dsDNA. Considering that the synthesis and purification of labeled DNA is more straightforward and that both single-stranded DNA and dsDNA have good solubility in buffered water (whereas with PNA, hydrophobic interactions are important), the EB intercalation approach is attractive. However, direct FRET from the CCP to EB was found to be inefficient despite the excellent spectral overlap between the donor emission and acceptor absorption. The absence of efficient FRET was attributed to the orthogonality of the transition dipoles of the CCP and EB in the self-assembled structure (see Scheme 1). When a FL molecule is linked to one terminus of the dsDNA as shown in Scheme 1B, the EB emission is significantly enhanced after excitation of the CCP. Because FL is attached by a flexible linker, the absorption and emission moments of the terminal FL are not orthogonal to either the CCP or the EB. Thus, FL functions as a “FRET gate” through which the excitation energy harvested by the conjugated polymer is transferred to the intercalated EB. This scheme was used to detect DNA with specific targeted sequences at concentrations as low as 2.5 nM.
Scheme 1.

Schematic representation of DNA sequence detection by FRET from CCP to intercalated dyes in the absence (A) and presence (B) of the FRET gate. The molecular structures of the CCP (PFP), FL, and EB are shown also.
We report here the results of an ultrafast spectroscopic study of the two-step FRET process from the CCP to EB by means of FL. The individual steps in the FRET-gate process were investigated with a combination of two time-resolved fluorescence spectroscopic techniques, pump-dump-emission spectroscopy (PDES) and time-correlated single-photon counting (TCSPC). TCSPC is well known as a sensitive method for lifetime measurements and is capable of measuring decay dynamics at very low concentrations (4). However, the temporal resolution of TCSPC is typically limited to ≈60 ps. PDES, which has been used to investigate the excited-state decay dynamics and the charge-generation mechanism in conjugated polymers (5–7), can extend the dynamical measurements at very low concentrations into the picosecond and even femtosecond regime with good signal-to-noise ratios (8). Both PDES and TCSPC have sensitivities sufficient to handle the low concentrations required for the DNA sensor (1, 2, 8). The combination of PDES and TCSPC enable time resolution of the FRET processes over the entire relevant range (femtosecond to nanosecond time scales).
Experiments and Methods
Both ultrafast PDES and TCSPC were used in the experiments. For PDES, we used a Spectra-Physics Ti:sapphire laser system (oscillator, regenerative amplifier, and optical parametric amplifier) that generates output pulses with a repetition rate of 1 kHz. The 400-nm pump beam is obtained from the second harmonic of the 800-nm pulse from the regenerative amplifier. The tunable output from the optical parametric amplifier (from UV to near IR) was used for the dump beam. The optical alignment for PDES has been described in detail (8) and will only be summarized briefly here. In PDES, the pump pulse first promotes a fraction of chromophore molecules into their excited state. The time-delayed beam then dumps the excited-state population by stimulated emission. The resulting excited-state population change is monitored by emission in the perpendicular direction, which is detected by a photomultiplier tube that is connected to a lock-in amplifier and computer. As a result, the signal (ΔI/I) is negative, because the dump beam decreases the excited-state population and thereby decreases the measured fluorescence. In this work, the data are presented as -ΔI/I for a straightforward description of the evolution of the excited-state population.
A Spectra-Physics Ti:sapphire oscillator (80 MHz) pulsed laser system was used for TCSPC. The repetition rate was reduced to 4 MHz with a pulse picker. The output was subsequently frequency-doubled to generate the 385- and 465-nm output for excitation beams. The decay trace of the emission was collected by the photon-counting system.
Time-dependent anisotropy measurements provide direct information on the relative orientation of the excitation and emission moments (4). For both PDES and TSCPC, the anisotropy is defined as:
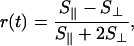 |
where S∥ and S⊥ are signal intensities for pump and emission (or dump beam for PDES) with parallel and perpendicular polarizations, respectively. In homogeneous solutions, r(0) = 0.4 for parallel absorption and emission moments, and r(0) = -0.2 for mutually orthogonal transition moments.
The optical properties of the CCP poly[9,9-bis(6′-N,N,N-trimethylammonium)-hexyl] fluorene phenylene (PFP), FL, and EB and the detail sequence of the DNA (20 bases) used have been described in detail (2). The PFP used here has an average length of 15 monomers with polydispersity of weight-average molecular weight/number-average molecular weight ≈ 1.9. Both steady-state and time-resolved measurements were performed in potassium phosphate–sodium hydroxide buffer solution (50 mM, pH 7.4), with [PFP] = 1.6 × 10-6 M (in repeat units), [dsDNA], [dsDNA-C] = 1.0 × 10-7 M, and [EB] = 6.7 × 10-7 M. The ratio of PFP and dsDNA chain numbers is ≈1:1, and the charge ratio is ≈0.8:1. Under these conditions, the approach shown in Scheme 1B displays excellent selectivity between complementary and noncomplementary DNA strands.
As shown in Fig. 1, there is only weak direct energy transfer from PFP to EB in the absence of the FL. When FL-labeled dsDNA is used, the EB emission is enhanced by a factor of >5 (the PFP is excited at 380 nm).
Fig. 1.

Emission spectra of PFP/dsDNA (i), PFP/dsDNA/EB (ii), PFP/dsDNA-FL (iii), and PFP/dsDNA-FL/EB (iv) complexes in potassium phosphate–sodium hydroxide buffer solution (50 mM, pH 7.4). [PFP] = 1.6 × 10-6 M (in repeat units), [dsDNA], [dsDNA-FL] = 1.0 × 10-7 M, and [EB] = 6.7 × 10-7 M. The excitation wavelength is 380 nm for all samples.
Results and Discussion
Direct FRET from PFP to EB. The long-range excitation energy transfer from donor to acceptor through the dipole–dipole interaction was described initially by Förster (9). For donor and acceptor separated by a distance rDA, the rate of energy transfer is given by (4):
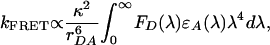 |
where κ2 is a factor describing the relative orientation of the transition dipoles of the donor and acceptor; the integral, 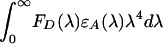 , expresses the degree of spectral overlap between the emission of the donor, FD(λ), and the absorption of the acceptor, εA(λ).
, expresses the degree of spectral overlap between the emission of the donor, FD(λ), and the absorption of the acceptor, εA(λ).
Note, however, that despite the excellent spectral overlap between donor emission and acceptor absorption (2) and short donor–acceptor distance brought about by the electrostatic attraction between PFP and dsDNA, the direct energy transfer from PFP to EB is not efficient (see Fig. 1). The inefficient energy transfer was identified as arising from the unfavorable relative orientation of transition dipoles of the donor and acceptor, i.e., a small κ2 in Eq. 2 (2). The absorption and emission transitions of the conjugated polymer are both parallel to the polymer chain. The emission transition of intercalated EB is nearly perpendicular to the axis of the double helix (dsDNA) with a small distribution of angles about the precisely orthogonal configuration (10, 11), a configuration that is unfavorable for FRET.
This proposed mechanism was examined for the PFP/DNA/EB complex in solution by time-resolved fluorescence lifetime and anisotropy measurements (Fig. 2). The sample was excited at 385 nm, and the emission was collected at 630 nm. The data can be approximately fit with the sum of three exponentials: 0.36 ns (49%), 2.1 ns (27%), and 16.5 ns (24%). The 0.36-ns component is likely due to contamination from the decay of the PFP emission. As a result of the limited temporal resolution of TCSPC, this time constant appears slower than the average emission lifetime of the PFP/DNA itself (the PFP emission is partially quenched by DNA and has an average lifetime of ≈90 ps). Only the slowest components of the PFP emission decay (including the emission of PFP molecules that do not bind to DNA and those in the excimer state) show up in TCSPC measurements. The relatively large contribution of the PFP emission results from the fact that the oscillator strength of PFP is much larger than that of EB, as evidenced by the large difference in their excited-state lifetimes (90–360 ps vs. ≈9 ns). Thus, even a small contribution of PFP emission in steady-state spectra (integrated) will cause significant contamination in the time-resolved data.
Fig. 2.

Emission decay (a) and anisotropy (b) at 630 nm in PFP/dsDNA/EB measured with TCSPC. The excitation wavelength is 385 nm.
The two slower components of the emission decay at 630 nm (2.1 and 16.5 ns) result from the decay of the emission from EB molecules that bind to DNA in different forms. EB is known to bind to dsDNA in two different forms: intercalated into the dsDNA and bound outside the dsDNA by electrostatic interactions (12, 13). Intercalated EB displays enhanced luminescence efficiency and a longer excited-state lifetime as a result of the protected environment, whereas EB bound outside the double helix is only weakly fluorescent (13) with a correspondingly shorter excited-state lifetime because it is exposed to the solvent environment. Fluorescence titration measurements indicated that the contribution from the electrostatic bound EB increased when higher EB concentrations were used. When more EB was added, the energy transfer from PFP to EB was significantly improved for both cases when DNA with complementary and noncomplementary base pairs was used. In the latter case, no dsDNA is formed and the intercalation is prohibited; EB is mainly bound to DNA by electrostatic interactions. Thus, the 2.1-ns component of the decay is attributed to the EB molecules that are electrostatically bound outside the dsDNA, and the 16.5-ns component is attributed to the emission from the intercalated EB. The 16.5-ns lifetime is comparable with the reported lifetime (22 ns) for EB intercalated in dsDNA (11).
The time-dependent anisotropy confirms the near orthogonality of the PFP and EB transition moments. As shown in Fig. 2, the anisotropy decays from an initial positive value (characteristic of emission from the conjugated polymer) to a local minimum of r ≈ -0.07 after energy transfer to the EB. The negative anisotropy indicates a large angle between the excitation (PFP) and emission (EB) moments (4). The anisotropy then relaxes from -0.07 to 0 with a time scale of >10 ns. Because of the limited signal-to-noise ratio, we did not attempt to fit the data; the red line through the data in Fig. 2b is simply a guide for the eye. We also did not attempt to extract the rate of energy transfer, which is expected to be complex because EB will be intercalated into the dsDNA at random positions and therefore cause a distribution of energy-transfer rates.
As noted above, the positive value at t = 0 results from the polymer emission (the polymer absorption and emission moments are parallel); the initial very fast decay of the anisotropy from positive values to -0.07 results from the energy transfer to the EB. The subsequent slow decay from -0.07 to 0 is caused by the reorientation of the entire complex.
Deviation of the measured value from r = -0.2 (perfect orthogonality between the excitation and emission moments) arises, at least in part, the from EB molecules that are not intercalated into a double helix. If we exclude the contribution to the emission from such randomly oriented EB outside the dsDNA with r = 0 (54% as implied by the preexponential factors), an anisotropy of r ≈ -0.15 is inferred for EB intercalated into dsDNA. This result supports the proposal that the PFP absorption moment and the emission moment of the intercalated EB are nearly orthogonal (i.e., as sketched schematically in Scheme 1, in which the complex is depicted as two oppositely charged rigid rods).
There are a number of reasons to expect deviations from perfect orthogonality as implied by the residual direct FRET from PFP to EB (in the absence of FL). For example, the two components (dsDNA and PFP) are not truly rigid. Moreover, the cloud of counter ions (14) from the DNA and PFP can partially screen the electrostatic interaction between the DNA and PFP. Thus, one would expect distributions in the PFP/DNA alignment within the complex. In addition, the EB can “wobble” around while intercalated inside the dsDNA, leading to deviations from the perfectly orthogonal orientation (10, 11). EB molecules outside the dsDNA can also relay the excitation energy to the intercalated EB and thereby contribute to the nonzero direct FRET from PFP to EB.
On the other hand, the relatively short lengths (15 repeat units for PFP and 20 bp for DNA) imply relatively rigid structures for both PFP and dsDNA. The combined effects of electrostatic and hydrophobic interactions between PFP and DNA will tend to favor the formation of the complex suggested by Scheme 1B. The reality of this basic structure is supported by the significant negative anisotropy demonstrated in Fig. 2.
The near orthogonal geometry between the transition moments of PFP and the intercalated EB is also supported by the fact that the energy transfer from conjugated polymer to EB can be enhanced significantly by purposely introducing a nonstraight moiety into the conjugated polymer backbone (3). Moreover, efficient energy transfer is observed (as expected) when PFP dendrimers are used as donors (S.W. and G.C.B., unpublished results).
Energy Transfer from PFP to FL. The efficient energy transfer from PFP to FL is evidenced by the reduced blue PFP emission and enhanced green FL emission in the presence of FL-labeled dsDNA. The significant reduction in the PFP emission implies that FRET takes place within a few tens of picoseconds, beyond the temporal resolution of TCSPC measurements. Thus, the energy-transfer process from PFP to FL was time-resolved with PDES.
The excited-state dynamics of the donor (PFP) in the presence and absence of acceptor (FL) were measured by using a pump wavelength of 400 nm, a dump wavelength of 450 nm, and an emission wavelength of 430 nm (hereafter denoted as P400 nm/D450 nm/E430 nm data). Consistent with the steady-state data, the donor excited state displays a significantly faster decay rate when the acceptor is present (Fig. 3a). In the absence of the acceptor (PFP/dsDNA), the donor decay can be fit with a double exponential: 50 ps (49%) and 130 ps (51%) with an average lifetime of ≈90 ps. In contrast, in the presence of the acceptor (PFP/DNA-FL), the excited-state decay of PFP can be fit with a double exponential decay [13 ps (47%) and 44 ps (53%)], which gives an average lifetime of ≈30 ps.
Fig. 3.

Ultrafast PDES measurements of the energy-transfer dynamics from PFP to FL in the PFP/dsDNA-FL complex in dilute solution. (a) The excited-state decay of the donor (PFP) in the absence (filled squares) and presence (open circles) of the acceptor (FL) measured at P400 nm/D450 nm/E430 nm. (b) PDES data for PFP/dsDNA-FL at P400 nm/D550 nm/E530 nm with parallel (filled squares) and perpendicular (open circles) pump/dump beam polarizations. (c) The excited-state decay of the donor caused by energy transfer (open circles, obtained from division of the two data sets shown in a, i.e., P400 nm/D450 nm/E430 nm data in the presence and absence of acceptor) and the simultaneous growth of the acceptor emission (filled squares, obtained by subtracting donor contamination from the P400 nm/D545 nm/E530 nm data, as described in Discussion) for PFP/dsDNA-FL complexes. (d) Comparison of anisotropy decay and energy-transfer dynamics. The anisotropy decay (filled squares) is calculated from data shown in b, and energy-transfer dynamics (open circles) are obtained from the division of the two data sets shown in a.
The excited-state dynamics of the acceptor (FL) were also measured with PDES with a pump wavelength of 400 nm, a dump wavelength of 550 nm, and an emission wavelength of 530 nm (P400 nm/D550 nm/E530 nm data). Measurements with parallel or perpendicular pump/dump beam polarizations are shown in Fig. 3b. The results indicate significant contamination from the donor emission that is partially suppressed when the perpendicular pump/dump beam polarizations are used. In other words, the signal is depolarized quickly as energy transfer occurs.
The net contribution from the acceptor emission is shown in Fig. 3c. Assuming that the signal at t = 0 originates totally from the donor emission (as implied by the anisotropy, see Fig. 2), the net acceptor contribution was obtained by subtracting the rescaled P400 nm/D450 nm/E430 nm data (by matching the signal levels of the two data sets at t = 0) from the P400 nm/D550 nm/D530 nm data. To remove any contribution from rotational depolarization, the S∥ + 2S⊥ data were used to obtain the results presented in Fig. 3c. The decay of the donor emission caused by FRET is indicted by the open circles and was obtained from the decay curves of the donor in the presence of the acceptor divided by that in the absence of the energy acceptor, i.e., the two data sets in Fig. 3a. Fig. 3c shows clearly that the growth of the acceptor emission is concomitant with the decay of the donor emission during the energy-transfer process as predicted.
The data in Fig. 3b indicate significant emission depolarization during the course of energy transfer. This is very different from the donor decay (P400 nm/D450 nm/E430 nm data), for which little depolarization was observed within the same time range. The time-dependent anisotropy for P400 nm/D550 nm/E530 nm data are calculated and shown in Fig. 3d (filled squares). The energy-transfer dynamics (the same data sets as in Fig. 3c) are plotted in the same graph for direct comparison. The anisotropy decay follows the general trend of energy-transfer dynamics, although it is somewhat slower than the latter. The anisotropy decay can be fit with a dominant fast component [65 ps (84%)] and a minor slow component [600 ps (16%)]. The energy-transfer decay is a double exponential with time constants of 10 ps (45%) and 90 ps (55%) and an average lifetime of 54 ps.
The fact that anisotropy follows the general trend of energy transfer indicates that the orientation of the FL (acceptor) dipole relative to PFP (donor) dipole is distributed over a range of angles. Energy transfer to a randomly oriented acceptor will cause the decay of fluorescence anisotropy at wavelengths at which the donor and acceptor emissions both contribute (15). The anisotropy is given by the additivity law (4):
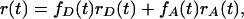 |
where fD(t) and fA(t) are fractional intensities of the donor and acceptor, respectively. In this equation, fD(t) and fA(t) are time-dependent during the energy-transfer process. At t = 0, before energy transfer, only the donor contributes to the emission at 550 nm (fD = 1 and fA = 0). Thus, the initial anisotropy is very close to r = 0.4, which is consistent with the general understanding that the absorption and emission transition moments of conjugated polymers are parallel to the polymer chain. As the FRET process proceeds, the donor contribution decreases and the acceptor contribution increases. The observation of emission depolarization caused by FRET implies that the FL transition moment is distributed randomly over a relatively wide range of angles. However, the residual anisotropy of ≈0.06 after FRET suggests that it is not totally random, consistent with fact that the FL is linked to one terminus of the DNA (the orientation of chromophore molecules linked to the terminus of DNA usually spans a cone-like distribution of angles) (16). Of course, the residual anisotropy is depolarized further as a result of reorientation of the entire complex.
The observation that the anisotropy is slightly slower than energy-transfer dynamics can also be understood within the same picture. The orientation angle between the donor and acceptor spans a range of distributions as illustrated in Scheme 2. Those donor–acceptor pairs with smaller orientation angles have faster energy-transfer rates, and those pairs with larger angles have slower energy-transfer rates. Those with faster energy-transfer rates are less depolarized, whereas those with slower energy-transfer rates are more depolarized. Thus, the emission-depolarization process lags behind the population transfer during FRET.
Scheme 2.

Schematic representation of relative orientations of the three optical components: conjugated polymer (PFP, donor), FL (FRET gate), and intercalated EB (acceptor).
The arguments discussed above are also supported by comparison of anisotropy decay of the FL emission observed at 530 nm under direct excitation compared with that by FRET (Fig. 4) for the PFP/DNA-FL complex. The energy transfer from PFP to FL accelerates the depolarization over that which would normally be observed because of rotational diffusion. When FL is directly excited at 465 nm, an initial anisotropy of r ≈ 0.35 is obtained. The anisotropy decays with two exponential components, a dominant fast component of 240 ps and a slower component with a minor time constant of ≈5 ns. The 240-ps component is caused by the restricted local motion of the FL molecules, whereas the 5-ns component arises from reorientation of the entire complex. These data are consistent with the picture of a small chromophore attached to the terminus of DNA: The FL chromophore both spins and wags up and down, forming a cone-like distribution at the end of the dsDNA. The 5-ns time constant is consistent with the reorientation time scales of the biomacromolecules with similar sizes (4, 17). From the relative contribution of this slow component (anisotropy amplitude of ≈0.06), the restricted local orientation of the FL molecules is estimated to be within a cone angle of ≈52° (17, 18). Previously, a cone angle of 60° was obtained for fluorophore molecules linked to biomacromolecules (16).
Fig. 4.

Time-dependent anisotropy of the FL emission at 530 nm under direct excitation (open circles, FL is directly excited at 465 nm) or by FRET (solid lines, PFP is excited at 385 nm instead). (Inset) The initial decay of the anisotropy as obtained by PDES is shown as the solid curve (open circles, FL is directly excited at 465 nm).
In contrast, the time-dependent anisotropy is very different in the energy-transfer case when the polymer is excited instead at 385 nm. The anisotropy data obtained by PDES (the anisotropy data in Fig. 3d) are used for the early decay of the anisotropy as shown in Fig. 4 Inset. The anisotropy decay is dominated by a fast component (≈50 ps) followed by a slow component (27 ns), with a very minor contribution on the intermediate time scale (≈280 ps). The fast FRET from PFP to FL with a range of orientation angles quickly brings the anisotropy down to a small finite value (≈0.07). The subsequent randomization process of the FL fragments causes little further depolarization of the emission, because it does not change the overall distribution of donor–acceptor orientation angles; as time progresses, some become more polarized and some become less polarized. The intermediate time scale is only responsible for the anisotropy decay with an amplitude of ≈0.01. Because FL is attached to the terminus of the DNA, its motion is limited. The FL emission is not fully depolarized after energy transfer. The residual anisotropy (≈0.06) is subsequently depolarized by reorientation of the entire complex on a nanosecond time scale (27 ns obtained from the fitting, but with large uncertainty because of the small time window limited by the FL-emission lifetime).
We emphasize that the description of the FRET from conjugated polymer to FL with a sum of exponentials is used simply for convenience. Donor–acceptor pairs with different orientation angles will have slightly different donor–acceptor distances, causing variations in energy-transfer rates. The cloud of counter ions around the polymers will also lead to a distribution of donor–acceptor separations (14). Thus, the FRET process is a superposition of many contributions that average into an approximate exponential decay. The biexponential energy-transfer dynamics from PFP to FL (10 and 90 ps) suggests the formation of different conformations of the self-assembled complex as a result of the coexistence of competing hydrophobic and electrostatic interactions (8, 19). By using the steady-state spectra and assuming almost randomly oriented donor–acceptor pairs (κ2 = 2/3), the Förster distance (R0) is estimated to be 33 Å. The energy-transfer rates of 10 and 90 ps correspond to donor–acceptor separations of 23 and 33 Å, respectively, which are very close to the values previously obtained for the similar PFP/PNA/DNA complex (8).
Energy Transfer from FL to EB. In the previous section we demonstrated efficient energy transfer from PFP to FL. Steady-state measurements (Fig. 1) indicate that the energy is subsequently transferred to the intercalated EB, which was confirmed by different excited-state dynamics of FL in the presence and absence of EB, as measured with TCSPC. The samples were excited at 385 nm, and the emission was monitored at 530 nm. As shown in Fig. 5, the decay of the FL emission is significantly faster in the presence of EB. The measured emission decays are fit with a sum of three exponential components: 0.35 ns (51%), 1.6 ns (45%), and 4.6 ns (4%) in the absence of EB and 0.08 ns (32%), 0.45 ns (62%), and 2.6 ns (6%) in the presence of EB. Excluding the fast component that arises from the PFP emission, the average FL excited-state lifetime was obtained to be ≈1.8 ns in the absence of EB and 0.64 ns in the presence of EB. This result confirms that the FL functions as a FRET gate that functions to relay the excitation energy from the light-harvesting conjugated polymer (PFP) to the intercalated fluorescent probe molecules (EB), as sketched in the DNA sensor approach shown in Scheme 1B.
Fig. 5.

Emission decay from FL emission at 530 nm (S∥ + 2S⊥ data) in PFP/dsDNA-FL in the presence (open circles) and absence (filled squares) of EB, measured by TCSPC. Both samples were excited at 385 nm.
FL as a FRET Gate. The lifetime and anisotropy results support the two-step energy-transfer process shown in Scheme 2: the transition moments of PFP and intercalated EB are nearly perpendicular to each other (see above), whereas the angle between the transition moments of PFP and the FL fragment spans a broad range. Therefore, the orientation of the FL transition moment typically lies between that of PFP and the EB in the same complex. Consequently, the FL molecule attached to one terminus of the dsDNA acts as a FRET gate, relaying the excitation energy from the energy antenna (PFP) to the intercalated EB.
Note that the FL excited-state lifetime in the presence of EB (≈640 ps) is longer than the time scale of local motion of the FL fragment (≈240 ps). Thus, the FL molecules dynamically wobble around the angular range within the excited-state lifetime, as shown in Scheme 2. This dynamic wobble also enables the two-step FRET process to relay the excitation energy.
The enhanced EB emission by means of FL might also partially arise from bridging the distance between the energy donor (PFP) and acceptor (EB) by FL. This type of sequential energy-transfer mechanism has been used to explain the efficient energy transfer in the photosynthetic light-harvesting system (20). The FL molecule is attached to a terminus of DNA by a flexible (CH2)6 linker, which may act a spatial bridge to relay the excitation energy between the PFP and intercalated EB. As demonstrated by previous work (16) and the anisotropy data presented here, however, the FL molecule linked to DNA is distributed within a 60° cone of angles. As a result, this spatial bridge effect is relatively unimportant. The orientation alignment of three components within the self-assembled complex is the dominant mechanism by which the FL relays the excitation energy from PFP to the intercalated EB.
Summary
We have investigated the two-step FRET-gate energy-transfer process from the CCP to the EB intercalated into dsDNA through a FL molecule linked to one terminus of the DNA, a scheme originally proposed for DNA sequence detection. PDES and TCSPC were used to monitor the entire energy-transfer process over times ranging from a subpicosecond to tens of nanoseconds. Time-resolved fluorescence anisotropy measurements were carried out to investigate the relative orientation of transition moments. The results are fully consistent with the FRET-gate energy-transfer mechanism shown in Scheme 2. The inefficient direct energy transfer from conjugated polymer to intercalated EB results from the near orthogonality of the orientations of the two transition moments. When dsDNA with a FL molecule linked to one terminus is used, the orientation of the FL molecules spans a range of angular distributions. The orientation of the transition moment of FL thus typically lies between that of conjugated polymer and EB. Consequently, the FL acts as a FRET gate to relay the excitation energy from the conjugated polymer to the intercalated EB.
Acknowledgments
This work is supported by funds from the National Science Foundation under Grants DMR 0099843 and DMR 0343312, the Air Force Office of Scientific Research under Grant F49620-02-1-0127, the National Institutes of Health under Grant GM62958-01, and the Army Institute for Collaborative Biotechnologies.
Abbreviations: CCP, cationic conjugated polymer; FL, fluorescein; PNA, peptide nucleic acid; EB, ethidium bromide; PDES, pump-dump-emission spectroscopy; TCSPC, time-correlated single-photon counting; PFP, poly(fluorene phenylene).
References
- 1.Gaylord, B. S., Heeger, A. J. & Bazan, G. C. (2002) Proc. Natl. Acad. Sci. USA 99, 10954-10957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang, S., Gaylord, B. S. & Bazan, G. C. (2004) J. Am. Chem. Soc. 126, 5446-5451. [DOI] [PubMed] [Google Scholar]
- 3.Liu, B., Wang, S., Bazan, G. C. & Mikhailovsky, A. (2003) J. Am. Chem. Soc. 125, 13306-13307. [DOI] [PubMed] [Google Scholar]
- 4.Lakowicz, J. R. (1999) Principles of Fluorescence Spectroscopy (Kluwer Academic/Plenum, New York).
- 5.Yan, M., Rothberg, L. J., Kwock, E. W. & Miller, T. M. (1995) Phys. Rev. Lett. 75, 1992-1995. [DOI] [PubMed] [Google Scholar]
- 6.Rothberg, L. J., Yan, M., Fung, A. W. P., Jedju, T. M., Kwock, E. W. & Galvin, M. E. (1997) Synth. Met. 84, 537-538. [Google Scholar]
- 7.Muller, J. G., Lemmer, U., Feldmann, J. & Scherf, U. (2002) Phys. Rev. Lett. 88, 147401. [DOI] [PubMed] [Google Scholar]
- 8.Xu, Q.-H., Gaylord, B. S., Wang, S., Bazan, G. C., Moses, D. & Heeger, A. J. (2004) Proc. Natl. Acad. Sci. USA 101, 11634-11639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Förster, T. (1948) Ann. Phys. 2, 55-75. [Google Scholar]
- 10.Magde, D., Zappala, M., Knox, W. H. & Nordlund, T. M. (1983) J. Phys. Chem. 87, 3286-3288. [Google Scholar]
- 11.Duhamel, J., Kanyo, J., DinterGottlieb, G. & Lu, P. (1996) Biochemistry 35, 16687-16697. [DOI] [PubMed] [Google Scholar]
- 12.Lepecq, J. B. & Paoletti, C. (1967) J. Mol. Biol. 27, 87-106. [DOI] [PubMed] [Google Scholar]
- 13.Angerer, L. M. & Moudrian, E. N. (1972) J. Mol. Biol. 63, 505-521. [DOI] [PubMed] [Google Scholar]
- 14.Korolev, N., Lyubartsev, A. P., Laaksonen, A. & Nordenskiold, L. (2003) Nucleic Acids Res. 31, 5971-5981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nguyen, T. Q., Wu, J. J., Doan, V., Schwartz, B. J. & Tolbert, S. H. (2000) Science 288, 652-656. [DOI] [PubMed] [Google Scholar]
- 16.Norman, D. G., Grainger, R. J., Uhrin, D. & Lilley, D. M. J. (2000) Biochemistry 39, 6317-6324. [DOI] [PubMed] [Google Scholar]
- 17.Parkhurst, L. J., Parkhurst, K. M., Powell, R., Wu, J. & Williams, S. (2002) Biopolymers 61, 180-200. [DOI] [PubMed] [Google Scholar]
- 18.Lipari, G. & Szabo, A. (1980) Biophys. J. 30, 489-506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu, B., Gaylord, B. S., Wang, S. & Bazan, G. C. (2003) J. Am. Chem. Soc. 125, 6705-6714. [DOI] [PubMed] [Google Scholar]
- 20.Fleming, G. R. & Van Grondelle, R. (1994) Phys. Today 47, 48-55. [Google Scholar]