

Abstract
Acute lymphoblastic leukemia (ALL) is the most common childhood cancer, and despite cure rates exceeding 90% in children, it remains an important cause of morbidity and mortality in children and adults. The past decade has been marked by extraordinary advances into the genetic basis of leukemogenesis and treatment responsiveness in ALL. Both B-cell and T-cell ALL comprise multiple subtypes harboring distinct constellations of somatic structural DNA rearrangements and sequence mutations that commonly perturb lymphoid development, cytokine receptors, kinase and Ras signaling, tumor suppression, and chromatin modification. Recent studies have helped to understand the genetic basis of clonal evolution and relapse and the role of inherited genetic variants in leukemogenesis. Many of these findings are of clinical importance, and ongoing studies implementing clinical sequencing in the management of leukemia are expected to improve diagnosis, monitoring of residual disease, and early detection of relapse and to guide precise therapies. Here, we provide a concise review of genomic studies in ALL and discuss the role of genomic testing in clinical management.
Acute lymphoblastic leukemia (ALL) is of B-cell precursor (BCP) lineage (BCP-ALL) or, less commonly, T-cell precursor lineage (T-ALL). Both comprise multiple subtypes commonly defined by structural chromosomal alterations that are initiating lesions, with secondary somatic (tumor-acquired) DNA copy number alterations and sequence mutations that contribute to leukemogenesis. Chromosomal alterations include aneuploidy and chromosomal rearrangements that result in oncogene deregulation or expression of chimeric fusion genes. The prevalence of these alterations varies according to age (Fig 1), and identification is important for diagnosis, risk classification, and, for some lesions, targeted therapy (Table 1).
Fig 1.

Age distribution of acute lymphoblastic leukemia (ALL) subtypes. The prevalence of ALL subtypes varies in children with standard-risk (SR) ALL (age 1 to 9 years and WBC count < 50 × 109/L), children with high-risk (HR) ALL (age 10 to 15 years and/or WBC count > 50 × 109/L), and adolescents (age 16 to 20 years), young adults (age 21 to 39 years), adults (age 40 to 59 years), and older adults (age 60 to 86 years) with ALL. Other, B-cell ALL lacking recurrent abnormalities; Ph, Philadelphia chromosome. Data adapted.1-3
Table 1.
Key Genetic Subtypes of ALL and Recurrent Genomic Features
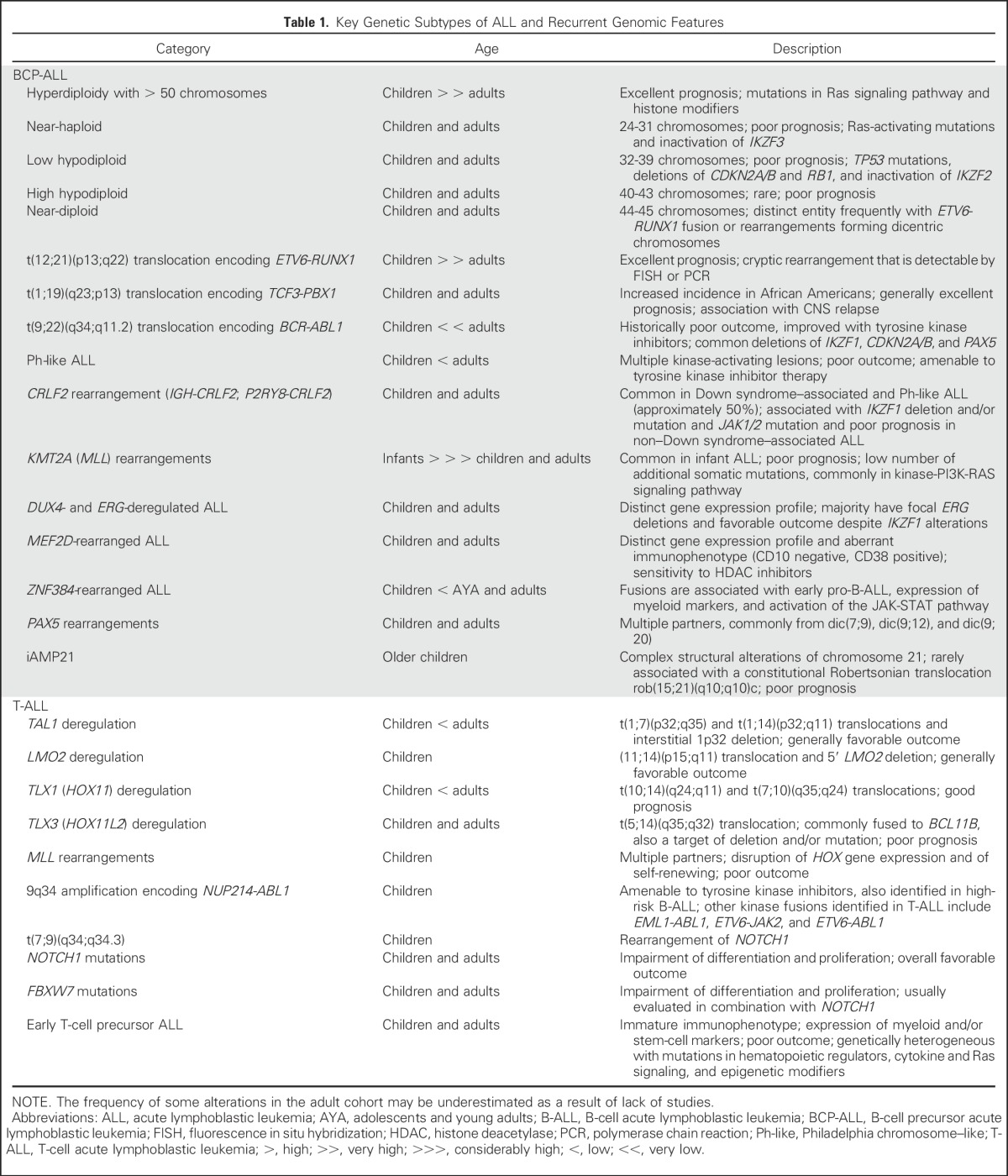
BCP-ALL WITH RECURRING CHROMOSOMAL ALTERATIONS
KMT2A (MLL) rearrangements, particularly the t(4;11)(q21;q23) translocation, are most frequent in infants (< 1 year of age) and are associated with poor outcome.4,5 High hyperdiploidy with gain of at least five chromosomes and ETV6-RUNX1 are each present in 25% to 30% of patients with childhood ALL but occur in less than 3% of young adults and are associated with favorable outcome. Conversely, BCR-ABL1 (Philadelphia [Ph] chromosome) –positive ALL composes 2% to 5% of childhood and 25% of adult ALL, and although historically associated with poor prognosis, outcomes have been markedly improved with the use of tyrosine kinase inhibitors (TKIs). The translocation t(1;19)(q23;p13) resulting in the TCF3-PBX1 fusion occurs in approximately 5% to 6% of childhood and adult BCP-ALLs.6,7 It was originally considered to be a high-risk subtype of ALL, but with contemporary therapy, it is now associated with a favorable outcome, although some studies have reported that it has an independent risk factor for CNS relapse.8 A variant of the t(1;19) translocation, t(17;19)(q23;p13), results in the TCF3-HLF fusion9 (< 1% of ALLs), which is associated with a poor prognosis.10
Complex intrachromosomal amplification of chromosome 21 (iAMP21) is most common in older children and is associated with poor prognosis, which is improved with intensive treatment.11 Hypodiploidy with less than 44 chromosomes occurs in 2% to 3% of patients and is a negative prognostic factor.12 Hypodiploid ALL itself comprises several subtypes with distinct transcriptional profiles and genetic alterations, including near-haploid cases (24 to 31 chromosomes) with Ras-activating mutations and IKZF3 alterations, and low hypodiploidy (32 to 39 chromosomes) with IKZF2 alterations and TP53 mutations that are frequently inherited.13
Secondary DNA deletions, gains, and mutations are characteristic of BCP-ALL, are important cooperating lesions in leukemogenesis, and may be acquired or enriched during disease progression. These include alterations of lymphoid transcription factors (IKZF1, PAX5, EBF1), cell-cycle regulation and tumor suppression (CDKN2A/CDKN2B, RB1), regulation of apoptosis, transcriptional regulation and coactivation (ETV6, ERG), and epigenetic alterations.14 The prevalence, type of alteration, and gene vary between subtypes. KMT2A-rearranged cases show low frequency of secondary somatic mutations, which are often subclonal, indicating that KMT2A rearrangement is sufficient to induce leukemia.5 IKZF1 alterations are a hallmark of BCR-ABL1–positive and Ph-like ALL and are associated with poor outcome1,15-18; in contrast, other members of the IKAROS transcription factor family, IKZF2 and IKZF3, are selectively mutated in hypodiploid ALL.13 In high hyperdiploid ALL, secondary events target genes in the Ras signaling pathway and in chromatin modifiers.19
NEW SUBTYPES OF BCP-ALL
Approximately 25% of childhood ALLs and a higher proportion of adult BCP-ALLs lack a unifying chromosomal alteration on cytogenetic analysis (Fig 1). Several new subtypes of ALL have been recently described that exhibit distinct leukemic-cell gene expression profiles but diverse, often cytogenetically cryptic, founding alterations.
Ph-Like ALL
The 2016 revision to the WHO classification of myeloid neoplasms and acute leukemia recognized BCR-ABL1–like or Ph-like ALL as a new leukemia entity of clinical importance due to its association with an adverse prognosis and responsiveness to TKIs.20 Ph-like ALLs harbor a gene-expression profile similar to BCR-ABL1–positive ALLs but lack BCR-ABL1.17,21 The prevalence of Ph-like ALL increases with age and varies from 10% in standard-risk childhood ALL to greater than 20% in adult ALL, with a peak prevalence of 27.9% in young adults (age 21 to 39 years).1,2 In both children and adults, Ph-like ALL is associated with high-risk clinical features, a poor response to induction chemotherapy, elevated minimal residual disease (MRD) levels, and/or poor survival.22
Common genomic features of BCR-ABL1–like ALL are alterations of B-lymphoid transcription factor genes (particularly IKZF1 deletions) and genetic alterations deregulating cytokine receptor and tyrosine kinase signaling. These include rearrangements and mutation of CRLF2 (approximately 50%), rearrangements of ABL-class tyrosine kinase genes (12%), rearrangements of JAK2 (7%) and the erythropoietin receptor gene (EPOR; 3% to 10%), mutations activating JAK-STAT signaling (11%) and Ras signaling (NRAS, KRAS, PTPN11, and NF1; 6%), and less common kinase alterations (FLT3, NTRK3, BLNK, TYK2, and PTK2B).1,2,23 All kinase fusions retain an intact tyrosine kinase domain and typically exhibit constitutive kinase activation (Fig 2). With the exception of EPOR and JAK2 rearrangements, which are increased in adult Ph-like ALL, there are no significant differences in the frequency of kinase subtypes across different age groups (Fig 3).
Fig 2.

Signaling pathways in Philadelphia chromosome (Ph) –like acute lymphoblastic leukemia (ALL). Deregulation of JAK2, ABL, or other (FLT3, NTRK3, BLNK, ABL, PTK2B) signaling pathways in Ph-like ALL is caused by activating mutations (lightning bolts), fusion genes, and/or genomic deletions (X) that are responsible for overexpression of cytokine receptors (eg, CRLF2, IL-7, and EPOR), expression of truncated receptors missing regulatory domains (eg, EPOR), cell delocalization, and constitutive activation of tyrosine kinases. Some downstream signaling pathways are shown. Dashed circles and line represent likely pathways activated by the kinase alterations and amenable to inhibition by kinase inhibitors, respectively. ABLi, Abelson murine leukemia viral oncogene homolog 1 inhibitor; BCL2i, B-cell lymphoma 2 inhibitor; FAKi, focal adhesion kinase inhibitor; FLT3i, Fms-related tyrosine kinase 3 inhibitor; JAKi, JAK inhibitor; MAPK, mitogen-activated protein kinase; MEKi, MAPK/ERK kinase inhibitor; mTORi, mammalian target of rapamycin inhibitor; PI3Ki, phosphoinositide 3-kinase inhibitor; TRKi, tropomyosin receptor kinase inhibitor; Y, tyrosine residue.
Fig 3.

Frequency of Philadelphia chromosome (Ph) –like acute lymphoblastic leukemia (ALL) subtypes across age. Prevalence of CRFL2-rearranged JAK mutant (mut), CRFL2-rearranged JAK wild-type (WT), JAK2 rearrangements (JAK2r), EPOR rearrangements (EPORr), other JAK-STAT alterations, ABL1-class fusions, all other kinase lesions, and unknown subtype in children, young adults, adults, and older adults. Data adapted.1,2,23
CRLF2 encodes cytokine receptor-like factor 2, also known as the thymic stromal-derived lymphopoietin receptor (TSLPR) that forms a heterodimeric receptor with the interleukin-7 receptor α chain (IL7Rα) for thymic stromal lymphopoietin (TSLP). CRLF2 is deregulated by translocation into the immunoglobulin heavy chain locus (IGH-CRLF2); focal deletion upstream of CRLF2, resulting in formation of a P2RY8-CRLF2 fusion; and less commonly, CRLF2 point mutations (F232C).24 CRLF2 rearrangements are most common in Ph-like and Down syndrome–associated ALL and are age dependent, with P2RY8-CRLF2 associated with young age and IGH-CRLF2 associated with older age and Hispanic ancestry.25,26 CRLF2 is overexpressed on the cell surface of leukemic lymphoblasts and detectable by flow cytometric immunophenotyping. The majority of CRLF2-rearranged ALLs have additional alterations driving JAK-STAT or Ras signaling, particularly activating JAK1 or JAK2 mutations, FLT3 and IL7R sequence mutations, SH2B3 deletions, TSLP rearrangements, and Ras mutations.1,2,27 In most studies, CRLF2 rearrangements are associated with poor prognosis, particularly in cases with concomitant IKZF1 alteration.28,29 Therapies targeting JAK-STAT, PI3K/mTOR, and BCL2 signaling alone or in combination have shown efficacy in preclinical models.30,31
Another major Ph-like ALL genetic subgroup involves ABL-class rearrangements, which include fusions to ABL1, ABL2, CSF1R (encoding the macrophage colony-stimulating factor receptor), PDGFRA, or PDGFRB that are all targetable by inhibitors of ABL1, such as imatinib and dasatinib.1,2,32,33
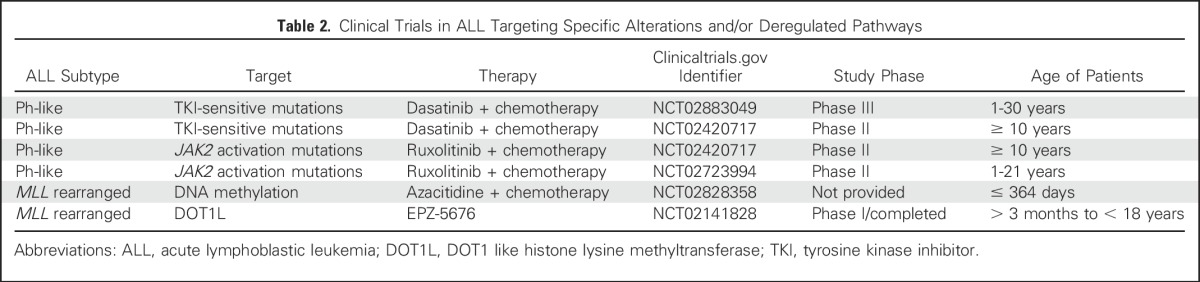
Genomic rearrangements that produce JAK2 fusion genes or rearrangements targeting EPOR are highly sensitive to JAK2 inhibitors, including ruxolitinib, in preclinical models. JAK2 is rearranged to at least 14 different partner genes in Ph-like ALL. EPOR rearrangements include reciprocal or cryptic translocations with immunoglobulin and other loci that deregulate receptor expression and also truncate the cytoplasmic tail of the receptor, resulting in augmented JAK-STAT signaling.1,2,23,32 The extensive preclinical data showing activation of signaling pathways, inhibition with JAK-STAT or ABL inhibitors, synergy with conventional chemotherapy, and anecdotal responsiveness to TKI therapy in patients with Ph-like ALL have led to the multiple prospective studies examining the efficacy of TKIs in Ph-like ALL (Table 2).
Table 2.
Clinical Trials in ALL Targeting Specific Alterations and/or Deregulated Pathways
DUX4- and ERG-Deregulated ALL
Several studies recently identified a subtype of BCP-ALL (up to 7% of BCP-ALLs) with a distinct immunophenotype and gene expression profile characterized by deregulation of the double homeobox 4 gene (DUX4) and the ETS transcription factor gene (ERG).34-37 DUX4 encodes a double homeobox transcription factor located in a macrosatellite D4Z4 repeat in the subtelomeric region of the long arm of chromosome 4.
DUX4 is not expressed in normal B cells, and translocation to IGH results in expression of a truncated DUX4 isoform in the B-cell lineage.34-37 Less commonly, ERG-DUX4 fusions have also been described.37 Prior studies had reported intragenic deletions of the ERG gene in approximately 5% of childhood ALLs, which are now known to be restricted to DUX4-rearranged cases. Notably DUX4-rearranged ALLs commonly express aberrant ERG transcripts and truncated C-terminal ERG proteins irrespective of the presence of ERG deletions. The basis for this association has now been elucidated (Fig 4). DUX4 rearrangement is an early initiating event in leukemogenesis, and aberrantly expressed DUX4 binds to an intragenic region of ERG, resulting in expression of a noncanonical first exon and transcript, ERGalt. This encodes a truncated C-terminal ERG protein that retains the DNA-binding and transactivating domains of ERG, inhibits wild-type ERG transcriptional activity, and is transforming.36 Notably, DUX4/ERG-deregulated ALL is associated with a favorable outcome, despite the presence of concomitant genetic alterations otherwise associated with a poor outcome, such as IKZF1 deletions, which are present in approximately 40% of patients.38,39 DUX4 rearrangement is not evident on karyotypic analysis but is important to identify by gene expression or sequencing approaches to accurately assign risk and guide therapy.
Fig 4.

Mechanism of leukemogenesis mediated by DUX4 and ERG deregulation. DUX4 rearrangements result in profound transcriptional deregulation of ERG and expression of a novel ERG isoform, ERGalt, and frequent RAG-mediated ERG deletions. ERGalt uses a noncanonical first exon whose transcription is initiated by DUX4 binding. It inhibits wild-type (WT) ERG transcriptional activity and is proleukemogenic.
MEF2D and ZNF384 Gene Fusions
Myocyte enhancer factor 2D (MEF2D) and zinc finger 384 (ZNF384) rearrangements characterize distinct B-ALL subtypes, accounting for approximately 3% to 4% and 3% of pediatric patients and approximately 6% and 7% of adult patients, respectively.3,34,35,40
MEF2D is rearranged to BCL9, HNRNPUL1, SS18, FOXJ2, CSF1R, and DAZAP1, with the most common target of rearrangement being BCL9. All fusions preserve the MEF2D MADS-box domain that mediates DNA binding, result in enhanced MEF2D transcriptional activity, and are transforming and leukemogenic in vitro and in vivo.3,35,41 MEF2D ALL is associated with older age of onset, an aberrant immunophenotype (CD10 negative, CD38 positive), and poor outcome. The deregulation of MEF2D target genes results in a therapeutic vulnerability, because one such gene is histone deacetylase 9 (HDAC9), and human xenografts of MEF2D ALL are exquisitely sensitive to histone deacetylase inhibitors, such as panobinostat.3
ZNF384 rearrangements involve the fusion of a 5′ partner gene, usually a transcriptional regulator or chromatin modifier (EP300, CREBBP, TAF15, SYNRG, EWSR1, TCF3, and ARID1B), to the entire coding region of ZNF384. ZNF384-rearranged ALLs are often diagnosed as B-ALLs with expression of myeloid antigens or as B/myeloid mixed-phenotype acute leukemias, suggesting transformation of a hematopoietic progenitor with B/myeloid potential. ZNF384-rearranged B-ALL has an intermediate prognosis and is characterized by upregulation of the JAK-STAT pathway, suggesting a potential benefit from treatment with inhibitors of this pathway.35
Other Rearrangements
Additional recurrent alterations in BCP-ALL include IGH translocations, including CRLF2 and EPOR in Ph-like ALL, CEBP gene family members, and ID4. Their frequency is low among children younger than 10 years old (< 3%) but considerably higher (10%) among adolescents and young adults (age 15 to 24 years), and prognosis is poor.42
PAX5 is also rearranged to a diverse range of fusion partners in approximately 2% of B-ALLs. These commonly result in the 5′ N-terminal DNA-binding domain of PAX5 fused to the 3′ C terminus of the partner gene, resulting in loss of the transactivating domain of PAX5. Several of these fusions inhibit the normal transcriptional activity of PAX5, although it remains to be directly shown whether these fusions promote leukemogenesis through haploinsufficiency of wild-type PAX543 or whether they are exerting an oncogenic effect.
GENETICS OF T-ALL
T-ALL is an aggressive and heterogeneous disease that accounts for approximately 15% and 25% of pediatric and adult ALLs, respectively. Approximately 50% of patients with T-ALL harbor chromosomal translocations that most commonly involve the 14q11 (T-cell receptor α and δ [TRA and TRD]) and 7q34 (TRB) regions, juxtaposing the T-cell receptor genes to pivotal transcription factor genes, such as TAL1, TAL2, LYL1, OLIG2, LMO1, LMO2, TLX1 (HOX11), TLX3 (HOX11L2), NKX2-1, NKX2-2, NKX2-5, HOXA genes, MYC, and MYB. In addition, T-ALLs may harbor cryptic rearrangements of ABL1 (NUP214-ABL1, EML1-ABL1, and ETV6-ABL1) that may be amenable to TKI therapy. Moreover, gene expression profiling studies have helped in the classification of T-ALL into molecular subgroups that are characterized by unique gene expression signatures and aberrant activation of specific T-ALL transcription factor oncogenes, including MEF2C, HOXA, TLX1, NKX2.1, TLX3, TAL1, LMO1, and LMO2.44,45
Sequence mutations and DNA copy number alterations include those in NOTCH1, FBXW7, and MYB; in genes involved in the JAK-STAT (IL7R, JAK1, JAK3, and STAT5B) and Ras/PI3K/AKT (NRAS/KRAS and PTEN) pathways; in epigenetic regulators (PHF6, SUZ12, EZH2, TET2, H3F3A, and KDM6A); in transcription regulators (LEF1, WT1, BCL11B, and ZEB2); and in genes involved in mRNA maturation and ribosome activity (CNOT3, RPL5, and RPL10). Activating NOTCH1 mutations and loss-of-function mutations of FBXW7, leading to inhibition of ubiquitin-mediated degradation of the activated form of NOTCH1, occur in more than 60% and 15% of T-ALLs, respectively.46 Despite promising preclinical studies inhibiting NOTCH signaling by γ-secretase inhibitors, severe GI toxicities and lack of cytotoxic antitumor responses still limit their direct translation into patient benefit.47 Given the role of MYC, a known FBXW7 substrate, in T-ALL leukemia initiation, inhibitors of the bromodomain and extraterminal (BET) family of proteins have shown antileukemic activities in in vitro and in vivo models of T-ALL.48
Early T-cell precursor (ETP) ALL is a distinct form of leukemia characterized by reduced expression of T-cell markers (CD1a, CD8, and CD5) and aberrant expression of myeloid or stem-cell markers.49 ETP-ALL has a poor outcome, although this is mitigated by contemporary risk-adapted therapy.50,51 ETP-ALL is genetically heterogeneous, with mutation of multiple cellular pathways including hematopoietic and lymphoid development (RUNX1, IKZF1, ETV6, GATA3, and EP300); Ras, cytokine receptor, and kinase signaling (NRAS, IL7R, KRAS, JAK1, JAK3, NF1, PTPN11, and SH2B3); and loss-of-function mutations targeting epigenetic regulators (EZH2, SUZ12, EED, and SETD2).52 The gene expression profile of ETP-ALL is similar to that of hematopoietic stem cells, suggesting that ETP-ALL may represent one of a spectrum of immature leukemias, rather than a true T-ALL. The involvement of JAK-STAT and PRC2 pathways in ETP-ALL suggests that JAK inhibition and/or chromatin-modifying agents may be therapeutically useful.53
Recent studies have identified pathogenic noncoding mutations in T-ALL, notably mutations upstream of the oncogene TAL1. These generate a binding site for the MYB transcription factor, thereby recruiting a protein complex including TAL1 and the H3K27 acetylator CREBBP, resulting in formation of an oncogenic superenhancer region with high levels of H3K27 acetylation.54
RELAPSED ALL
Relapsed ALL has a poor outcome with conventional therapy and is more common with increasing age, so there is great interest in characterizing genetic drivers of relapse. Genomic studies have shown that leukemia evolution leading to relapse usually does not proceed in a sequential linear fashion but, instead, follows a complex branched pathway. Although primary chromosome translocations are retained, the majority of patients who experience relapse also exhibit new secondary genetic alterations or, commonly, relapse-acquired lesions frequently arising from a minor clone at diagnosis. Genetic lesions driving clonal evolution may arise from cooperation between recombination-activating genes (RAG1 and RAG2) and activation-induced cytidine deaminase (AID).15,55,56 Mutations influencing drug sensitivity and proliferation in particular stroma or environments will outgrow and become dominant.
Common relapse-acquired lesions include mutations in the transcriptional coactivator and acetyl transferase CREBBP (CREB-binding protein [CBP]), which occur in up to 20% of relapsed ALLs and impair sensitivity to glucocorticoid therapy,57-59 and mutations in the 5′-nucleotidase catalytic enzyme II (NT5C2) gene, which confer increased resistance to purine analogs.60,61 Other recurrent somatic mutations in relapsed ALL include deletions in the DNA mismatch repair gene mut-S homolog 6 (MSH6) and the glucocorticoid receptor NR3C1 and mutations in the H3K36 trimethyltransferase SETD2, the lysine-specific demethylase KDM6, and the epigenetic regulator MLL2.62,63
Ras pathway mutations (eg, KRAS, NRAS, FLT3, and PTPN11) are often selected for or acquired during treatment and thus predominate in the relapsed leukemic clone. They are associated with high-risk features and poor prognosis, but treatment with MEK inhibitors has been reported to offer clinical benefit in vitro and in xenograft models.64
INHERITED VARIANTS AND RISK FOR ALL DEVELOPMENT
Inherited variants and rare deleterious mutations have been shown to play a role in the risk of developing ALL. Some of these variants are in IKZF1 (7p12.2), CDKN2A/CDKN2B (9p21), ARID5B (10q21.2), CEBPE (14q11.2), PIP4K2A (10p12.2), and GATA3 (10p14). ARID5B and PIP4K2A genotypes are associated with risk of hyperdiploid ALL, whereas the risk allele in GATA3 has been associated with Ph-like ALL.
TP53 alterations occur in 91% of low-hypodiploid ALLs in children, 43% of which are found in nontumor cells, suggesting that low-hypodiploid ALL represents a manifestation of Li-Fraumeni syndrome.13 Risk of developing ALL is increased by 20-fold in patients with Down syndrome, whereas the rare constitutional Robertsonian translocation, rob(15;21)(q10;q10)c, is associated with an approximately 2,700-fold increased risk of developing iAMP21-ALL compared with the general population.
There are recent reports of several families with deleterious inherited mutations in the ETS domain of ETV6, which affect DNA binding efficiency and altered intracellular localization of the protein,65,66 and with a mutation in PAX5 (p.Gly183Ser), which attenuates the transcriptional activity of PAX5.67
CLINICAL IMPLICATIONS
Accurate, comprehensive identification of the full range of genetic alterations in ALL is important for diagnosis, risk stratification, implementation of targeted therapy, and sensitive monitoring of treatment response. This is now possible but poses logistic, financial, and ethical challenges.
Ideally, diagnostic testing should detect all types of alterations of clinical relevance, including nucleotide substitutions, insertion/deletion mutations, DNA copy number alterations, and chromosomal rearrangements. The choice of the optimal detection method will depend on the number of genes to be screened and the desire to detect genomic rearrangements, as well as the desired sensitivity. Sequencing-based assays that use DNA or RNA of sets of genes are able to accurately detect mutations and rearrangements in a clinically relevant time frame68 but may not detect all focal deletions characteristic of ALL. It is likely that unbiased approaches such as transcriptome, exome, and whole-genome sequencing will be increasingly used. Sequencing-based approaches have also been used successfully to analyze antigen receptor rearrangements and quantitate MRD more sensitively than flow cytometric or conventional polymerase chain reaction–based approaches.69 Active areas of investigation include the use of similar approaches to quantitate specific mutations and rearrangements that facilitate resistance to therapy (eg, IKZF1, CREBBP, and NT5C2, and ABL in BCR-ABL1–positive ALL) and to incorporate these into clinical trials and management and the use of such results to change therapy when a mutation that confers resistance to a specific agent emerges.
These approaches are also important to identify mutated genes and deregulated pathways amenable to inhibition by targeted therapies, either at initial diagnosis or at relapse, particularly for high-risk ALL subtypes. This includes treatment of Ph-like ALL with kinase-activating mutations that have shown evidence of activity in case reports and are now are being tested in clinical trials (Table 2). It is likely that genomic data will also inform the use of immunotherapeutic approaches. For example, T cells engineered with a chimeric antigen receptor targeting the thymic stromal lymphopoietin receptor encoded by CRLF2 have demonstrated potent activity in preclinical models of Ph-like ALL.70
ACKNOWLEDGMENT
We thank our colleagues at St Jude Children’s Research Hospital, the Children’s Oncology Group, and the National Cancer Institute Therapeutically Applicable Research to Generate Effective Treatments (TARGET) Consortium (https://ocg.cancer.gov/programs/target). Several of the studies described were supported by the American Lebanese Syrian Associated Charities of St Jude Children’s Research Hospital, the National Cancer Institute of the US National Institutes of Health, Alex’s Lemonade Stand Foundation, the American Association for Cancer Research, the American Society of Hematology, the Henry Schueler 41 & 9 Foundation, the Leukemia and Lymphoma Society, the National Health and Medical Research Council (Australia), the Pew Charitable Trusts, Stand Up To Cancer, the St Baldrick’s Foundation, and the Lady Tata Memorial Trust. We thank B. Stelter of Biomedical Communications at St Jude Children’s Research Hospital for assistance with figure preparation and apologize to the authors of the many articles that could not be cited as a result of space considerations.
AUTHOR CONTRIBUTIONS
Conception and design: All authors
Collection and assembly of data: All authors
Data analysis and interpretation: All authors
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Genetic Basis of Acute Lymphoblastic Leukemia
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/site/ifc.
Ilaria Iacobucci
No relationship to disclose
Charles G. Mullighan
Honoraria: Incyte, Amgen
Consulting or Advisory Role: Incyte
Speakers' Bureau: Amgen
Research Funding: Loxo
Patents, Royalties, Other Intellectual Property: Inventor on a pending patent application related to gene expression signatures for detection of underlying Philadelphia chromosome–like events and therapeutic targeting in leukemia (PCT/US2012/069228).
Travel, Accommodations, Expenses: Incyte, Amgen
REFERENCES
- 1.Roberts KG, Li Y, Payne-Turner D, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med. 2014;371:1005–1015. doi: 10.1056/NEJMoa1403088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roberts KG, Gu Z, Payne-Turner D, et al: High frequency and poor outcome of Philadelphia chromosome-like acute lymphoblastic leukemia in adults. J Clin Oncol 35:394-401, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gu Z, Churchman ML, Roberts KG, et al: Genomic analyses identify recurrent MEF2D fusions in acute lymphoblastic leukaemia. Nat Commun 7:13331, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meyer C, Hofmann J, Burmeister T, et al. The MLL recombinome of acute leukemias in 2013. Leukemia. 2013;27:2165–2176. doi: 10.1038/leu.2013.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andersson AK, Ma J, Wang J, et al. The landscape of somatic mutations in infant MLL-rearranged acute lymphoblastic leukemias. Nat Genet. 2015;47:330–337. doi: 10.1038/ng.3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barber KE, Harrison CJ, Broadfield ZJ, et al. Molecular cytogenetic characterization of TCF3 (E2A)/19p13.3 rearrangements in B-cell precursor acute lymphoblastic leukemia. Genes Chromosomes Cancer. 2007;46:478–486. doi: 10.1002/gcc.20431. [DOI] [PubMed] [Google Scholar]
- 7.Burmeister T, Gökbuget N, Schwartz S, et al. Clinical features and prognostic implications of TCF3-PBX1 and ETV6-RUNX1 in adult acute lymphoblastic leukemia. Haematologica. 2010;95:241–246. doi: 10.3324/haematol.2009.011346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jeha S, Pei D, Raimondi SC, et al. Increased risk for CNS relapse in pre-B cell leukemia with the t(1;19)/TCF3-PBX1. Leukemia. 2009;23:1406–1409. doi: 10.1038/leu.2009.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Inaba T, Roberts WM, Shapiro LH, et al. Fusion of the leucine zipper gene HLF to the E2A gene in human acute B-lineage leukemia. Science. 1992;257:531–534. doi: 10.1126/science.1386162. [DOI] [PubMed] [Google Scholar]
- 10.Hunger SP. Chromosomal translocations involving the E2A gene in acute lymphoblastic leukemia: Clinical features and molecular pathogenesis. Blood. 1996;87:1211–1224. [PubMed] [Google Scholar]
- 11.Moorman AV, Robinson H, Schwab C, et al. Risk-directed treatment intensification significantly reduces the risk of relapse among children and adolescents with acute lymphoblastic leukemia and intrachromosomal amplification of chromosome 21: A comparison of the MRC ALL97/99 and UKALL2003 trials. J Clin Oncol. 2013;31:3389–3396. doi: 10.1200/JCO.2013.48.9377. [DOI] [PubMed] [Google Scholar]
- 12.Nachman JB, Heerema NA, Sather H, et al. Outcome of treatment in children with hypodiploid acute lymphoblastic leukemia. Blood. 2007;110:1112–1115. doi: 10.1182/blood-2006-07-038299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holmfeldt L, Wei L, Diaz-Flores E, et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet. 2013;45:242–252. doi: 10.1038/ng.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mullighan CG, Goorha S, Radtke I, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007;446:758–764. doi: 10.1038/nature05690. [DOI] [PubMed] [Google Scholar]
- 15.Mullighan CG, Miller CB, Radtke I, et al. BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature. 2008;453:110–114. doi: 10.1038/nature06866. [DOI] [PubMed] [Google Scholar]
- 16.Iacobucci I, Storlazzi CT, Cilloni D, et al. Identification and molecular characterization of recurrent genomic deletions on 7p12 in the IKZF1 gene in a large cohort of BCR-ABL1-positive acute lymphoblastic leukemia patients: On behalf of Gruppo Italiano Malattie Ematologiche dell’Adulto Acute Leukemia Working Party (GIMEMA AL WP) Blood. 2009;114:2159–2167. doi: 10.1182/blood-2008-08-173963. [DOI] [PubMed] [Google Scholar]
- 17.Mullighan CG, Su X, Zhang J, et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med. 2009;360:470–480. doi: 10.1056/NEJMoa0808253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martinelli G, Iacobucci I, Storlazzi CT, et al. IKZF1 (Ikaros) deletions in BCR-ABL1-positive acute lymphoblastic leukemia are associated with short disease-free survival and high rate of cumulative incidence of relapse: A GIMEMA AL WP report. J Clin Oncol. 2009;27:5202–5207. doi: 10.1200/JCO.2008.21.6408. [DOI] [PubMed] [Google Scholar]
- 19.Paulsson K, Lilljebjörn H, Biloglav A, et al. The genomic landscape of high hyperdiploid childhood acute lymphoblastic leukemia. Nat Genet. 2015;47:672–676. doi: 10.1038/ng.3301. [DOI] [PubMed] [Google Scholar]
- 20.Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–2405. doi: 10.1182/blood-2016-03-643544. [DOI] [PubMed] [Google Scholar]
- 21.Den Boer ML, van Slegtenhorst M, De Menezes RX, et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: A genome-wide classification study. Lancet Oncol. 2009;10:125–134. doi: 10.1016/S1470-2045(08)70339-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roberts KG, Pei D, Campana D, et al. Outcomes of children with BCR-ABL1–like acute lymphoblastic leukemia treated with risk-directed therapy based on the levels of minimal residual disease. J Clin Oncol. 2014;32:3012–3020. doi: 10.1200/JCO.2014.55.4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iacobucci I, Li Y, Roberts KG, et al. Truncating erythropoietin receptor rearrangements in acute lymphoblastic leukemia. Cancer Cell. 2016;29:186–200. doi: 10.1016/j.ccell.2015.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yoda A, Yoda Y, Chiaretti S, et al. Functional screening identifies CRLF2 in precursor B-cell acute lymphoblastic leukemia. Proc Natl Acad Sci USA. 2010;107:252–257. doi: 10.1073/pnas.0911726107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Russell LJ, Capasso M, Vater I, et al. Deregulated expression of cytokine receptor gene, CRLF2, is involved in lymphoid transformation in B-cell precursor acute lymphoblastic leukemia. Blood. 2009;114:2688–2698. doi: 10.1182/blood-2009-03-208397. [DOI] [PubMed] [Google Scholar]
- 26.Mullighan CG, Collins-Underwood JR, Phillips LA, et al. Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia. Nat Genet. 2009;41:1243–1246. doi: 10.1038/ng.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mullighan CG, Zhang J, Harvey RC, et al. JAK mutations in high-risk childhood acute lymphoblastic leukemia. Proc Natl Acad Sci USA. 2009;106:9414–9418. doi: 10.1073/pnas.0811761106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harvey RC, Mullighan CG, Chen IM, et al. Rearrangement of CRLF2 is associated with mutation of JAK kinases, alteration of IKZF1, Hispanic/Latino ethnicity, and a poor outcome in pediatric B-progenitor acute lymphoblastic leukemia. Blood. 2010;115:5312–5321. doi: 10.1182/blood-2009-09-245944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cario G, Zimmermann M, Romey R, et al. Presence of the P2RY8-CRLF2 rearrangement is associated with a poor prognosis in non-high-risk precursor B-cell acute lymphoblastic leukemia in children treated according to the ALL-BFM 2000 protocol. Blood. 2010;115:5393–5397. doi: 10.1182/blood-2009-11-256131. [DOI] [PubMed] [Google Scholar]
- 30.Maude SL, Tasian SK, Vincent T, et al. Targeting JAK1/2 and mTOR in murine xenograft models of Ph-like acute lymphoblastic leukemia. Blood. 2012;120:3510–3518. doi: 10.1182/blood-2012-03-415448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Waibel M, Solomon VS, Knight DA, et al. Combined targeting of JAK2 and Bcl-2/Bcl-xL to cure mutant JAK2-driven malignancies and overcome acquired resistance to JAK2 inhibitors. Cell Reports. 2013;5:1047–1059. doi: 10.1016/j.celrep.2013.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roberts KG, Morin RD, Zhang J, et al. Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell. 2012;22:153–166. doi: 10.1016/j.ccr.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schwab C, Ryan SL, Chilton L, et al. EBF1-PDGFRB fusion in pediatric B-cell precursor acute lymphoblastic leukemia (BCP-ALL): Genetic profile and clinical implications. Blood. 2016;127:2214–2218. doi: 10.1182/blood-2015-09-670166. [DOI] [PubMed] [Google Scholar]
- 34.Liu YF, Wang BY, Zhang WN, et al. Genomic profiling of adult and pediatric B-cell acute lymphoblastic leukemia. EBioMedicine. 2016;8:173–183. doi: 10.1016/j.ebiom.2016.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yasuda T, Tsuzuki S, Kawazu M, et al. Recurrent DUX4 fusions in B cell acute lymphoblastic leukemia of adolescents and young adults. Nat Genet. 2016;48:569–574. doi: 10.1038/ng.3535. [DOI] [PubMed] [Google Scholar]
- 36.Zhang J, McCastlain K, Yoshihara H, et al. Deregulation of DUX4 and ERG in acute lymphoblastic leukemia. Nat Genet. 2016;48:1481–1489. doi: 10.1038/ng.3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lilljebjörn H, Henningsson R, Hyrenius-Wittsten A, et al. Identification of ETV6-RUNX1-like and DUX4-rearranged subtypes in paediatric B-cell precursor acute lymphoblastic leukaemia. Nat Commun. 2016;7:11790. doi: 10.1038/ncomms11790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harvey RC, Mullighan CG, Wang X, et al. Identification of novel cluster groups in pediatric high-risk B-precursor acute lymphoblastic leukemia with gene expression profiling: Correlation with genome-wide DNA copy number alterations, clinical characteristics, and outcome. Blood. 2010;116:4874–4884. doi: 10.1182/blood-2009-08-239681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Clappier E, Auclerc MF, Rapion J, et al. An intragenic ERG deletion is a marker of an oncogenic subtype of B-cell precursor acute lymphoblastic leukemia with a favorable outcome despite frequent IKZF1 deletions. Leukemia. 2014;28:70–77. doi: 10.1038/leu.2013.277. [DOI] [PubMed] [Google Scholar]
- 40.Suzuki K, Okuno Y, Kawashima N, et al. MEF2D-BCL9 fusion gene is associated with high-risk acute B-cell precursor lymphoblastic leukemia in adolescents. J Clin Oncol. 2016;34:3451–3459. doi: 10.1200/JCO.2016.66.5547. [DOI] [PubMed] [Google Scholar]
- 41.Prima V, Hunger SP. Cooperative transformation by MEF2D/DAZAP1 and DAZAP1/MEF2D fusion proteins generated by the variant t(1;19) in acute lymphoblastic leukemia. Leukemia. 2007;21:2470–2475. doi: 10.1038/sj.leu.2404962. [DOI] [PubMed] [Google Scholar]
- 42.Russell LJ, Enshaei A, Jones L, et al. IGH@ translocations are prevalent in teenagers and young adults with acute lymphoblastic leukemia and are associated with a poor outcome. J Clin Oncol. 2014;32:1453–1462. doi: 10.1200/JCO.2013.51.3242. [DOI] [PubMed] [Google Scholar]
- 43.Dang J, Wei L, de Ridder J, et al. PAX5 is a tumor suppressor in mouse mutagenesis models of acute lymphoblastic leukemia. Blood. 2015;125:3609–3617. doi: 10.1182/blood-2015-02-626127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ferrando AA, Neuberg DS, Staunton J, et al. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell. 2002;1:75–87. doi: 10.1016/s1535-6108(02)00018-1. [DOI] [PubMed] [Google Scholar]
- 45.Durinck K, Goossens S, Peirs S, et al. Novel biological insights in T-cell acute lymphoblastic leukemia. Exp Hematol. 2015;43:625–639. doi: 10.1016/j.exphem.2015.05.017. [DOI] [PubMed] [Google Scholar]
- 46.Weng AP, Ferrando AA, Lee W, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306:269–271. doi: 10.1126/science.1102160. [DOI] [PubMed] [Google Scholar]
- 47.Papayannidis C, DeAngelo DJ, Stock W, et al. A phase 1 study of the novel gamma-secretase inhibitor PF-03084014 in patients with T-cell acute lymphoblastic leukemia and T-cell lymphoblastic lymphoma. Blood Cancer J. 2015;5:e350. doi: 10.1038/bcj.2015.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roderick JE, Tesell J, Shultz LD, et al. c-Myc inhibition prevents leukemia initiation in mice and impairs the growth of relapsed and induction failure pediatric T-ALL cells. Blood. 2014;123:1040–1050. doi: 10.1182/blood-2013-08-522698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Coustan-Smith E, Mullighan CG, Onciu M, et al. Early T-cell precursor leukaemia: A subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncol. 2009;10:147–156. doi: 10.1016/S1470-2045(08)70314-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Patrick K, Wade R, Goulden N, et al. Outcome for children and young people with early T-cell precursor acute lymphoblastic leukaemia treated on a contemporary protocol, UKALL 2003. Br J Haematol. 2014;166:421–424. doi: 10.1111/bjh.12882. [DOI] [PubMed] [Google Scholar]
- 51.Wood B, Winter S, Dunsmore K, et al. Patients with early T-cell precursor (ETP) acute lymphoblastic leukemia (ALL) have high levels of minimal residual disease (MRD) at the end of induction: A Children’s Oncology Group (COG) study. Blood. 2009;114:9. abstr. [Google Scholar]
- 52.Zhang J, Ding L, Holmfeldt L, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481:157–163. doi: 10.1038/nature10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Maude SL, Dolai S, Delgado-Martin C, et al. Efficacy of JAK/STAT pathway inhibition in murine xenograft models of early T-cell precursor (ETP) acute lymphoblastic leukemia. Blood. 2015;125:1759–1767. doi: 10.1182/blood-2014-06-580480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mansour MR, Abraham BJ, Anders L, et al. Oncogene regulation: An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science. 2014;346:1373–1377. doi: 10.1126/science.1259037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Papaemmanuil E, Rapado I, Li Y, et al. RAG-mediated recombination is the predominant driver of oncogenic rearrangement in ETV6-RUNX1 acute lymphoblastic leukemia. Nat Genet. 2014;46:116–125. doi: 10.1038/ng.2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Swaminathan S, Klemm L, Park E, et al. Mechanisms of clonal evolution in childhood acute lymphoblastic leukemia. Nat Immunol. 2015;16:766–774. doi: 10.1038/ni.3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kino T, Nordeen SK, Chrousos GP. Conditional modulation of glucocorticoid receptor activities by CREB-binding protein (CBP) and p300. J Steroid Biochem Mol Biol. 1999;70:15–25. doi: 10.1016/s0960-0760(99)00100-4. [DOI] [PubMed] [Google Scholar]
- 58.Lambert JR, Nordeen SK. CBP recruitment and histone acetylation in differential gene induction by glucocorticoids and progestins. Mol Endocrinol. 2003;17:1085–1094. doi: 10.1210/me.2001-0183. [DOI] [PubMed] [Google Scholar]
- 59.Mullighan CG, Zhang J, Kasper LH, et al. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature. 2011;471:235–239. doi: 10.1038/nature09727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Meyer JA, Wang J, Hogan LE, et al. Relapse-specific mutations in NT5C2 in childhood acute lymphoblastic leukemia. Nat Genet. 2013;45:290–294. doi: 10.1038/ng.2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tzoneva G, Perez-Garcia A, Carpenter Z, et al. Activating mutations in the NT5C2 nucleotidase gene drive chemotherapy resistance in relapsed ALL. Nat Med. 2013;19:368–371. doi: 10.1038/nm.3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ma X, Edmonson M, Yergeau D, et al. Rise and fall of subclones from diagnosis to relapse in pediatric B-acute lymphoblastic leukaemia. Nat Commun. 2015;6:6604. doi: 10.1038/ncomms7604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mullighan CG, Phillips LA, Su X, et al. Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science. 2008;322:1377–1380. doi: 10.1126/science.1164266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Irving J, Matheson E, Minto L, et al. Ras pathway mutations are prevalent in relapsed childhood acute lymphoblastic leukemia and confer sensitivity to MEK inhibition. Blood. 2014;124:3420–3430. doi: 10.1182/blood-2014-04-531871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang MY, Churpek JE, Keel SB, et al. Germline ETV6 mutations in familial thrombocytopenia and hematologic malignancy. Nat Genet. 2015;47:180–185. doi: 10.1038/ng.3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Noetzli L, Lo RW, Lee-Sherick AB, et al. Germline mutations in ETV6 are associated with thrombocytopenia, red cell macrocytosis and predisposition to lymphoblastic leukemia. Nat Genet. 2015;47:535–538. doi: 10.1038/ng.3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shah S, Schrader KA, Waanders E, et al. A recurrent germline PAX5 mutation confers susceptibility to pre-B cell acute lymphoblastic leukemia. Nat Genet. 2013;45:1226–1231. doi: 10.1038/ng.2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.He J, Abdel-Wahab O, Nahas MK, et al. Integrated genomic DNA/RNA profiling of hematologic malignancies in the clinical setting. Blood. 2016;127:3004–3014. doi: 10.1182/blood-2015-08-664649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Faham M, Zheng J, Moorhead M, et al. Deep-sequencing approach for minimal residual disease detection in acute lymphoblastic leukemia. Blood. 2012;120:5173–5180. doi: 10.1182/blood-2012-07-444042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Qin H, Cho M, Haso W, et al. Eradication of B-ALL using chimeric antigen receptor-expressing T cells targeting the TSLPR oncoprotein. Blood. 2015;126:629–639. doi: 10.1182/blood-2014-11-612903. [DOI] [PMC free article] [PubMed] [Google Scholar]