

SUMMARY
Blood neutrophils perform an essential host-defense function by directly migrating to bacterial invasion sites to kill bacteria. The mechanisms mediating the transition from the migratory to bactericidal phenotype remain elusive. Here, we demonstrate that TRPM2, a trp superfamily member, senses neutrophil-generated reactive oxygen species and restrains neutrophil migration. The inhibitory function of oxidant sensing by TRPM2 requires the oxidation of Cys549, which then induces TRMP2 binding to formyl peptide receptor 1 (FPR1) and subsequent FPR1 internalization and signaling inhibition. The oxidant sensing-induced termination of neutrophil migration at the site of infection permits a smooth transition to the subsequent microbial killing phase.
In Brief
The molecular switches that turn a chemotactic neutrophil into a microbial killing machine are still unclear. Wang et al. show that, upon reaching the site of infection, increased ROS production by neutrophils—via a respiratory burst sensed by neutrophil TRPM2—stops cell migration and promotes a switch to microbial killing.
INTRODUCTION
Inflammation is an evolutionarily conserved process that protects the host from microbial pathogens and repairs injury-induced damage in tissues (Nathan, 2002). The evolutionary process has inextricably linked inflammation to its own resolution by generating necessary and highly efficient mechanisms to avoid tissue destruction, healing impairment and chronic inflammation (Nathan, 2002). Neutrophils, the first responders during inflammation, are essential for killing microbes, but they can also damage tissue (Kolaczkowska and Kubes, 2013). The orchestrated recruitment of neutrophils to sites of inflammation involves discrete steps, consisting of migration (diapedesis and chemotaxis) and microbial killing (phagocytosis and oxidative burst) (Butcher, 1991; Kolaczkowska and Kubes, 2013; Ley et al., 2007; Muller, 2011). Each step is precisely regulated by positive and negative feedback pathways to achieve efficient bacterial killing while minimizing damage to the host (Foxman et al., 1997; Heit et al., 2002; Liu et al., 2012, 2015; Lokuta and Huttenlocher, 2005). The signals that regulate the transition from the migratory phase (a crucial step in which neutrophils precisely reach the site of infection) to the subsequent bacterial killing phase remain unknown.
On reaching the site of infection, neutrophils engulf and kill pathogens primarily through the generation of reactive oxygen species (ROS) (Kolaczkowska and Kubes, 2013). Although ROS are bactericidal at high levels, they regulate neutrophil migration at low concentrations (Nathan and Cunningham-Bussel, 2013; Niethammer et al., 2009; Yoo et al., 2011). This was demonstrated in zebrafish by the observation that an extracellular gradient of epithelial cell-generated hydrogen peroxide (H2O2) at an injury site induces neutrophil recruitment to the wound site (Niethammer et al., 2009). In this system, the Src kinase Lyn senses locally generated H2O2 in tissue and directs a group of neutrophils to the injury site (Yoo et al., 2011). However, neutrophil-generated ROS were shown to be neither required for the extracellular H2O2 gradient nor sufficient for neutrophil recruitment to the wound (Niethammer et al., 2009). In addition, studies have demonstrated that mice lacking the p47phox or gp91phox subunits of NADPH oxidase exhibit higher neutrophil infiltration levels than WT mice (Gao et al., 2002; Pollock et al., 1995). However, directional neutrophil migration is either unaffected or reduced when ROS generation by neutrophils is inhibited (Hattori et al., 2010; Zicha et al., 1997). Thus, while neutrophil-generated ROS can regulate neutrophil migration, the underlying signaling mechanisms and ROS substrates that mediate neutrophil migration in response to chemoattractants have not been identified.
Transient receptor potential melastatin 2 (TRPM2) is an intracellular ROS sensor that transduces information to activate Ca2+ influx and regulate the membrane potential (Clapham, 2003). TRPM2 plays a role in regulating several key functions of inflammation, including specific aspects of the innate immune response (Knowles et al., 2011; Yamamoto et al., 2008), endothelial barrier integrity (Di et al., 2012; Dietrich and Gudermann, 2008; Hecquet et al., 2010), and apoptosis (Sumoza-Toledo et al., 2011). Binding of the second messengers ADP ribose (ADPR), nicotinamide adenine dinucleotide (NAD), and H2O2 to TRPM2 activates gating through the TRPM2 channel (Hara et al., 2002; Perraud et al., 2001; Sano et al., 2001; Wehage et al., 2002). Importantly, these activating signals are linked to the cellular redox state (Heiner et al., 2003). Other studies have also indicated that NAD or H2O2 may not activate TRPM2 directly (Buelow et al., 2008; Perraud et al., 2005; Toth and Csanady, 2010; Toth et al., 2015). Here, we addressed the role of the ROS-sensing function of TRPM2 in regulating neutrophil migration. We demonstrated that TRPM2-mediated ROS sensing negatively regulated neutrophil migration in a manner that was independent of its channel function. Oxidation of Cys549 in the TRPM2 N-terminal tail promoted ROS sensing, resulting in TRPM2 binding to formyl peptide receptor 1 (FPR1) and subsequent receptor internalization and migratory arrest. Importantly, this mechanism also applied to other tested chemoattractants. Thus, oxidation sensing by TRPM2 is crucial for mediating the transition from neutrophil migration to microbial killing, and mitigates tissue injury and inflammation induced by inappropriate neutrophil infiltration.
RESULTS
Deletion of TRPM2 Augments Neutrophil Transmigration and Neutrophilic Inflammation
Using the modified local Shwartzman reaction (LSR) (Brozna, 1990; Liu et al., 2015; Qian et al., 2009), we determined the function of TRPM2 in neutrophil-mediated vascular inflammation. Through subcutaneous dorsal skin injections, wild-type (WT) and Trpm2−/− mice were administered 80 μg of lipopolysaccharide (LPS) (Figure 1A, right side of each panel) or PBS as a control (Figure 1A, left side of each panel). After 24 hr, an equal volume of either tumor necrosis factor α (TNF-α) (0.2 μg) or PBS was injected subcutaneously at the same site that received LPS. LPS and TNF-α induced skin lesions resembling thrombo-hemorrhagic vasculitis, characterized by hemorrhage and dermal tissue necrosis at the injection sites (Figure 1C, right side of each panel). Compared with WT mice, Trpm2−/− mice displayed enhanced responses (Figures 1A and 1B). Histological examination showed increased erythrocyte extravasation and microcapillary thrombus formation at the LPS injection site in the dermal layers of Trpm2−/− mice compared with WT mice (Figure 1C). There was significantly increased neutrophil accumulation at the LPS injection site in Trpm2−/− mice, as measured by tissue myeloperoxidase (MPO) activity, compared with WT mice (Figure 1D, p < 0.01). Vascular injuries and skin lesions were not observed at the PBS injection sites in these mice.
Figure 1. Deletion of TRPM2 Enhances Neutrophil-Mediated Vascular Injury.

(A) LPS-induced microvascular injury in the classical local Shwartzman reaction (LSR) induced by consecutive injections of LPS followed by TNF-α. n = 3–4 mice per condition. Scale bar, 5 mm.
(B) The extent of hemorrhage was determined in WT and Trpm2−/− mice through densitometric analysis of skin samples receiving either LPS or PBS injections. Mean ± SEM. *p < 0.05.
(C) H&E-stained skin sections from WT (upper panels) and Trpm2−/− (lower panels) mice treated with LPS or PBS, as indicated, are shown. Erythrocyte extravasation, thrombus formation, and neutrophil accumulation are evident in the images from LPS-treated Trpm2−/− mice. Scale bar, 100 μm.
(D) Tissue MPO activity in the skin of mice from the WT and Trpm2−/− groups. Mean ± SEM. **p < 0.01.
(E) Tissue MPO activity in the lungs of WT and Trpm2−/− mice 4 hr after fMLF stimulation. Mean ± SEM. *p < 0.05 (Student’s t test). n = 3–4 mice per condition.
(F) Naphthol AS-D chloroacetate staining of lung sections from WT and Trpm2−/− mice obtained after fMLF challenge. No neutrophils were observed in untreated mice (left panels). fMLF-treated Trpm2−/− mice (right panels) showed greater neutrophil infiltration in the lungs. Arrowheads indicate infiltrating neutrophils. Scale bar, 50 μm.
Data are representative of (A), (C), and (F) or are from (B), (D), and (E) three independent experiments.
Next, we examined the role of TRPM2 in direct neutrophil infiltration in vivo. Intratracheal injection of a bacterial formyl peptide, fMet-Leu-Phe (fMLF), significantly increased neutrophil infiltration into the lungs, as determined by increased lung tissue MPO activity (Figure 1E). While basal MPO activity in Trpm2−/− lungs remained equal to that of WT mice, fMLF induced significantly greater MPO activity in Trpm2−/− lungs than in WT lungs (p < 0.05, Figure 1E). Histological sections of untreated lungs from either genotype showed normal structures and no evidence of neutrophil infiltration (Figure 1F). In response to fMLF challenge, Trpm2−/− mice, compared with WT mice, exhibited markedly greater increases in neutrophil sequestration in their lungs (Figure 1F). We observed similar effects following intratracheal injection of other attractants, including leukotriene B4 (LTB4) (Figure S2A), chemotactic factor C5a (Figure S2B), and inter-leukin-8 (IL-8) (Figure S2C), or direct injection of Pseudomonas aeruginosa (Figure S2D) bacteria.
We also performed adoptive transfer experiments in which both WT and Trpm2−/− neutrophils were isolated (the TRPM2 deletion in the murine neutrophils is shown in Figure S1A). Neutrophils were labeled with fluorescent dyes (carboxyfluorescein diacetate, succinimidyl ester) and injected intravenously into WT mice. Trpm2−/− neutrophil sequestration in the lungs was significantly higher than that of WT neutrophils after fMLF challenge (Figure S2E). Because WT mice were used in both groups, the difference represented the enhanced transmigratory ability of Trpm2−/− neutrophils.
TRPM2 Modulates Neutrophil Chemotaxis
We determined whether TRPM2 directly regulated neutrophil adhesion and migration, both of which are required for effective tissue infiltration by neutrophils. WT and Trpm2−/− neutrophils exhibited a capacity for adhesion similar to that of cultured WT murine lung vascular endothelial cells (Figure 2A). Studies show that formyl peptides induce neutrophils to adopt a polarized morphology and initiate migration after adhesion to endothelial cells or immobilized proteins, such as fibrinogen (Xu et al., 2003, 2008). WT and Trpm2−/− neutrophils exhibited normal polarized morphologies with filamentous actin (f-actin) accumulation in their pseudopods after formyl peptide treatments (fMLF or fMet-Ile-Phe-Leu [fMIFL], Figure 2B). The increment of f-actin was also not significantly different between the two groups of cells (Figure S1B). Thus, deletion of TRPM2 did not affect adhesion, polarization, or actin polymerization in neutrophils.
Figure 2. TRPM2 Inhibits Neutrophil Chemotaxis.
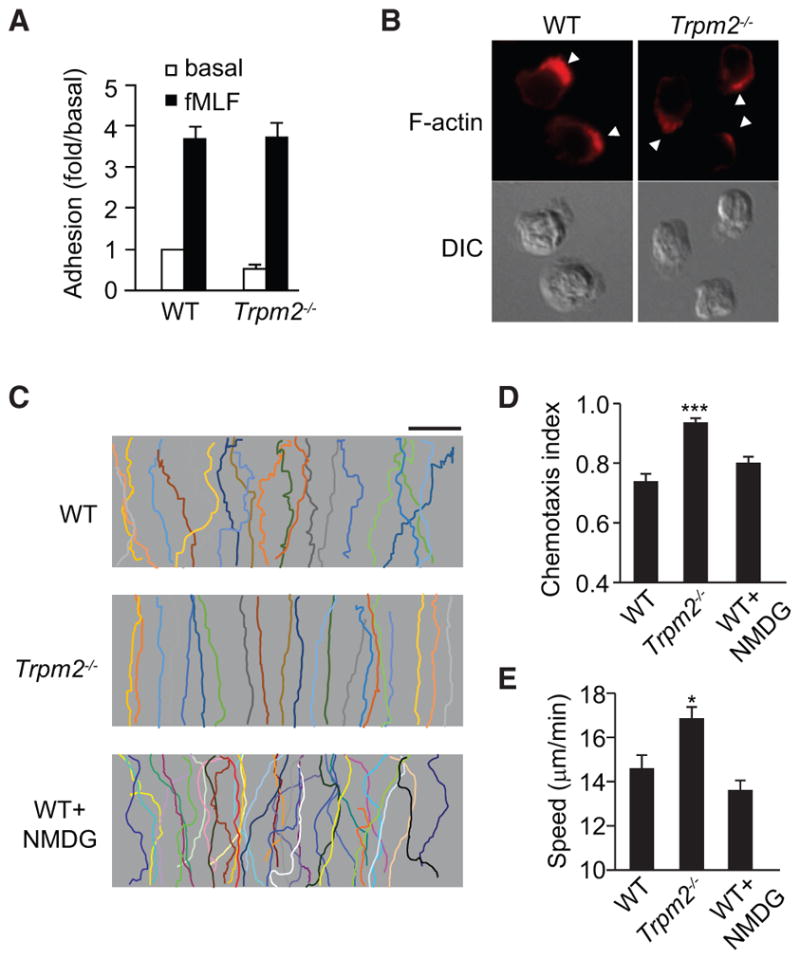
(A) Adhesion of WT and Trpm2−/− neutrophils to MLVECs after fMLF stimulation.
(B) Fluorescence microscopy (top row) and differential interference contrast (DIC) images (bottom row) of WT and Trpm2−/− neutrophils (n > 30 per group) stimulated with 100 nM fMLF for 2 min. Arrowheads indicate leading edges.
(C) Trajectories of WT and Trpm2−/− neutrophils or WT neutrophils treated with NMDG (145 mM) in an fMIFL gradient of 100 nM (>30 cells per condition). Each trace represents the trajectory of one cell. Scale bar, 100 μm.
(D and E) CI (D) and migration speed (E) of cells treated as described in (C). *p < 0.05, ***p < 0.01 compared with WT cells (Student’s t test).
Data are representative of (B) and (C) or are from (A), (D), and (E) three independent experiments mean and SEM in (A), (D), and (E).
We examined the migration of WT and Trpm2−/− neutrophils following the application of an fMIFL concentration gradient generated using an EZ-TAXIScan device. At the initial fMIFL concentration (10 nM), WT neutrophils migrated upward and reached the top of the attractant gradient (Figure 2C). However, compared with WT cells, Trpm2−/− neutrophils showed significantly enhanced directional migration with a straighter migration pattern comprising fewer zig-zags (Figure 2C). The chemotaxis index (CI) (the ratio of net migration in the correct direction to the total migration length [Xu et al., 2005]) was significantly higher in Trpm2−/− neutrophils than in WT cells (p < 0.001, Figure 2D). Similar results were obtained when cells were pre-primed with LPS (Figure S2F), challenged with different attractants, such as C5a (Figure S2G) and IL-8 (Figure S2H), or challenged with higher doses of formyl peptides, which normally induce early migratory arrest (Liu et al., 2012) (Figures S2I and S2J), or when cellular TRPM2 was knocked down (TRPM2 RNAi-treated HL60 cells, Figure S1C). However, deletion of another TRP channel, TRPC4, had no effect on neutrophil migration (Figure S1D).
TRPM2 Regulation of Cell Migration Is Independent of Channel Function
To address whether the TRPM2 cation channel function was responsible for the increased neutrophil migration observed in Trpm2−/− neutrophils, we blocked cation transportation through TRPM2 using an organic monovalent cation, N-methyl-D-glucamine (NMDG) (145 mM) (Di et al., 2012). WT cells treated with NMDG exhibited a CI and migration speed similar to that of untreated control cells (Figures 2C–2E), suggesting that the inhibitory function of TRPM2 in relation to neutrophil infiltration and migration was not dependent on the TRPM2 channel function.
To address the channel-independent function of TRPM2 in mediating neutrophil migration, we also recorded the fMLF-induced whole-cell cation current using the patch-clamp technique. The results indicated that the high concentration of fMLF (10 μM) induced a nonselective cation current, but the low concentration (100 nM, which was sufficient for neutrophil migration) failed to induce the current (Figures 3A–3C). We also observed similar patterns of Ca2+ release and Ca2+ influx in WT and Trpm2−/− neutrophils stimulated with fMLF (Figure 3D), further showing that TRPM2 regulated neutrophil migration through a Ca2+-channel-independent mechanism.
Figure 3. Channel-Independent Function of TRPM2 in Regulating Neutrophil Migration.
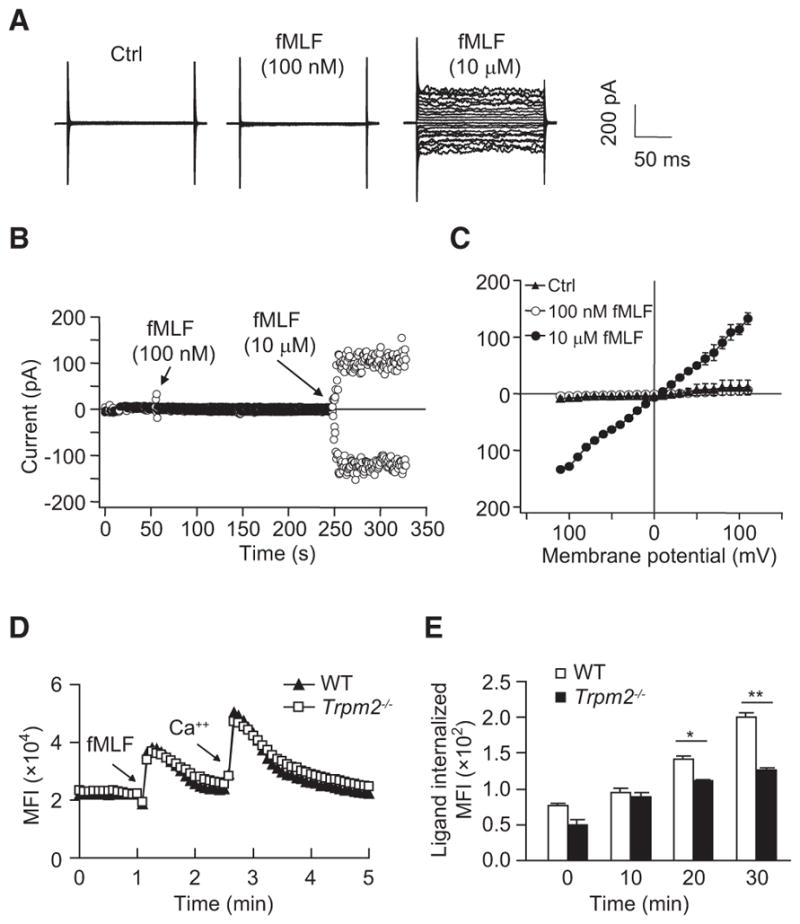
(A) High fMLF concentration-induced nonselective cation current in HL60 cells. The raw whole-cell current induced with fMLF (100 nM or 10 μM) is shown.
(B) Time course of the fMLF (100 nM or 10 μM)-induced current under a clamped membrane potential (+100 mV for outward current and −100 mV for inward current). The low fMLF (100 nM) concentration did not induce a cation current under these experimental conditions.
(C) Summary of the results obtained in (A) and (B). The summary is presented as the current amplitudes for different testing voltages (I–V plot). Mean ± SEM.
(D) Effects of fMLF on [Ca2+]i in WT and Trpm2−/− neutrophils. The extracellularCa2+ concentration was 1.26 mM. Arrows indicate when fMLF (100 nM) or Ca2+ (2 mM) was added.
(E) Receptor internalization in WT and Trpm2−/− neutrophils stimulated with 100 nM fMLF at various time points, presented as the mean fluorescence intensity (MFI) of the internalized ligand. Mean ± SEM. *p < 0.05, **p < 0.01 (Student’s t test).
Data are representative of (A), (B), and (D) or are from (C) and (E) three independent experiments.
Chemotactic receptor internalization is an important mechanism for suppressing signals that mediate neutrophil migration (Liu et al., 2012). We showed that the inhibition of this mechanism promotes chemotactic migration (Liu et al., 2012), similar to the response observed in the TRPM2 knockout cells described above (Figure 2). To further determine how TRPM2 regulates neutrophil migration, we examined receptor internalization dynamics in WT and Trpm2−/− neutrophils. We found the typical attractant-induced FPR1 internalization pattern in WT neutrophils, but receptor internalization was markedly reduced in Trpm2−/− neutrophils (Figure 3E). These findings suggest an important role for TRPM2 in regulating neutrophil migration by controlling FPR cell surface expression.
The Interaction of TRPM2 with FPR1 Downregulates Receptor Signaling
To investigate the role of TRPM2 in regulating FPR1 cell surface localization, we assessed the interaction of TRPM2 with FPR1. We found that endogenous TRPM2 interacted with FPR1 in HL60 cells (Figure 4A). To determine the sites responsible for the TRPM2 interaction with FPR1, we constructed hemagglutinin (HA)-tagged N-terminal (amino acids [aa] 1–752) and C-terminal (aa 1,047–1,503) fragments of TRPM2 and co-expressed these fragments with FLAG-tagged FPR1 in HEK293T cells for immunoprecipitation assays. FPR1 precipitated with the TRPM2 N-terminal fragment but not with the C-terminal fragment (Figure 4B).
Figure 4. TRPM2 Induces FPR1 Internalization.

(A) Immunoblot (IB) analysis of the endogenous FPR1 and TRPM2 interaction in HL60 cells.
(B) IB analysis of the interaction between HA-FPR1 and the FLAG-tagged full-length, N-terminal, or C-terminal fragment of TRPM2 in HEK293 cells.
(C) Fluorescence images of HEK293 cells expressing FPR1-YFP and CFP-TRPM2 show acceptor (YFP), donor (CFP), and net FRET signals before and after treatment with 100 nM fMLF. The scale for the net FRET images was color coded to represent a pixel intensity unit range of 0–661. Images were collected and analyzed as described in Experimental Procedures. The images shown are 90 μm2.
(D and E) Mean FRET intensity (D) and ratio of the mean FRET intensity (E) in the cytosol versus the plasma membrane before and after fMLF, as shown in (C). Mean ± SEM. **p < 0.01, ***p < 0.001 (Student’s t test).
Data are representative of three independent experiments (A–C) or three experiments (D and E).
We also performed a fluorescence resonance energy transfer (FRET) analysis using HEK293 cells expressing TRPM2 with a CFP tag on its N-terminal tail (CFP-TRPM2, donor) and FPR1 with a YFP tag on its C terminus (FPR1-YFP, receptor). After calculating the contribution from the crosstalk of each fluorophore in the FRET channel, we performed imaging of cells before and after stimulation with 100 nM fMLF for 5 min. The FRET intensity was localized to the cell membrane prior to fMLF stimulation (77% of the total FRET intensity, Figure 4C), indicating that most FPR1-TRPM2 interactions were localized to the cell membrane. However, after stimulation with fMLF, the FRET signal at the cell membrane was weaker (28% of the total FRET intensity), and the signal became diffuse throughout the cell body (Figure 4C), although the overall FRET intensity was increased (Figure 4D). The FRET signal ratio (cytosol/membrane) was significantly increased in cells exposed to fMLF compared with that in unstimulated cells (p < 0.001, Figure 4E), indicating that the interacting TRPM2-FPR1 proteins translocated from the membrane to the cytosol after fMLF stimulation. Similar results were obtained in HL60 cells using the proximity ligation assay (PLA) (Figures 5A–5C).
Figure 5. Proximity Ligation Assay Assessment of the Interaction between TRPM2 and FPR1.
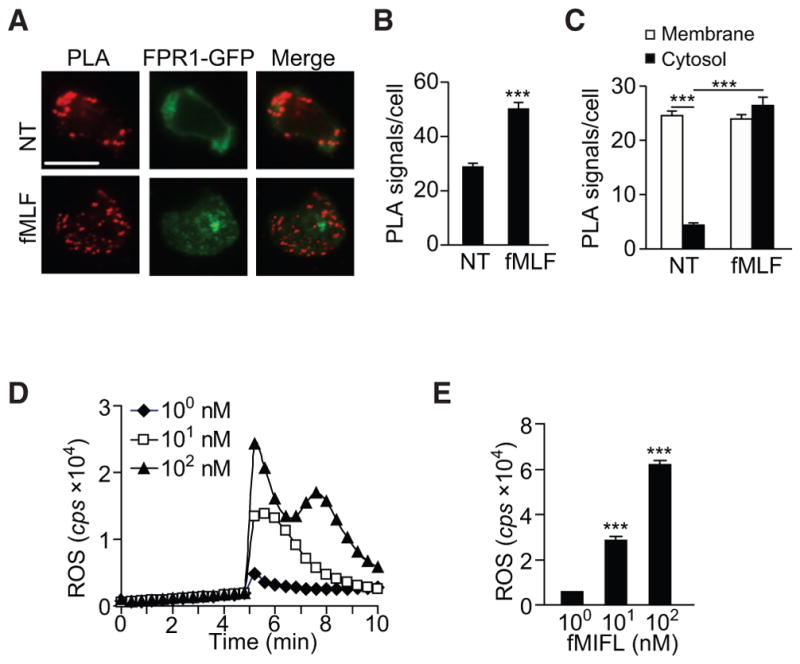
(A) Proximity ligation assay (PLA) detection of the TRPM2 and FPR1 interaction in stable FPR1-GFP-expressing HL60 cells before and after fMLF stimulation. Each red dot represents one FPR1 and TRPM2 interaction in HL60 cells. Scale bar, 10 μm.
(B and C) Mean PLA signal/cell (B) and the ratio of the mean PLA signal in the cytosol versus the plasma membrane (C) before and after fMLF, as shown in (A). Mean ± SEM. ***p < 0.001 (Student’s t test).
(D and E) Superoxide generation in WT mouse neutrophils after stimulation with different concentrations of fMIFL (1, 10, or 100 nM), determined in real time (D) or in total (E) based on luminol-ECL. cps, counts per second of light emitted. ROS, reactive oxygen species. Mean ± SEM. ***p < 0.001.
Data are representative of three independent experiments (A and D) or three experiments (B, C, and E).
The FRET experiments described above were supported by several controls. FRET images obtained from cells expressing CFP-TRPM2 or FPR1-YFP alone showed no FRET signal at the cell membrane (Figure S3). In addition, placement of the CFP tag on the C terminus (TRPM2-CFP) followed by co-expression with FRP1-YFP also resulted in no appreciable FRET signal (Figure S3). This finding confirmed the importance of the TRPM2 N terminus in mediating the TRPM2 interaction with FPR1.
TRPM2 Senses ROS through Oxidation at Cys549
Because TRPM2 is an ROS sensor (Clapham, 2003), we examined whether TRPM2 senses neutrophil-generated ROS to induce FPR1 internalization and thereby halt migration. Neutrophil ROS generation increased when neutrophils were stimulated with increasing concentrations of the formyl peptides (Figures 5D and 5E). After inhibiting cellular ROS with the NADPH oxidase inhibitor, diphenyleneiodonium (DPI) (10 μM for 30 min), we observed that the FRET signal corresponding to the TRPM2-FPR1 interaction persisted at the cell membrane after stimulation (Figure 6A). The FRET signal ratio (cytosol/membrane) was significantly lower in cells treated with DPI compared with untreated control cells (p < 0.05, Figure 6B). Thus, inhibition of ROS production prevented attractant-induced receptor internalization. This finding was reinforced by the direct measurement of receptor internalization in WT neutrophils treated with DPI (Figure 6C).
Figure 6. TRPM2 Senses ROS through Oxidation of Cys549.

(A) FRET images of HEK293 cells expressing FPR1-YFP and CFP-TRPM2 in the presence and absence of DPI (10 mM, 30 min) or expressing FPR1-YFP and CFP-TRPM2-C549A before and after treatment with 100 nM fMLF. The scale in the net FRET images was color coded to represent a pixel intensity unit range of 0–661. The images shown are 90 μm2.
(B) The ratio of the mean FRET intensity in the cytosol versus the plasma membrane before and after fMLF, as shown in (A). Mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 (Student’s t test).
(C) Receptor internalization in neutrophils treated with DPI (10 μM, 30 min) and stimulated with 100 nM fMLF is presented as the counts per minute (cpm) of internalized ligand. Mean ± SEM. *p < 0.05 (Student’s t test).
(D) Receptor internalization in WT-TRPM2- or TRPM2-C549A-expressing HL60 cells stimulated with 100 nM fMLF for 20 min is presented as the counts per minute (cpm) of internalized ligand. Mean ± SEM. ***p < 0.001 (Student’s t test).
(E and F) CI (E) and migration speed (F) of WT-TRPM2- or TRPM2-C549A-expressing HL60 cells migrating in an fMLF gradient of 100 nM. Mean ± SEM. **p < 0.01 compared with WT cells (Student’s t test).
Data are representative of three independent experiments (A) or from three experiments (B–F).
Next, we conducted experiments to characterize the TRPM2 oxidant-sensing domain responsible for stopping migration. A previous study showed that a TRPM2 mutant with a 20-aa deletion in the N-terminal region (aa 537–556) did not response to ROS, and its channel function was abolished (Wehage et al., 2002). Because the aa 537–556 sequence contains one cysteine residue (Cys549) that may be reversibly oxidized by H2O2, we mutated Cys549 to Ala and determined whether the mutated TRPM2 (TRPM2-C549A) affected FPR1 internalization. While FRET showed that TRPM2-C549A still interacted with FPR1 at the cell membrane, the signal failed to translocate to the cytosol after fMLF stimulation (Figure 6A). The FRET signal ratio (cytosol/membrane) was significantly lower in cells expressing TRPM2-C549A than in cells expressing WT TRPM2 (p < 0.01, Figure 6B). Therefore, the TRPM2 C549A mutation prevented attractant-induced receptor internalization, indicating that Cys549-mediated ROS sensing was required for attractant receptor internalization and neutrophil arrest.
We validated these results by directly measuring receptor internalization in HL60 cells expressing WT-TRPM2 or TRPM2-C549A (Figure 6D). Importantly, the cells expressing TRPM2-C549A exhibited enhanced cell migration, with a higher CI and speed than that of cells expressing WT-TRPM2 (Figures 6E and 6F). Therefore, prevention of TRPM2 Cys549 oxidation inhibited receptor internalization and enhanced cell migration, an outcome similar to that in the TRPM2 knockout cells.
H2O2 Induces Oxidation of TRPM2 Cys549
To assess the nature of H2O2-induced oxidation, we purified TRPM2 and treated it with different concentrations of H2O2 (0, 300, and 900 μM). The samples were analyzed for potential oxidation sites by mass spectrometry (MS). The -SH group on a cysteine is initially oxidized to sulfenic acid (SOH) and then di-oxidized into sulfinic acid (SO2H), or tri-oxidized into sulfonic acid (SO3H) (Reddie and Carroll, 2008). H2O2-mediated stimulation induced both di-oxidation and tri-oxidation of Cys549 (Figure 7A). Tri-oxidation was dominant in the tested samples, partly because there was an interference peak when di-oxidation was detected. Increasing the concentration of H2O2 increased the tri-oxidation of Cys549 (Figure 7B). Mono-oxidation was not detected. The fairly reactive state of the mono-oxidized cysteine may have facilitated its reduction to nonoxidized cysteine, induced by the reducing protocol used for sample preparation.
Figure 7. H2O2 Induces Oxidation of TRPM2 Cys549.

(A) Tandem MS (MS/MS) spectra of peptides with Cys549 oxidation. The upper panel is an example of Cys549 di-oxidation, and the lower panel an example of Cys549 tri-oxidation. The peptide sequence is shown (V539–R555). Red and blue bars represent the peptide’s N-terminal (b series) and C-terminal (y series) fragment ions, respectively, within the MS/MS spectrum.
(B) Relative abundance of Cys549 tri-oxidations in samples treated with 0, 300, and 900 μM H2O2. The area of each peak denotes the peptide intensity in the MS.
(C) Whole-cell currents induced by H2O2 or ADPR were recorded in HL60 cells transfected with WT-TRPM2 or TRPM2-C594A using the patch-clamp technique. H2O2 was added to the bath solution (300 μM) and ADPR was added to the pipette solution (500 μM). Mean ± SEM.
Data are representative of (A) and (B) or are from three independent experiments (C).
To address the role of Cys549 in regulating TRPM2 channel function, we recorded the H2O2-induced whole-cell cation current in HL60 cells expressing WT-TRPM2 or TRPM2-C549A. H2O2 (300 μM) induced a nonselective cation current in HL60 cells expressing WT-TRPM2 (Figure 7C). However, cells expressing TRPM2-C549A hardly showed any H2O2-induced currents compared with those of the WT controls (Figure 7C). The ADPR-induced current was not altered in either WT or mutant cells because the ADPR-responsive region was located in the TRPM2 C-terminal domain (Figure 7C).
DISCUSSION
We have previously described the importance of a neutrophil “stop” mechanism that is activated in response to chemoattractant during bacterial killing (Liu et al., 2012). This “stop” mechanism enables precise migration of neutrophils to sites of infection, but its mechanistic basis has remained unknown. The TRPM subfamily of trp channels is named for melastatin (TRPM1), a tumor-suppressor protein (Duncan et al., 1998). Here, we reveal a role for TRPM2 in an anti-inflammatory mechanism through oxidation on its N-terminal Cys549 residue. The sensing of ROS (generated in response to chemoattractant stimulation) by TRPM2 halted neutrophil migration through the interaction of oxidatively modified TRPM2 with the chemoattractant receptor FPR1, subsequently inducing receptor internalization and inactivating the signaling process responsible for neutrophil migration. We observed that oxidant production sensed by TRPM2 inhibited the migration of neutrophils in response to formyl peptides, IL-8, C5a, LTB4, and bacteria. The TRPM2 oxidant-sensing machinery functioned regardless of the chemo-attractants used here, suggesting that oxidant sensing by TRPM2 is a general mechanism through which neutrophils can precisely target sites of infection.
We showed that the function of TRPM2 in regulating neutrophil migration was independent of its cation channel activity. TRPM2 channel activation requires a high ROS concentration, usually within the micromolar to millimolar range (Hara et al., 2002; Wehage et al., 2002). This concentration is only achieved during an oxidative burst in the late stage of neutrophil activation (Heiner et al., 2003). The low concentrations of attractant (e.g., 10–100 nM fMLF) used in our experiments were sufficient to induce robust neutrophil transmigration and chemotaxis (Liu et al., 2012; Xu et al., 2003, 2008) but did not induce the characteristic oxidative burst (Bender et al., 1983; Herrmann et al., 2007; Sullivan et al., 1987).
A previous study revealed decreased migration of murine neutrophils with functionally defective TRPM2 channels (Yamamoto et al., 2008). The explanation for this discrepancy probably lies in the different mouse models used (TRPM2 knockout mice versus mice expressing functionally defective TRPM2). Although TRPM2 channel activity was nonexistent in both groups of mice, TRPM2 protein expression differed. Using an antibody against the TRPM2 N terminus, we detected low TRPM2 levels in neutrophils obtained from the Trpm2−/− mice used in this study (Figure S1A), whereas TRPM2 was still expressed in the mice used in the previous study (Yamamoto et al., 2008 and our unpublished data). It is possible that residual TRPM2 (with a defective channel function) is sufficient to impair migration. Using HL60 cells expressing the N-terminal region (aa 1–752) of TRPM2 (without the channel domains), we also observed increased receptor internalization and impaired cell migration (our unpublished data), similar to the previously reported phenotypes (Yamamoto et al., 2008). These findings together are consistent with the model in which TRPM2 senses ROS to modulate neutrophil migration through a channel-independent mechanism. However, we cannot rule out the possibility that the TRPM2 channel function also plays a critical role in the microbial killing phase. Upon reaching the site of infection, high concentrations of chemoattractants or ROS (generated during the respiratory burst) activate the TRPM2 channel (Hara et al., 2002; Wehage et al., 2002), thereby inducing calcium influx to activate additional bactericidal pathways (Knowles et al., 2011).
Although TRPM2 is a known oxidant sensor (Clapham, 2003), its ROS-sensing sites had not been identified until now. Additional mechanisms have also been proposed by which oxidants may activate TRPM2 channel through ADPR (Buelow et al., 2008; Perraud et al., 2005). Our data highlight the importance of the Cys549 residue in oxidant sensing to regulate neutrophil migration. The Cys549 to Ala mutation enhanced neutrophil migration and inflammation and abolished the H2O2-induced current through TRPM2, suggesting a key role for this residue. Using MS, we observed that Cys549 was tri-oxidized upon H2O2 stimulation. Tri-oxidation was also observed on Cys693, yet mutation of Cys693 to Ala had little or no effect on neutrophil migration (our unpublished data). Although TRPM2 interacted with FPR1, only Cys549-oxidized TRPM2 promoted receptor internalization. We also observed that membrane-bound GRK2 (active state) was lower in TRPM2-knockout neutrophils and TRPM2-C549A-expressing HL60 cells than in the WT controls (our unpublished data), suggesting that oxidatively modified TRPM2 may induce receptor internalization through a mechanism involving GRK2. In addition, these results did not rule out the possibility that ADPR is also involved in ROS-mediated TRPM2 activation. For example, fMLF or ROS may stimulate ADPR release (Heiner et al., 2006), and thus it is also possible that TRPM2 can be co-activated by the subsequently released ADPR.
ROS have variable effects on cell migration that are dependent on ROS levels (Kuiper et al., 2011; Nathan and Cunningham-Bussel, 2013). Low ROS levels may promote cell migration by inactivating the phosphatase PTEN, allowing phosphatidylinositol-3,4,5-trisphosphate to accumulate at the leading edge of migrating cells (Chaubey et al., 2013; Kuiper et al., 2011). In addition, H2O2 generated by epithelial cells in zebrafish mediates neutrophil recruitment to wounds (Niethammer et al., 2009; Yoo et al., 2011). The present results show a causal relationship between neutrophil-generated ROS, sensed by TRPM2, and the suppression of neutrophil migration. Although we propose that the actions of ROS on TRPM2 are mechanistically important for the suppression of neutrophil migration, the hormetic nature of ROS makes it impossible to rule out other inhibitory pathways activated by different ROS concentrations. For example, high levels of ROS promote the accumulation of glutathionylated actin and may subsequently impair actin polymerization and cell migration (Dalle-Donne et al., 2003; Nathan and Cunningham-Bussel, 2013; Wang et al., 2001).
In conclusion, neutrophil-generated ROS, sensed by TRPM2, elicited a crucial signal to halt neutrophil migration, enabling these cells to begin the bacterial killing phase. Thus, our findings suggest that the important immunomodulatory role of TRPM2 represents a target for optimizing the bactericidal function of neutrophils. It is possible that pharmacologically enhancing this function would have the potential to mitigate neutrophil-induced inflammatory injury.
EXPERIMENTAL PROCEDURES
Antibodies, Reagents, and Mice
Rabbit anti-TRPM2 antibody (ab87050) was from Abcam (against N-terminal amino acids 432–481 of TRPM2). Mouse anti-GRK2 monoclonal antibody (C-9, sc-13143) and rabbit polyclonal antibody for FPR1 (H-230, sc-30016) were purchased from Santa Cruz Biotechnology. Rabbit monoclonal antibody to phosphorylated p38 MAPK (3D7, #9215) was from Cell Signaling Technology. The green-fluorescent fluorescein-conjugated hexapeptide of formyl-Nle-Leu-Phe-Nle-Tyr-Lys (F1314) was from Life Technologies. fMLF, fMIFL, protease inhibitors, phosphatase inhibitor “cocktails,” and normal molecular chemicals were purchased from Sigma-Aldrich. The FLIPR Calcium 5 Assay Kit was from Molecular Devices.
WT C57BL/6 mice were from Charles River Laboratories. Trpm2−/− mice (obtained from B.A. Miller) were generated and originally provided by GlaxoSmithKline (Knowles et al., 2011). Mice were bred and housed in a pathogen-free animal care facility at the University of Illinois at Chicago. All animal experiments were performed under the protocol approved by The Institutional Animal Care and Use Committee of the University of Illinois at Chicago.
Cell Migration and Adhesion Assays
Live cells were imaged after stimulation with either a uniform concentration of fMLF or a concentration gradient generated by the EZ-TAXIScan device. EZ-TAXIScan assay was described previously (Liu et al., 2012). In brief, cells migrated over a 50-μg/mL fibronectin or fibrinogen-coated coverglass on a horizontal glass surface under a silicon chip. Cells were washed with RPMI, 25 mM HEPES (pH 7.0), and 0.1% BSA, and resuspended in a solution containing RPMI, 25 mM HEPES, and 0.1% BSA. Cells were loaded on the bottom of the chip, and the chemoattractant was added to the top of the chip to generate a chemoattractant gradient. Cells migrated for 30 min, and images were recorded with the EZ-TAXIScan software and then analyzed using ImageJ software.
For neutrophil transmigration, age-matched male WT and TRPM2 knockout mice were anesthetized with intraperitoneal injection of a mixture of ketamine (100 mg/kg) and xylazine (10 mg/kg). The trachea was exposed and injected with 30 μL of 10 nM fMLF or 10 nM fMIFL in PBS. Control group mice were injected with the same volume of PBS. The cut skin was closed with suture. Four hours later, the mice were anesthetized again and the lung tissues were flushed and removed for staining or MPO activity assay.
For cell adhesion, mouse lung vascular endothelial cells (MLVECs) were isolated and grown to confluence in 96-well gelatin-coated plates. Neutrophils loaded with calcein-AM were added to MLVECs and the assays for polymorphonuclear leukocyte adhesion and migration on endothelial cells was performed as described by Xu et al. (2008).
Whole-Cell Recordings
Whole-cell current induced by fMLF was measured using an EPC-10 patch-clamp amplifier (HEKA) using the Pulse V 8.8 acquisition program (HEKA). Currents were elicited by using a series of test pulses ranging from −110 to +110 mV with holding potential set at 0 mV (test pulses were 200 ms in duration and delivered at 2-s intervals). The pipette solution contained 135 mM CsSO3CH3, 8 mM NaCl, 2 mM MgCl2, and 10 mM HEPES (pH 7.2); the bath solution comprised 145 mM NaCl, 2 mM CaCl2, 1 mM MgCl2, and 10 mM HEPES (pH 7.4); and the NMDG-containing bath solution consisted of 150 mM NMDG-Cl, 1 mM MgCl2, and 10 mM HEPES (pH 7.4). Whole-cell currents were analyzed using IGOR software (WaveMetrics).
FRET Analysis
Images were obtained using a Zeiss LSM 510 Meta confocal microscope equipped with an 80-mW UV laser and a 25-mW Kr/Ar laser, mounted on an Axiovert 200M microscope stand, and using an alpha Plan-Fluar 100× oil objective. For emission ratio imaging, the following filter sets were used: EYFP (514 nm excitation) HFT 458/514, NFT 515, LP 530; FRET (458 nm excitation) HFT 454/514, MFT 515, LP 530; CFP (458 nm excitation) HFT 454/514, NFT 515, BP 500/20 IR. Cells were imaged in a single focal plane at a rate of 7.86 s/scan for each channel.
For image analysis, the contribution of spectral overlap of EYFP and ECFP fluorescence into the FRET channel were quantified using singly transfected constructs (YFP-FPR or CFP-TRPM2). Using MetaMorph software, cell membranes were traced and the total integrated pixel intensity was quantified for this zone in CFP, FRET, and YFP channels. The ratio of FRET intensity to fluorophore intensity was σ1 = 0.50 ± 0.024 for CFP and σ2 = 0.30 ± 0.03 for YFP. For doubly transfected cells, the net FRET signal was quantified and visualized as FRETnet = σ1FRETtotal − (σ1FRETCFP − σ2FRETYFP). Relative distribution of FRET intensity on the membrane and in the cytosol was determined by tracing cell membranes using MetaMorph software, and the integrated pixel intensity per unit area was determined for the cytosol and membrane and plotted as the ratio.
PLA between TRPM2 and FPR1 in HL60 cells was carried out according to the protocol as described by Soderberg et al. (2006) using a Duolink PLA kit (Sigma).
Modified Local Shwartzman Reaction
Age-matched 8- to 12-week-old WT and Trpm2−/− mice were anesthetized by intraperitoneal injection of ketamine (100 mg/kg) and xylazine (10 mg/kg), and the dorsal skin was shaved. Eighty micrograms of LPS (O555:B5, Sigma) in 80 μL of PBS was injected into the right dorsum. As a negative control, 80 μL of PBS was injected into the left dorsum. Twenty-four hours later, 0.2 μg of TNF-α in 80 μL of PBS was injected into the same point on the right dorsum and 80 μL of PBS into the left dorsum. At 24 hr post TNF-α injection, the mice were euthanized and the interior of the dorsal skin was exposed for macroscopic examination. The tissues were either fixed in 10% formalin for histological analysis with H&E staining or frozen at −80°C for MPO activity assay.
Lung Tissue Myeloperoxidase Activity
Lung tissue MPO activity was measured as described by Garrean et al. (2006). In brief, skin or lung tissues were flushed free of blood by PBS and homogenized in 50 mM phosphate buffer (PB) (pH 6.0). The homogenates were centrifuged at 40,000 × g at 4°C for 30 min. After discarding the supernatants, the pellets were resuspended in PB buffer containing 0.5% hexadecyl trimethylammonium bromide and vigorously vibrated to break up the large pellets. The pellets were then frozen at −70°C for 30 min and thawed at 37°C. Subsequently the pellets were homogenized and centrifuged a second time. Thereafter the supernatants were used for MPO activity assay with a kinetics reading at 460 nm for 5 min. Neutrophil sequestration was quantified as MPO activity normalized by tissue weight and the data presented as Vmax value per gram of tissue.
Cell Culture, Transfection, and Isolation of Mouse Bone Marrow Neutrophils
HL60 cells were cultured in RPMI 1640 with 10% fetal bovine serum. For experiments, HL60 cells were differentiated by adding 1.3% DMSO into the medium for 7 days. To establish the stable cell lines, we subcloned TRPM2 N terminus, TRPM2 C terminus, TRPM2L, and TRPM2 mutant genes into lentivirus expression vectors. After virus packaging in HEK293T cells, HL60 cells were infected and screened by puromycin and further sorted by flow cytometry.
For bone marrow neutrophil isolation, mice were euthanized, and the femurs and tibias taken out and flushed by a 27-gauge needle with a 10-mL syringe filled with calcium and magnesium-free Hank’s balanced salt solution (HBSS) plus 0.1% BSA. Cells were then centrifuged and resuspended in HBSS. After filtering with a 40-μm strainer, cells in 3 mL of HBSS were loaded onto a pre-prepared gradient solution (3 mL of Nycoprep on the top and 3 mL of 72% Percoll on the bottom). The samples were centrifuged at 2,400 rpm at room temperature for 20 min with the brake off. The middle layer was collected and washed once in HBSS. Sterilized distilled water (9 mL) was added for 22 s and thereafter 1 mL of 10×PBS to remove the red blood cells. Finally, the cells were collected and resuspended in HBSS or medium (neutrophil purity >90%).
Ligand Internalization Assay
Mouse bone morrow neutrophils were isolated and incubated in modified HBSS (mHBSS) for ligand internalization assay. Cells were stimulated with fluorescein-labeled or 125I-labeled fMLF for the intended time points. A 50-fold volume of ice-cold PBS was added to the tube to stop the reaction. Following centrifugation and discarding the supernatant, 2 mL of pre-chilled stripping buffer was added and mixed for 5 min on ice to remove any peptide left on the cell surface. The samples were then washed three times with PBS and prepared for flow cytometry assay or radioactive count.
Neutrophil Superoxide Production
Neutrophils (1 × 106) in HBSS containing 0.5% BSA were seeded into a white, 96-well, flat-bottomed tissue culture dish (E&K Scientific). Superoxide generation by neutrophils was measured by using iso-luminol-enhanced chemiluminescence (Gao et al., 2007). In brief, iso-luminol was added to the cell suspension to a final concentration of 100 μM, and horseradish peroxidase was added to a final concentration of 40 units/mL. The cell suspension was then incubated at 37°C for 5 min. Superoxide production was determined by a WALLAC 1420 Multilabel Counter before and after cells were stimulated with fMLF. The relative concentration of oxidant in the culture medium was expressed as counts per second.
Calcium Assay
The FLIPR Calcium 5 assay kit from Molecular Devices was used in the calcium flux assay. Mouse bone marrow neutrophils were centrifuged and resuspended into mHBSS (20 mM HEPES, 10 mg/mL glucose, and 1% BSA) to a concentration of 2 × 106/mL. Cell suspension (100 μL) was seeded to each well of the 96-well plate, then the plate was centrifuged at 100 × g for up to 3 min with the brake off. After loading an equal volume of the dye, the plate was incubated in the incubator (37°C, 5% CO2) for 1 hr. To analyze the extracellular influx, we added 2.5 mM EGTA directly before reading on a flex station device.
Mass Spectrometry
FLAG-tagged TRPM2 was expressed in HEK293 cells and purified with the anti-FLAG M2 affinity gel, then treated with H2O2 (0, 300, and 900 μM) for 90 min. One microgram of protein sample for each concentration was taken out and prepared for MS analysis. Protein samples were sequentially reduced with DTT and alkylated with iodoacetamide, then digested with trypsin at 37°C overnight on Millipore Microcon YM-10 filters (10-kDa cutoff) based on filter-aided sample preparation protocol (Wisniewski et al., 2009). The digest was separated with a nanoLC system and detected in data-dependent analysis mode on an LTQ Orbitrap Fusion (Thermo Fisher Scientific). The liquid chromatography-MS data were searched against the SwissProt Human database using Sequest HT algorithm on the Proteome Discoverer 1.4 platform (Thermo Fisher). Searching parameters were set as such precursor mass tolerance at 20 ppm, fragment ion mass tolerance at 0.05 Da, trypsin enzyme with three miscleavages, variable modifications of oxidation at methionine, and oxidation, di-oxidation, tri-oxidation, and carbamidomethylation at cysteine. Peptides identified from the database search were filtered at 0.01 false discovery rate. Relative quantitation of oxidized cysteine-containing peptides was calculated based on areas under the curve of corresponding peptides from control and treated samples. Scaffold 4 was also used to visualize spectra.
Immunoprecipitation and Immunofluorescence Assays
Immunoprecipitation and immunofluorescence were performed as described by Xu et al. (2008). Densitometry of bands on autoradiography was performed with scanned X-ray films and ImageJ software. Results of at least three independent experiments are presented as a bar graph using a.u. to compare the intensity of the bands.
Statistical Analysis
All experiments were performed at least three times. Representative data are shown throughout. For comparisons in independent experiments a two-tailed Student’s t test was used in the statistical assay. Significant difference between samples was set at p < 0.05.
Supplementary Material
Highlights.
TRPM2 sensing of ROS shifts neutrophils from migration to bacterial killing
ROS sensing by TRPM2 is a central stop mechanism for neutrophil migration
TRPM2 suppresses neutrophil migration through a channel-independent mechanism
Cys549 is the ROS-sensing site on TRPM2
Acknowledgments
We thank GlaxoSmithKline for permission to use the Trmp2−/− mouse.
Footnotes
Supplemental Information includes three figures and can be found with this article online at http://dx.doi.org/10.1016/j.devcel.2016.07.014.
AUTHOR CONTRIBUTIONS
G.W., L.C., X.L., N.A.S., A.D., and J.X. designed research; G.W., L.C., X.L., N.A.S., A.D., S.T., and X.H. performed research; X.W., Y.C., and T.J. performed MS analysis; G.W., L.C., X.L., N.A.S., A.D., and J.X. analyzed data; C.T., Y.Z., Y.S., X.G., and C.B. contributed new reagents and tools; N.A.S., A.D., A.B.M., and J.X. wrote the paper.
References
- Bender JG, McPhail LC, Van Epps DE. Exposure of human neutrophils to chemotactic factors potentiates activation of the respiratory burst enzyme. J Immunol. 1983;130:2316–2323. [PubMed] [Google Scholar]
- Brozna JP. Shwartzman reaction. Semin Thromb Hemost. 1990;16:326–332. doi: 10.1055/s-2007-1002685. [DOI] [PubMed] [Google Scholar]
- Buelow B, Song Y, Scharenberg AM. The Poly(ADP-ribose) polymerase PARP-1 is required for oxidative stress-induced TRPM2 activation in lymphocytes. J Biol Chem. 2008;283:24571–24583. doi: 10.1074/jbc.M802673200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher EC. Leukocyte-endothelial cell recognition: three (or more) steps to specificity and diversity. Cell. 1991;67:1033–1036. doi: 10.1016/0092-8674(91)90279-8. [DOI] [PubMed] [Google Scholar]
- Chaubey S, Jones GE, Shah AM, Cave AC, Wells CM. Nox2 is required for macrophage chemotaxis towards CSF-1. PLoS One. 2013;8:e54869. doi: 10.1371/journal.pone.0054869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapham DE. TRP channels as cellular sensors. Nature. 2003;426:517–524. doi: 10.1038/nature02196. [DOI] [PubMed] [Google Scholar]
- Dalle-Donne I, Giustarini D, Rossi R, Colombo R, Milzani A. Reversible S-glutathionylation of Cys 374 regulates actin filament formation by inducing structural changes in the actin molecule. Free Radic Biol Med. 2003;34:23–32. doi: 10.1016/s0891-5849(02)01182-6. [DOI] [PubMed] [Google Scholar]
- Di A, Gao XP, Qian F, Kawamura T, Han J, Hecquet C, Ye RD, Vogel SM, Malik AB. The redox-sensitive cation channel TRPM2 modulates phagocyte ROS production and inflammation. Nat Immunol. 2012;13:621. doi: 10.1038/ni.2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich A, Gudermann T. Another TRP to endothelial dysfunction: TRPM2 and endothelial permeability. Circ Res. 2008;102:275–277. doi: 10.1161/CIRCRESAHA.107.170548. [DOI] [PubMed] [Google Scholar]
- Duncan LM, Deeds J, Hunter J, Shao J, Holmgren LM, Woolf EA, Tepper RI, Shyjan AW. Down-regulation of the novel gene mel-astatin correlates with potential for melanoma metastasis. Cancer Res. 1998;58:1515–1520. [PubMed] [Google Scholar]
- Foxman EF, Campbell JJ, Butcher EC. Multistep navigation and the combinatorial control of leukocyte chemotaxis. J Cell Biol. 1997;139:1349–1360. doi: 10.1083/jcb.139.5.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao XP, Standiford TJ, Rahman A, Newstead M, Holland SM, Dinauer MC, Liu QH, Malik AB. Role of NADPH oxidase in the mechanism of lung neutrophil sequestration and microvessel injury induced by Gram-negative sepsis: studies in p47phox−/− and gp91phox−/− mice. J Immunol. 2002;168:3974–3982. doi: 10.4049/jimmunol.168.8.3974. [DOI] [PubMed] [Google Scholar]
- Gao XP, Zhu X, Fu J, Liu Q, Frey RS, Malik AB. Blockade of class IA phosphoinositide 3-kinase in neutrophils prevents NADPH oxidase activation- and adhesion-dependent inflammation. J Biol Chem. 2007;282:6116–6125. doi: 10.1074/jbc.M610248200. [DOI] [PubMed] [Google Scholar]
- Garrean S, Gao XP, Brovkovych V, Shimizu J, Zhao YY, Vogel SM, Malik AB. Caveolin-1 regulates NF-kappaB activation and lung inflammatory response to sepsis induced by lipopolysaccharide. J Immunol. 2006;177:4853–4860. doi: 10.4049/jimmunol.177.7.4853. [DOI] [PubMed] [Google Scholar]
- Hara Y, Wakamori M, Ishii M, Maeno E, Nishida M, Yoshida T, Yamada H, Shimizu S, Mori E, Kudoh J, et al. LTRPC2 Ca2+-permeable channel activated by changes in redox status confers susceptibility to cell death. Mol Cell. 2002;9:163–173. doi: 10.1016/s1097-2765(01)00438-5. [DOI] [PubMed] [Google Scholar]
- Hattori H, Subramanian KK, Sakai J, Jia Y, Li Y, Porter TF, Loison F, Sarraj B, Kasorn A, Jo H, et al. Small-molecule screen identifies reactive oxygen species as key regulators of neutrophil chemotaxis. Proc Natl Acad Sci USA. 2010;107:3546–3551. doi: 10.1073/pnas.0914351107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecquet CM, Ahmmed GU, Malik AB. TRPM2 channel regulates endothelial barrier function. Adv Exp Med Biol. 2010;661:155–167. doi: 10.1007/978-1-60761-500-2_10. [DOI] [PubMed] [Google Scholar]
- Heiner I, Eisfeld J, Luckhoff A. Role and regulation of TRP channels in neutrophil granulocytes. Cell Calcium. 2003;33:533–540. doi: 10.1016/s0143-4160(03)00058-7. [DOI] [PubMed] [Google Scholar]
- Heiner I, Eisfeld J, Warnstedt M, Radukina N, Jungling E, Luckhoff A. Endogenous ADP-ribose enables calcium-regulated cation currents through TRPM2 channels in neutrophil granulocytes. Biochem J. 2006;398:225–232. doi: 10.1042/BJ20060183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heit B, Tavener S, Raharjo E, Kubes P. An intracellular signaling hierarchy determines direction of migration in opposing chemotactic gradients. J Cell Biol. 2002;159:91–102. doi: 10.1083/jcb.200202114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann JM, Bernardo J, Long HJ, Seetoo K, McMenamin ME, Batista EL, Jr, Van Dyke TE, Simons ER. Sequential chemotactic and phagocytic activation of human polymorphonuclear neutrophils. Infect Immun. 2007;75:3989–3998. doi: 10.1128/IAI.00388-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles H, Heizer JW, Li Y, Chapman K, Ogden CA, Andreasen K, Shapland E, Kucera G, Mogan J, Humann J, et al. Transient Receptor Potential Melastatin 2 (TRPM2) ion channel is required for innate immunity against Listeria monocytogenes. Proc Natl Acad Sci USA. 2011;108:11578–11583. doi: 10.1073/pnas.1010678108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013;13:159–175. doi: 10.1038/nri3399. [DOI] [PubMed] [Google Scholar]
- Kuiper JW, Sun C, Magalhaes MA, Glogauer M. Rac regulates PtdInsP(3) signaling and the chemotactic compass through a redox-mediated feedback loop. Blood. 2011;118:6164–6171. doi: 10.1182/blood-2010-09-310383. [DOI] [PubMed] [Google Scholar]
- Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7:678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- Liu X, Ma B, Malik AB, Tang H, Yang T, Sun B, Wang G, Minshall RD, Li Y, Zhao Y, et al. Bidirectional regulation of neutrophil migration by mitogen-activated protein kinases. Nat Immunol. 2012;13:457–464. doi: 10.1038/ni.2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Yang T, Suzuki K, Tsukita S, Ishii M, Zhou S, Wang G, Cao L, Qian F, Taylor S, et al. Moesin and myosin phosphatase confine neutrophil orientation in a chemotactic gradient. J Exp Med. 2015;212:267–280. doi: 10.1084/jem.20140508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lokuta MA, Huttenlocher A. TNF-alpha promotes a stop signal that inhibits neutrophil polarization and migration via a p38 MAPK pathway. J Leukoc Biol. 2005;78:210–219. doi: 10.1189/jlb.0205067. [DOI] [PubMed] [Google Scholar]
- Muller WA. Mechanisms of leukocyte transendothelial migration. Annu Rev Pathol. 2011;6:323–344. doi: 10.1146/annurev-pathol-011110-130224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan C. Points of control in inflammation. Nature. 2002;420:846–852. doi: 10.1038/nature01320. [DOI] [PubMed] [Google Scholar]
- Nathan C, Cunningham-Bussel A. Beyond oxidative stress: an immunologist’s guide to reactive oxygen species. Nat Rev Immunol. 2013;13:349–361. doi: 10.1038/nri3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niethammer P, Grabher C, Look AT, Mitchison TJ. A tissue-scale gradient of hydrogen peroxide mediates rapid wound detection in zebra-fish. Nature. 2009;459:996–999. doi: 10.1038/nature08119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perraud AL, Fleig A, Dunn CA, Bagley LA, Launay P, Schmitz C, Stokes AJ, Zhu Q, Bessman MJ, Penner R, et al. ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature. 2001;411:595–599. doi: 10.1038/35079100. [DOI] [PubMed] [Google Scholar]
- Perraud AL, Takanishi CL, Shen B, Kang S, Smith MK, Schmitz C, Knowles HM, Ferraris D, Li W, Zhang J, et al. Accumulation of free ADP-ribose from mitochondria mediates oxidative stress-induced gating of TRPM2 cation channels. J Biol Chem. 2005;280:6138–6148. doi: 10.1074/jbc.M411446200. [DOI] [PubMed] [Google Scholar]
- Pollock JD, Williams DA, Gifford MA, Li LL, Du X, Fisherman J, Orkin SH, Doerschuk CM, Dinauer MC. Mouse model of X-linked chronic granulomatous disease, an inherited defect in phagocyte superoxide production. Nat Genet. 1995;9:202–209. doi: 10.1038/ng0295-202. [DOI] [PubMed] [Google Scholar]
- Qian F, Deng J, Cheng N, Welch EJ, Zhang Y, Malik AB, Flavell RA, Dong C, Ye RD. A non-redundantrolefor MKP5inlimitingROS production and preventing LPS-induced vascular injury. EMBO J. 2009;28:2896–2907. doi: 10.1038/emboj.2009.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddie KG, Carroll KS. Expanding the functional diversity of proteins through cysteine oxidation. Curr Opin Chem Biol. 2008;12:746–754. doi: 10.1016/j.cbpa.2008.07.028. [DOI] [PubMed] [Google Scholar]
- Sano Y, Inamura K, Miyake A, Mochizuki S, Yokoi H, Matsushime H, Furuichi K. Immunocyte Ca2+ influx system mediated by LTRPC2. Science. 2001;293:1327–1330. doi: 10.1126/science.1062473. [DOI] [PubMed] [Google Scholar]
- Soderberg O, Gullberg M, Jarvius M, Ridderstrale K, Leuchowius KJ, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson LG, Landegren U. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods. 2006;3:995–1000. doi: 10.1038/nmeth947. [DOI] [PubMed] [Google Scholar]
- Sullivan R, Griffin JD, Simons ER, Schafer AI, Meshulam T, Fredette JP, Maas AK, Gadenne AS, Leavitt JL, Melnick DA. Effects of recombinant human granulocyte and macrophage colony-stimulating factors on signal transduction pathways in human granulocytes. J Immunol. 1987;139:3422–3430. [PubMed] [Google Scholar]
- Sumoza-Toledo A, Lange I, Cortado H, Bhagat H, Mori Y, Fleig A, Penner R, Partida-Sanchez S. Dendritic cell maturation and chemotaxis is regulated by TRPM2-mediated lysosomal Ca2+ release. FASEB J. 2011;25:3529–3542. doi: 10.1096/fj.10-178483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth B, Csanady L. Identification of direct and indirect effectors of the transient receptor potential melastatin 2 (TRPM2) cation channel. J Biol Chem. 2010;285:30091–30102. doi: 10.1074/jbc.M109.066464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth B, Iordanov I, Csanady L. Ruling out pyridine dinucleotides as true TRPM2 channel activators reveals novel direct agonist ADP-ribose-2′-phosphate. J Gen Physiol. 2015;145:419–430. doi: 10.1085/jgp.201511377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Boja ES, Tan W, Tekle E, Fales HM, English S, Mieyal JJ, Chock PB. Reversible glutathionylation regulates actin polymerization in A431 cells. J Biol Chem. 2001;276:47763–47766. doi: 10.1074/jbc.C100415200. [DOI] [PubMed] [Google Scholar]
- Wehage E, Eisfeld J, Heiner I, Jungling E, Zitt C, Luckhoff A. Activation of the cation channel long transient receptor potential channel 2 (LTRPC2) by hydrogen peroxide. A splice variant reveals a mode of activation independent of ADP-ribose. J Biol Chem. 2002;277:23150–23156. doi: 10.1074/jbc.M112096200. [DOI] [PubMed] [Google Scholar]
- Wisniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nat Methods. 2009;6:359–362. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- Xu J, Wang F, Van Keymeulen A, Herzmark P, Straight A, Kelly K, Takuwa Y, Sugimoto N, Mitchison T, Bourne HR. Divergent signals and cytoskeletal assemblies regulate self-organizing polarity in neutrophils. Cell. 2003;114:201–214. doi: 10.1016/s0092-8674(03)00555-5. [DOI] [PubMed] [Google Scholar]
- Xu J, Wang F, Van Keymeulen A, Rentel M, Bourne HR. Neutrophil microtubules suppress polarity and enhance directional migration. Proc Natl Acad Sci USA. 2005;102:6884–6889. doi: 10.1073/pnas.0502106102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Gao XP, Ramchandran R, Zhao YY, Vogel SM, Malik AB. Nonmuscle myosin light-chain kinase mediates neutrophil transmigration in sepsis-induced lung inflammation by activating beta2 integrins. Nat Immunol. 2008;9:880–886. doi: 10.1038/ni.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto S, Shimizu S, Kiyonaka S, Takahashi N, Wajima T, Hara Y, Negoro T, Hiroi T, Kiuchi Y, Okada T, et al. TRPM2-mediated Ca2+ influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nat Med. 2008;14:738–747. doi: 10.1038/nm1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo SK, Starnes TW, Deng Q, Huttenlocher A. Lyn is a redox sensor that mediates leukocyte wound attraction in vivo. Nature. 2011;480:109–112. doi: 10.1038/nature10632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zicha D, Dunn GA, Segal AW. Deficiency of p67phox, p47phox or gp91phox in chronic granulomatous disease does not impair leucocyte chemotaxis or motility. Br J Haematol. 1997;96:543–550. doi: 10.1046/j.1365-2141.1997.d01-2080.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.