

Abstract
Mutations in the mtDNA have been found to fulfill all of the criteria expected for pathogenic mutations causing prostate cancer. Focusing on the cytochrome oxidase subunit I (COI) gene, we found that 11–12% of all prostate cancer patients harbored COI mutations that altered conserved amino acids (mean conservation index = 83%), whereas <2% of no-cancer controls and 7.8% of the general population had COI mutations, the latter altering less conserved amino acids (conservation index = 71%). Four conserved prostate cancer COI mutations were found in multiple independent patients on different mtDNA backgrounds. Three other tumors contained heteroplasmic COI mutations, one of which created a stop codon. This latter tumor also contained a germ-line ATP6 mutation. Thus, both germ-line and somatic mtDNA mutations contribute to prostate cancer. Many tumors have been found to produce increased reactive oxygen species (ROS), and mtDNA mutations that inhibit oxidative phosphorylation can increase ROS production and thus contribute to tumorigenicity. To determine whether mutant tumors had increased ROS and tumor growth rates, we introduced the pathogenic mtDNA ATP6 T8993G mutation into the PC3 prostate cancer cell line through cybrid transfer and tested for tumor growth in nude mice. The resulting mutant (T8993G) cybrids were found to generate tumors that were 7 times larger than the wild-type (T8993T) cybrids, whereas the wild-type cybrids barely grew in the mice. The mutant tumors also generated significantly more ROS. Therefore, mtDNA mutations do play an important role in the etiology of prostate cancer.
Keywords: cybrid, oxidative phosphorylation, inherited predisposition
There is increasing evidence that mitochondrial gene mutations are associated with various cancers, but their pathophysiological significance remains unclear. The first clear demonstration that mtDNA mutations in cancer could have functional significance came with the report of a middle-aged woman with a renal adenocarcinoma that was heteroplasmic (mixture of mutant and normal mtDNAs) for a deletion of 294-nucleotide pairs (nps) in the mtDNA oxidative phosphorylation (OXPHOS) gene ND1 (1). Subsequently, a variety of mtDNA coding region and control region mutations have been reported in colon cancer cells (2), prostate cancer (3–5), and a variety of other solid tumors (6).
Mutations in nuclear DNA (nDNA)-encoded mitochondrial genes have also been linked to cancer. Mutations in the nDNA-encoded succinate dehydrogenase (SDH) B, SDHC, and SDHD subunits of OXPHOS complex II have been linked to paragangliomas (7–10). However, mutations in SDHA, the succinate-binding subunit, have been linked to Leigh Syndrome (11), not paraganglioma, demonstrating that transformation due to complex II mutants is not simply the result of energy deficiency. An alternative mitochondrial contribution to tumorigenicity could be increased reactive oxygen species (ROS) production. This possibility is supported by the observation that the mev-1 mutation in the SDHC gene of Caenorhabditis elegans markedly increases ROS production (12, 13), and increased ROS production has been proposed to be an important factor in tumor formation in association with inactivation of p16ink4a and p53 (14).
ROS are generated as a toxic by-product of mitochondrial OXPHOS. OXPHOS is composed of the electron transport chain (ETC), encompassing complexes I, II, III, and IV and the H+-transporting ATP synthase, complex V. Reducing equivalents (electrons) from dietary calories are passed down the ETC, where they ultimately reduce O2 (four electrons) to generate H2O. The energy that is released is used to pump protons out across the mitochondrial inner membrane through complexes I, III, and IV to create an electrochemical gradient (ΔP = ΔΨ + ΔμH+). ΔP is then used as a source of potential energy by complex V to condense ADP and phosphate to generate ATP, and ATP is exchanged across the mitochondrial inner membrane for spent cytosolic ADP by the adenine nucleotide translocators (15).
When calories are plentiful, the ETC becomes more reduced and electrons from complexes I and III can be donated directly to O2 to generate superoxide anion 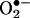 , the first of the ROS.
, the first of the ROS. 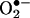 is converted to H2O2 by mitochondrial manganese superoxide dismutase, and H2O2 can be converted to water by glutathione peroxidase or catalase. H2O2 can also acquire an additional electron from a reduced transition metal to generate the highly reactive hydroxyl radical •OH. H2O2, which is semistable, can also diffuse out of the mitochondrion and into the cytosol and the nucleus, where it can act (15).
is converted to H2O2 by mitochondrial manganese superoxide dismutase, and H2O2 can be converted to water by glutathione peroxidase or catalase. H2O2 can also acquire an additional electron from a reduced transition metal to generate the highly reactive hydroxyl radical •OH. H2O2, which is semistable, can also diffuse out of the mitochondrion and into the cytosol and the nucleus, where it can act (15).
At high levels ROS are toxic, but at low levels they are mitogenic, presumably interacting with various nuclear regulatory factors (AP-I, NF-κB APE/ref-1) (16), regulatory kinases (Src kinase, protein kinase C, mitogen-activated protein kinase), receptor tyrosine kinases (17), protein-tyrosine phosphatases (18), and angiogenic factors (19, 20). Consistent with mitochondrial ROS being important in tumor formation, mitochondrial manganese superoxide dismutase is reduced in many types of tumors including prostate cancer, mutations in the mitochondrial manganese superoxide dismutase gene promoter have been observed in a number of tumors, and transformation of certain tumors with the mitochondrial manganese superoxide dismutase cDNA can reverse the malignant phenotype (17, 21–23).
Mitochondrial OXPHOS is assembled from multiple polypeptides, some encoded by the mtDNA and others by the nDNA. In addition to the 12S and 16S rRNAs and 22 tRNAs for mitochondrial protein synthesis, the mtDNA encodes for 13 polypeptides, 7 (ND1, ND2, ND3, ND4L, ND4, ND5, and ND6) of 46 polypeptides of complex I, 1 (cytochrome b) of 11 polypeptides of complex III, 3 [cytochrome oxidase subunit (CO) I, II, and III] of 13 polypeptides of complex IV, and two (ATP6 and ATP8) of 16 polypeptides of complex V. COI is the main catalytic subunit of cytochrome c oxidase (complex IV), and ATP6 is central to the proton channel of the ATP synthase (complex V) (15).
The inhibition of OXPHOS that increases ROS production has been confirmed in mice in which the heart/muscle isoform gene of the adenine nucleotide translocator gene (Ant1) was inactivated. This inactivation resulted in the hyperpolarization of ΔP, increased ROS generation, and elevated mtDNA damage (24, 25).
If mitochondrial dysfunction and ROS production contribute to cancer, then this finding would elicit two hypotheses. First, cancers should harbor both germ-line and somatic mtDNA mutations, which should partially inhibit OXPHOS and, thus, increase ROS production. Second, mtDNA mutations that increase ROS production should stimulate tumor growth. We have tested both of these predictions on prostate cancer and confirmed their validity. Therefore, this article supports the conclusion that mitochondrial defects contribute to the etiology of cancer.
Materials and Methods
Patient Materials. All patient studies were implemented under Emory University Institutional Review Board approved protocols. Histologically confirmed prostate cancer samples were selected from our collection of radical prostatectomies, institutional tissue resources, and microdissected samples prepared between 1995 and 2002. The “no-cancer” control group was assembled from subjects who had undergone prostate biopsy and had been found to be free of prostate cancer. These individuals were all at least 50 years old and had a <4 ng/ml prostate-specific antigen.
Sequencing the mtDNA COI Gene. The mtDNA region encompassing COI was amplified between nucleotides 5772–7600. Both strands of the PCR product were sequenced by using nested primers (6080F, 6930F, 6340R, and 7150R). The templates were denatured at 96°C and primers extended in the presence of “Big Dye Terminators” for 25 cycles of 96°C for 10 sec, then 55°C for 5 sec, and 60°C for 4 min. The reactions were chilled to 4°C, and the excess dye terminators removed by Centri-sep columns. The trace files were determined by using an Applied Biosystems PRISM 3100 genetic analyzer, analyzed by using sequencher 4.1 gene analysis software (Gene Codes, Ann Arbor, MI), and interpreted within the context of mitomap (www.mitomap.org) and our current collection of 1,338 complete mtDNA sequences (26). To assure that the sequence variants found were not due to the spurious amplification of nDNA pseudogenes, we scanned our mtDNA pseudogene database (27) for any pseudogene that might have been amplified. Only one (Emb/AL359496.30/on chromosome 6) matched the primers used and could have potentially been amplifiable in the above experiments. However, this pseudogene could not have contributed to the current results because it lacks all of the COI mutations that we found in the prostate cancer samples.
Histopathological and Molecular Analysis of Laser Capture Microdissection (LCM)-Isolated Prostate Cancer Epithelium. Normal and tumor prostate epithelial cells were collected by LCM. Fresh radical prostatectomy specimens, embedded in Tissue-Tek OCT compound (Sakura Finetek, Torrance, CA), were frozen at -80°C and sectioned by cryostat (Shandon, Pittsburgh) to yield 7-μm sections. Each section was fixed in 70% alcohol and hematoxylin/eosin-stained (28). Before microdissection, cut sections were dehydrated in graded alcohols followed by xylene and air-dried for 5 min. Benign and malignant epithelial regions were selected by a pathologist, and at least 5,000 cells each were collected by LCM by using a Pixcell II LCM system (Arcturus Engineering, Mountain View, CA).
DNA was extracted from the microdissected tissue by transfer to 0.04% proteinase K/10 mM Tris·HCL (pH 8.0)/1 mM EDTA/1% Tween-20 and digested overnight. Organic (phenol/chloroform) extraction was followed by ethanol/sodium acetate precipitation. DNA pellets were suspended in distilled water.
The entire mtDNA of tumor 18 was amplified in 1- to 2-kb overlapping fragments, the fragments purified by using Centricon-100 concentrator columns, and then cycle sequenced. The dried sequencing reactions were resuspended, heated at 94°C for 2 min, loaded on “Long Ranger” DNA sequencing gels, and the sequencing traces determined by using an Applied Biosystems PRISM 377 automated DNA sequencer.
The heteroplasmy of the COI G5949A mutation was analyzed by RFLP analysis. The mutant region was PCR-amplified by using a forward primer in which an A at np 5946 was changed to C, underlined (5′-CTCTACAAACCACAAAGACCTT-3′). The combination of the patient's G5949A mutation plus the introduced C generates a new DdeI restriction site.
The presence of the COI polypeptide in prostate tumor 18 tissue was analyzed by immunohistochemistry. Formalin-fixed, paraffin-embedded sections (5 μm) from tissue adjacent to the frozen section block used for LCM-directed DNA sequencing were deparaffinized and rehydrated, then steamed in citrate buffer (pH 6) for 20 min and cooled for 10 min. After exposure to 3% hydrogen peroxide for 5 min at room temperature, mouse monoclonal anti-human COI antibody (Molecular Probes) at a dilution of 1:250 was reacted for 25 min, followed by 25 min each of biotinylated secondary linking antibody and a streptavidin bound peroxidase enzyme complex. The sections were then stained with diaminobenzidine as chromogen for 5 min and counterstained with hematoxylin for 1 min. Between incubations, the sections were washed with Tris-buffered saline.
PC3(mtDNA T8993T/G) Cybrids. Transmitochondrial cybrids were prepared by treating PC3 prostate cancer cells with 5 μg/ml rhodamine 6-G in culture medium for 7 days to cure them of their resident mitochondria (29). Then 3 × 106 of the rhodamine 6-G-treated PC3 cells were fused by electric shock to ≈2 × 107 cytoplasts obtained by Ficoll-cytochalasin B step-gradient enucleation of homoplasmic mutant (T8993G) or homoplasmic wild-type (T8993T) 143B(TK-) cell lines (29). The homoplasmic T8993G mutant or T8993T wild-type donor cells were cybrids that had been previously prepared by fusing 143B(TK-) cells devoid of mtDNA (ρ0 cells) with cytoplasts from the lymphoblastoid cell line of a patient that was heteroplasmic for the mtDNA NARP/Leigh ATP6 T8993T/G mutation (30). Rhodamine 6-G PC3 cell to T8993G/T cytoplast fusions were induced in a BTX PN453 slide chamber with a 3.2-mm electrode gap by using a BTX Electro Cell Manipulator 200 equipped with an optimizer (Biotechnologies & Experimental Research, San Diego). The fusion mixture was plated in DMEM containing 10% FCS, 4.5 mg/ml glucose, 1 mM pyruvate, and hypoxanthine/aminopterin/thymidine but without uridine (29). The PC3 nuclear origin of the PC3(mtDNA T8993T/G) cybrid clones was confirmed by using the insulin gene variable number tandem repeats (31) and the chromosome number confirmed by karyotyping. The mtDNA genotype was confirmed by HpaII digestion (29).
In Vivo Tumorigenesis of PC3(mtDNA T8993T/G) Cybrids. All animal experiments were carried out as part of an Institutional Animal Care and Use Committee-approved protocol. Six-week-old male athymic (nude) mice were purchased from Harlan Labs (Indianapolis) or Charles River Laboratories and injected s.c. over the scapula with 106 viable cybrid cells. Animals were maintained in sterile housing, four animals to a cage, and observed on a daily basis. At 10-day intervals, the tumors were measured by using calipers, and the volumes of the tumors were calculated by using V = (L × W2)/2. Animals were euthanized with carbon monoxide. ROS Production in Tumors. The endogenous in vivo 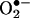 production of the PC3(mtDNA T8993T/G) tumors was determined by staining tumor slices with dihydroethidium (Molecular Probes). Tumor masses, dissected out of the nude mice, were placed in OCT compound and flash-frozen in a methylbutane-chilled bath, then placed in liquid nitrogen. Cryostat slices (30 μm) were stained in the dark for 30 min at 37°C in a humidified 5% CO2 incubator with a 1 μM solution of dihydroethidium. Samples were analyzed by using confocal microscopy with an argon laser by using 510 nm of excitation and 595 nm of emission.
production of the PC3(mtDNA T8993T/G) tumors was determined by staining tumor slices with dihydroethidium (Molecular Probes). Tumor masses, dissected out of the nude mice, were placed in OCT compound and flash-frozen in a methylbutane-chilled bath, then placed in liquid nitrogen. Cryostat slices (30 μm) were stained in the dark for 30 min at 37°C in a humidified 5% CO2 incubator with a 1 μM solution of dihydroethidium. Samples were analyzed by using confocal microscopy with an argon laser by using 510 nm of excitation and 595 nm of emission.
Results
Identification of the mtDNA Variants in LCM-Isolated Prostate Cancer Epithelium. To determine whether mtDNA mutations were associated with the prostate cancer, we used LCM to isolate prostate cancer epithelial cells from several prostate tumors and sequenced segments of their mtDNAs. This experiment revealed a variety of potentially pathogenic mtDNA mutations. As a example, for tumor 18, the entire mtDNA was sequenced in a series of overlapping segments. This finding revealed 38 base substitutions relative to the Cambridge Reference Sequence (mitomap), including 31 previously reported polymorphisms and 7 previously uncharacterized mutations; the latter including 1 ribosomal RNA mutation and 3 missense mutations. The three new amino acid substitution mutations included a chain termination mutation in COI (G5949A) and two missense mutations, one in cytb (A14769G) and the other in ATP6 (C8932T).
The G5949A mutation introduced a stop codon into COI at amino acid 16 (G16X) (Fig. 1A). To determine the origin of this mutation, we developed a restriction fragment length polymorphism test for the mutation and tested the cancerous and normal epithelium from this prostate. This experiment revealed that the cancerous epithelial cells of tumor 18 were homoplasmic mutant, whereas the adjacent normal epithelial cells were homoplasmic wild-type (Fig. 1B). Hence, this mutation must have arisen during the genesis of the cancer cell and then segregated to a pure mutant in the malignant cells.
Fig. 1.

Tumor 18 COI G16X mutation in LCM-isolated prostate cancer epithelium. (A) mtDNA sequencing chromatograms of LCM-collected pure prostate cancer epithelium (upper chromatogram) reveals only the mutant 5949T (G5949A, opposite strand, black letters above), mtDNAs and pure normal epithelium (lower chromatogram) reveals only the wild-type 5949C (G5949G, opposite strand, black letters above) mtDNAs. (B) RFLP analysis of the G5949A mutation detected through the creation of an intentionally introduced DdeI restriction site. Lanes 1–4, DdeI digests. Lanes: 1, pure cancer epithelium (pure mutant); 2, pure normal epithelium (pure wild-type); 3 and 4, wild-type controls; 5, molecular weight markers. (C) Immunohistochemical staining of a section from tumor 18 stained with a COI-specific antibody. M, malignant glands; B, benign glands.
To determine whether the G16X mutation actually eliminated the COI protein from the prostate cancer cells, we performed immunohistochemistry on tumor sections (Fig. 1C). The normal epithelial cells proved to be strongly positive for COI, whereas the adjacent cancer cells were completely negative.
The cytb A14769G mutation in this patient altered an amino acid (N8S) with a relatively low interspecific amino acid conservation index (CI) of 20.5%, indicating that this variant probably had limited effect on the cellular physiology. By contrast, the ATP6 C8932T mutation altered an amino acid (P136S) with a CI of 64%, which could be functionally significant. Therefore, both germ-line and somatic mtDNA mutations may have contributed to the formation of tumor 18.
COI Mutations in Prostate Cancer. The loss of the mtDNA-encoded COI subunit in tumor 18 is consistent with proteomic surveys of LCM-isolated prostate cancer epithelia, which revealed that the ratio of nDNA-encoded complex IV subunits (COX IV, Vb, and VIc) to mtDNA-encoded subunits (COI and COII) was increased in most prostate tumors (32, 33). Hence, deficiency of mtDNA COI, COII, and COIII subunits might be a common feature of prostate cancer.
To determine whether COI was in fact deficient, we sequenced the COI genes from multiple prostate cancer tumors and controls. We chose to study only the COI genes because this permitted us to survey a large number of tumor and control samples, thus making statistical evaluation feasible. Moreover, COI mutations have been observed in the mtDNAs in colon cancer cell lines (2), colonic crypt cells (34), and sideroblastic anemia patients (35, 36). However, polymorphisms and pathogenic mutations in COI are relatively uncommon (mitomap) (26, 37).
DNA was extracted and the COI gene sequenced from prostatectomy specimens or peripheral blood cells taken from 260 European and African American patients with pathologically confirmed prostate cancer and from the lymphocytes of 54 “no-cancer” (prostate cancer negative) controls. COI missense mutations (Table 1) were found in 12% of the prostate cancer patients but in only 1.9% of the no-cancer controls, a significant increase in frequency (P = 0.023) (Table 2). Furthermore, in a population sample of 1,019 European and African mtDNA sequences, 7.8% had COI mutations, which was also significantly lower than that of cancerous prostates (P = 0.015) (Table 2). Because COI missense polymorphisms are more common in African mtDNAs of macrohaplogroup L than in the rest of the world (37), we also analyzed only patients and controls of European ancestry. From this group, COI missense mutations were found in 11% of the prostate cancer specimens, 0% of no-cancer controls (P = 0.016), and 6.5% in a population sample of 898 Europeans (P = 0.025) (Table 2). Thus, COI mutations are significantly increased in prostate cancer over the no-cancer controls and the general population.
Table 1. All prostate cancer-associated COI mutations.
| Mutant | Amino acid | Cl | Haplogroup | Prostate | Blood | Case |
|---|---|---|---|---|---|---|
| C59111 | A3V | 13 | U | M | M | 18 |
| G5913A | D4N | 18 | K | M | M | 8 |
| A5935G | N11S | 100 | N | M | M* | 29 |
| G5949A | G16X | — | U | M/W | W | 18 |
| G5973A | A24T | 92 | H | M | M | 3 |
| G6081A | A60T | 97 | L2 | M | M | 31 |
| G6150A | V83I | 95 | L1 | M | M | 21 |
| T6124C | M74T | 95 | T | M/W | M/W | 19 |
| T6253C | M117T | 69 | H | M | M | 1, 2, 4 |
| G6261A | A120T | 97 | J, T, L1, N | M | M | 6, 7, 17, 26, 27, 30 |
| G6267A | A122T | 92 | L1 | M | M | 24 |
| G6285A | V128I | 100 | H | M | M | 16 |
| C6340T | T146I | 79 | H, N | M | M* | 5, 23 |
| G6480A | V193I | 87 | I | M | M | 14 |
| A6663G | I254V | 95 | O, L2 | M | M | 12, 13, 20, 22, 25 |
| G6924T | A341S | 100 | K | M | W | |
| G7041A | V380I | 100 | T | M | M | 9 |
| T7080C | F393L | 97 | U | M | M* | 10 |
| A7083G | I395V | 33 | H | M | M | 15 |
| A7158G | I419V | 21 | N | M | M* | 28 |
| A7305C | M468L | 90 | U | M | M | 11 |
M, mutant; W, wild type.
Four cases had no blood available. DNA sequencing of a separate organ (seminal vesicle) in these four cases confirmed these as germ-line mutations. Variants 7389 and 7146, defining L0 and L0L1, respectively, were not considered
Table 2. Frequency of COI mutations in prostate cancer patients, controls, and the general population.
| n | COI mutant | Frequency, % | P | |
|---|---|---|---|---|
| Cancer | 260 | 31 | 11.9ab | |
| EA | 180 | 19 | 10.6cd | |
| AA | 80 | 12 | 15.0 | |
| No cancer | 54 | 1 | 1.9a | 0.023 |
| EA | 46 | 0 | 0c | 0.016 |
| AA | 8 | 1 | 12.5 | 0.674 |
| Population | 1338 | 104 | 7.8 | |
| EA + AA | 1019 | 79 | 7.8b | 0.015 |
| EA | 898 | 58 | 6.5d | 0.025 |
| AA | 121 | 21 | 17.4 | 0.432 |
| Non(EA + AA) | 319 | 25 | 7.8 |
Variants 7389 and 7146, defining L0 and L0L1, respectively, were not considered. EA, European ancestry; AA, African ancestry. Frequencies with the same superscript were compared, and the P value (from Fisher's exact test) is represented in the right column.
The interspecific conservation of the altered COI amino acids in prostate cancer was also significantly higher than that in the general population. The average CI for the prostate cancer mutations was 83 ± 25%, whereas that for a general population sample of 1,338 mtDNA sequences was 71 ± 35 (P = 0.029). The CI of the prostate cancer COI mutations was comparable with the CI observed for global human mtDNA “adaptive mutations” (85 ± 9%) and far above the CI of global “neutral polymorphisms” (23 ± 15%) (26). Thus, the prostate cancer COI mutations must be functionally significant.
Three of the prostate cancer COI mutations had the characteristics of new somatic pathogenic mutations, being heteroplasmic and changing highly conserved amino acids. The first of these mutations was the tumor 18 chain-termination mutation, G5949A (G16X). The second was a T6124C mutation (M74T) with a CI of 95% that was heteroplasmic in both the prostate tissue and blood cells. The third mutant was C6924T (A341S), which had a CI of 100% and was primarily a mutant in the prostate tissue but wild-type in blood (Table 1).
Four other prostate cancer COI mutations were found in more than one patient and in each case was associated with prostate cancer. The T6253C mutation (CI = 69%) was found in three independent cases, all on haplogroup H, the most common European haplogroup. The C6340T mutation (CI = 79%) was observed in two patients on two different haplogroup backgrounds (H and N). The G6261A mutation (CI = 97%) was observed in six patients on four different haplogroups (J, T, L1, and N). Finally, the A6663G mutation (CI = 95%) was observed in five patients on two different haplogroups (L2 and unclassified) (Table 1).
Because the T6253C, C6340T, G6261A, and A6663G mutations were homoplasmic in these patient's lymphocytes, they must have arisen in the female germ-line. The importance of germ-line mutations in prostate cancer was supported by the observation that none of the European descent no-cancer control men had COI mutations, yet 6.5% of the European population had COI mutations and 11% of the European prostate cancer patient men had COI mutations. Because the frequency of COI mutations is significantly different between the noncancer controls and the general population (P = 0.05) and between the general population and the prostate cancer-positive men (P = 0.016), it follows that men that harbor germ-line COI mutations must have a substantially increased risk of developing prostate cancer. Therefore, both somatic and germ-line COI mutations are associated with prostate cancer, and COI mutations must be a significant risk factor for prostate cancer.
We have exhaustively surveyed only COI mutations in this study. However, it is likely that additional mtDNA polypeptide mutations can contribute to the etiology of prostate cancer (5). This possibility is supported by the ATP6 C8932T (P136S) observed in tumor 18 as well as the presence of two previously uncharacterized missense mutations in the complete mtDNA sequence of the PC3 tumor cell line, which is haplogroup U5, and contained a np T11120C (ND4/F121L; CI = 12.8%) variant of unlikely functional significance and a np C13802T (ND5/T489M; CI = 62.5%) variant that could be functionally relevant. Additional mtDNA missense mutations were found in other tumors analyzed from LCM material (data not shown).
Cybrid Studies Reveal That mtDNA Mutations Enhance Cancer Cell Growth. To investigate the functional importance of mtDNA mutations for prostate cancer, we chose to model the tumor 18 germ-line ATP6 C8932T (P136S) mutation by using the well characterized pathogenic ATP6 np 8993G L156R mutation (38). The ATP6 np 8993G mutation is just 20 amino acids away from the P136S mutation and is known to cause a 70% inhibition in ADP-stimulated respiration (30) and an increase in mitochondrial ROS production (39).
We introduced mtDNAs harboring the ATP6 mutant T8993G or wild-type T8993T base into a prostate cancer cell line through transmitochondrial cybrids. The mitochondrial donor cells were derived from the same heteroplasmic patient (30) and were homoplasmic for either the T8993G or T8993T mtDNA. These cells were enucleated and the cytoplasts fused to PC3 cells that had been cured of their resident mtDNAs by using rhodamine 6-G (29). Six PC3 cybrids that were homoplasmic for the mutant mtDNA (T8993G) [PC3(mtDNA T8993G) nos. 2, 5, 8, 20, 21, and 26] and four cybrids that were homoplasmic wild-type (T8993T) [PC3(mtDNA T8993T) WT nos. 1, 3, 5, and 10] were isolated and studied further.
The PC3(mtDNA ATP6 T8993T) and PC3(mtDNA ATP6 T8993G) cybrids were injected s.c. into nude mice in four separate experiments conducted on both early passage (at about passage 6) and late-passage (at about passage 25) cultures. The results from all four experiments encompassing multiple trials for both the T8993G versus the T8993T clones were combined for each time point and the values averaged and plotted (Fig. 2). These experiments revealed that the average tumor volume of the mutant PC3(mtDNA T8993G) cybrids was significantly higher than that of the wild-type PC3(mtDNA T8993T) cybrids at every time point (P < 0.026 by Mann–Whitney test) (Fig. 2). Indeed, the PC3(mtDNA T8993T) wild-type cybrids barely grew at all in the mice. By day 110, the average tumor volume of the PC3(mtDNA T8993T) wild-type cybrids was 0.11 ml, whereas that of the PC3(mtDNA T8993G) mutant cybrids was 0.78 ml, more than a 7-fold increase. Moreover, this result is an underestimate of the differential growth rate of the PC3(mtDNA T8993G) tumors because mice with rapidly growing tumors had to be euthanized throughout the various experiments. This decision removed the fastest growing PC3(mtDNA T8993G) tumors from the later average tumor-size calculations, resulting in an uneven mutant cybrid curve with a reduced average slope (Fig. 2). Hence, the tumor growth rate of the PC3 prostate cancer cell nucleus was enhanced by the introduction of an ATP6 mutation known to reduce ATP synthase activity and increase mitochondrial ROS production (30, 39).
Fig. 2.

Increased tumor growth of PC3(mtDNA T8993G versus T8993T) cybrids. This graph is a composite of four independent experiments in which nude mice were injected s.c. with six different mutant PC3 cybrid clones [PC3(mtDNA T8993G)] and four different wild-type cybrid clones [PC3(mtDNA T8993T)]. All six mutant and four wild-type clones were tested at an early passage (at about passage 6), and two of the mutant clones and one wild-type clone were again tested at a later passage (at about passage 25). A total of 91 animals were injected with the mutant clones, 71 with the early passage clones and 20 with the late-passage clones; whereas 55 mice were injected with the wild-type clones, 45 with the early passage and 10 with the late-passage. Each injected mouse was measure for tumor volume every 10 days, with the final data set encompassing 11 time points. However, in experiments where the animals receiving the mutant clones developed debilitating tumors, the mice had to be euthanized before the maximum experimental time point. This decision resulted in a final total of 615 determinations of tumor volume for mice harboring the mutant clones and 378 determinations for those harboring the wild-type clones.
To confirm that the mutant PC3(mtDNA T8993G) cybrids generated more ROS, we tested the tumor cells for superoxide anion production by staining tumor sections with dihydroethidium (Fig. 3). The nonfluorescent dihydroethidium is oxidized to fluorescent ethidium by  . The average fluorescence pixel density of the PC3(mtDNA T8993G) mutant tumor cells was 71.2 ± 9.2 (n = 3; MT5, MT8, and MT20), whereas that of the PC3(mtDNA T8993T) wild-type tumor cells was 46.7 ± 4.2 (n = 2, WT3 and WT10). Thus, there was significantly more ROS produced by the PC3(mtDNA T8993G) mutant tumors (P = 0.013 by t test). Therefore, prostate cancer cells that harbor mtDNA mutations, which increase ROS production, show increased tumor growth.
. The average fluorescence pixel density of the PC3(mtDNA T8993G) mutant tumor cells was 71.2 ± 9.2 (n = 3; MT5, MT8, and MT20), whereas that of the PC3(mtDNA T8993T) wild-type tumor cells was 46.7 ± 4.2 (n = 2, WT3 and WT10). Thus, there was significantly more ROS produced by the PC3(mtDNA T8993G) mutant tumors (P = 0.013 by t test). Therefore, prostate cancer cells that harbor mtDNA mutations, which increase ROS production, show increased tumor growth.
Fig. 3.

Dihydroethidium evaluation of PC3(mtDNA T8993T) versus PC3(mtDNA T8993G) tumor ROS production. (A) Relative fluorescence level seen in PC3(mtDNA T8993T) tumor sections, representative of wild-type clones 3 and 10. (B) Relative fluorescence level seen in PC3(mtDNA T8993G) tumor sections, representative of mutant clones 5, 8, and 20.
Discussion
The current study provides convincing evidence that mtDNA mutations play an important role in the etiology of prostate cancer. Prostate cancers have a significantly increased frequency of functionally important COI mutations, and the introduction into prostate cancer cells of a mtDNA mutation, which inhibits OXPHOS and increases ROS production, increased their in vivo growth.
The COI mutations that we identified in prostate cancer fulfilled all of the criteria expected for mtDNA mutations that cause this disease. The COI mutations were significantly more frequent in prostate cancer patients than in no-cancer controls or in the general population. The COI mutations altered significantly more conserved amino acids, and they included both new heteroplasmic somatic and recurrent homoplasmic germ-line mutations. Hence, mtDNA COI mutations appear to be a causal factor in the etiology of prostate cancer.
Germline COI mutations were also found to be an important risk factor for developing prostate cancer. COI missense mutations were common in the general population (7.8%), yet virtually absent (<2%) in cancer-negative controls. Thus, most men harboring COI missense mutations must move into the prostate cancer category by late middle age. The association between germ-line COI mtDNA mutations and prostate cancer risk might also explain why African American men are more prone to prostate cancer than European American men (40). Overall, COI variants are relatively common in African mtDNA (17.4%, Table 2), in part due to certain African mtDNA lineages harboring ancient COI protein polymorphisms (e.g., the np 7389 and 7146 variants in African lineages L0 and L0L1) (26). Therefore, these ancient COI protein polymorphisms may be contributing to an increased predisposition to prostate cancer in African American men today. However, a much larger study will be required to test the statistical validity of this proposition.
Given that highly conserved COI mtDNA missense variants (CI = 70%) are so common in the general population (7.8%), we wonder why COI mutations aren't more commonly found in neuromuscular disease. One possibility is that the human cell has the capacity to partially compensate for complex IV defects by changing the expression of the COX subunits. Mice lacking liver adenine nucleotide translocators were found to selectively upregulate complex IV by 2-fold (41). Hence, the biochemical effects of partial complex IV defects might be ameliorated by altered complex IV gene expression. This result would be consistent with the observation that prostate cancers have increased levels of nDNA-encoded to mtDNA-encode complex IV subunits (32, 33). Even so, the COI mutations would inhibit the ETC, and this result could chronically increase mitochondrial ROS production and stimulate cell proliferation (15, 17, 23).
If prostate cancer is the most common clinical consequence of COI mutations, then this finding may explain why COI mutations are so common in the general population. Prostate cancer kills middle aged or older males, but the mtDNA is exclusively maternally inherited. Hence, deleterious COI mutations that cause prostate cancer would have minimal effect on the genetic fitness of the mutant mtDNA.
Mutations in the mtDNA that inhibit the ETC and increase ROS production could act as both tumor promoters and tumor initiators. The fact that mtDNA mutations which increase ROS production can be potent tumor promoters was demonstrated by our introduction of the pathogenic mtDNA ATP6 T8993G mutation (30, 39) into PC3 cells and showing a dramatic increased tumor growth rate in association with increased cellular ROS production. Moreover, the lack of tumor growth observed for the PC3(mtDNA T8993T) wild-type cybrids might also support this conclusion because it is well established that PC3 cells readily form tumors in nude mice. Because the PC3 mtDNA was found to harbor a conserved ND5 np C13802T (T489M) mutation, it is possible that removal of this mutation reduced the tumorigenic potential of the PC3 cells.
Whether mtDNA mutations might also serve as tumor initiators was suggested by tumor 18, which harbored both a germ-line ATP6 P136S and a somatic COI G16X mutation. Because the germ-line ATP6 mutation must have preceded that COI G16X mutation, it is possible that ROS generated as a result of the ATP6 P136S mutation could have damaged to the mtDNA and caused the COI G16X mutation.
These observations also indicate that mtDNA variants could have accounted for earlier somatic cell genetic observations that the cybrid transfer of chloramphenicol resistant (CAPR) mtDNAs from a nontumorigenic Chinese hamster cell line into a tumorigenic cell line suppressed tumorigenesis (42), whereas the reciprocal transfer had no effect. Similarly, the transfer of CAPR mtDNAs from the near euploid HT1080 cells into the aneuploid HeLa cells suppressed growth, but the reciprocal transfer caused no change (43).
Our demonstration that partial defects in OXPHOS, which increase ROS, can contribute to cancer now provides an explanation for the observation of Otto Warburg >70 years ago that solid tumors have a high rate of “aerobic-glycolysis” (44). Mutations that inhibit OXPHOS would not only make more ROS, they would oxidize less pyruvate and NADH. The pyruvate and NADH would be converted to lactate by lactate dehydrogenase, resulting in excessive lactate production during aerobic respiration, aerobic-glycolysis, a physiological state that has been documented for cells harboring the ATP6 T8993G mutation (45). If mitochondrial ROS production is essential for solid tumor promotion, then aerobic-glycolysis should be a common feature of solid tumors, which Warburg noted.
In conclusion, this study has revealed that mtDNA mutations are not only associated with a predisposition to neuromuscular disease but also a predisposition to cancer. Therefore, we can now add cancer to the list of mitochondrial diseases.
Acknowledgments
We thank the Emory Urology Trust for Urologic Research for longitudinal support of our research; Jonathan Simons and the Winship Cancer Institute for the provision of critical infrastructure; and patients who so willingly agreed to participate. This work was supported by U.S. Department of Defense Grant DAMNS17-00-1-0080 (to J.A.P.), National Institutes of Health Grants CA96994 (to J.A.P.), CA98912 (to J.A.P.), NS21328 (to D.C.W.), and AG13154 (to D.C.W.), and an Ellison Medical Foundation Senior Investigator Grant (to D.C.W.).
Author contributions: J.A.P. and D.C.W. designed research; A.K.B., C.Q.S., J.H., SD.L., M.M.I., S.H.H., and D.C.W. performed research; E.R.-P., M.B.A., F.F.M., and D.C.W. contributed new reagents/analytic tools; J.A.P., E.R.-P., W.D.F., and D.C.W. analyzed data; J.A.P. and D.C.W. wrote the paper; E.R.-P. developed and utilized mtDNA population variation data.; and F.F.M. provided clinical materials.
Abbreviations: CI, conservation index; CO, cytochrome oxidase subunit; ETC, electron transport chain; LCM, laser capture microdissection; nDNA, nuclear DNA; np, nucleotide pair; OXPHOS, oxidative phosphorylation; ROS, reactive oxygen species; SDH, succinate dehydrogenase.
References
- 1.Horton, T. M., Petros, J. A., Heddi, A., Shoffner, J., Kaufman, A. E., Graham, S. D., Jr., Gramlich, T. & Wallace, D. C. (1996) Genes Chromosomes Cancer 15, 95-101. [DOI] [PubMed] [Google Scholar]
- 2.Polyak, K., Li, Y., Zhu, H., Lengauer, C., Willson, J. K., Markowitz, S. D., Trush, M. A., Kinzler, K. W. & Vogelstein, B. (1998) Nat. Genet. 20, 291-293. [DOI] [PubMed] [Google Scholar]
- 3.Chen, J. Z., Gokden, N., Greene, G. F., Mukunyadzi, P. & Kadlubar, F. F. (2002) Cancer Res. 62, 6470-6474. [PubMed] [Google Scholar]
- 4.Chinnery, P. F., Samuels, D. C., Elson, J. & Turnbull, D. M. (2002) Lancet 360, 1323-1325. [DOI] [PubMed] [Google Scholar]
- 5.Jeronimo, C., Nomoto, S., Caballero, O. L., Usadel, H., Henrique, R., Varzim, G., Oliveira, J., Lopes, C., Fliss, M. S. & Sidransky, D. (2001) Oncogene 20, 5195-5198. [DOI] [PubMed] [Google Scholar]
- 6.Copeland, W. C., Wachsman, J. T., Johnson, F. M. & Penta, J. S. (2002) Cancer Invest. 20, 557-569. [DOI] [PubMed] [Google Scholar]
- 7.Astuti, D., Latif, F., Dallol, A., Dahia, P. L., Douglas, F., George, E., Skoldberg, F., Husebye, E. S., Eng, C. & Maher, E. R. (2001) Am. J. Hum. Genet. 69, 49-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baysal, B. E., Ferrell, R. E., Willett-Brozick, J. E., Lawrence, E. C., Myssiorek, D., Bosch, A., van der Mey, A., Taschner, P. E., Rubinstein, W. S., Myers, E. N., et al. (2000) Science 287, 848-851. [DOI] [PubMed] [Google Scholar]
- 9.Niemann, S. & Muller, U. (2000) Nat. Genet. 26, 268-270. [DOI] [PubMed] [Google Scholar]
- 10.Vanharanta, S., Buchta, M., McWhinney, S. R., Virta, S. K., Peczkowska, M., Morrison, C. D., Lehtonen, R., Januszewicz, A., Jarvinen, H., Juhola, M., et al. (2004) Am. J. Hum. Genet. 74, 153-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bourgeron, T., Rustin, P., Chretien, D., Birch-Machin, M., Bourgeois, M., Viegas-Pequignot, E., Munnich, A. & Rotig, A. (1995) Nat. Genet. 11, 144-149. [DOI] [PubMed] [Google Scholar]
- 12.Ishii, N., Fujii, M., Hartman, P. S., Tsuda, M., Yasuda, K., Senoo-Matsuda, N., Yanase, S., Ayusawa, D. & Suzuki, K. (1998) Nature 394, 694-697. [DOI] [PubMed] [Google Scholar]
- 13.Senoo-Matsuda, N., Yasuda, K., Tsuda, M., Ohkubo, T., Yoshimura, S., Nakazawa, H., Hartman, P. S. & Ishii, N. (2001) J. Biol. Chem. 276, 41553-41558. [DOI] [PubMed] [Google Scholar]
- 14.Arbiser, J. L. (2004) Semin. Cancer Biol. 14, 81-91. [DOI] [PubMed] [Google Scholar]
- 15.Wallace, D. C. & Lott, M. T. (2002) in Emery and Rimoin's Principles and Practice of Medical Genetics, eds. Rimoin, D. L., Connor, J. M., Pyeritz, R. E. & Korf, B. R. (Churchill Livingstone, London), pp. 299-409.
- 16.Evans, A. R., Limp-Foster, M. & Kelley, M. R. (2000) Mutat. Res. 461, 83-108. [DOI] [PubMed] [Google Scholar]
- 17.McCord, J. M. (2000) Am. J. Med. Genet. 108, 652-659. [DOI] [PubMed] [Google Scholar]
- 18.Lee, S. R., Kwon, K. S., Kim, S. R. & Rhee, S. G. (1998) J. Biol. Chem. 273, 15366-15372. [DOI] [PubMed] [Google Scholar]
- 19.Haddad, J. J. (2002) Eur. Cytokine Network 13, 250-260. [PubMed] [Google Scholar]
- 20.Michiels, C., Minet, E., Mottet, D. & Raes, M. (2002) Free Radical Biol. Med. 33, 1231-1242. [DOI] [PubMed] [Google Scholar]
- 21.Baker, A. M., Oberley, L. W. & Cohen, M. B. (1997) Prostate 32, 229-233. [DOI] [PubMed] [Google Scholar]
- 22.Bostwick, D. G., Alexander, E. E., Singh, R., Shan, A., Qian, J., Santella, R. M., Oberley, L. W., Yan, T., Zhong, W., Jiang, X., et al. (2000) Cancer 89, 123-134. [PubMed] [Google Scholar]
- 23.Xu, Y., Krishnan, A., Wan, X. S., Majima, H., Yeh, C. C., Ludewig, G., Kasarskis, E. J. & St. Clair, D. K. (1999) Oncogene 18, 93-102. [DOI] [PubMed] [Google Scholar]
- 24.Esposito, L. A., Melov, S., Panov, A., Cottrell, B. A. & Wallace, D. C. (1999) Proc. Natl Acad. Sci. USA 96, 4820-4825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Graham, B., Waymire, K., Cottrell, B., Trounce, I. A., MacGregor, G. R. & Wallace, D. C. (1997) Nat. Genet. 16, 226-234. [DOI] [PubMed] [Google Scholar]
- 26.Ruiz-Pesini, E., Mishmar, D., Brandon, M., Procaccio, V. & Wallace, D. C. (2004) Science 303, 223-226. [DOI] [PubMed] [Google Scholar]
- 27.Mishmar, D., Ruiz-Pesini, E., Brandon, M. & Wallace, D. C. (2004) Hum. Mutat. 23, 125-133. [DOI] [PubMed] [Google Scholar]
- 28.Goldsworthy, S. M., Stockton, P. S., Trempus, C. S., Foley, J. F. & Maronpot, R. R. (1999) Mol. Carcinog. 25, 86-91. [PubMed] [Google Scholar]
- 29.Trounce, I. A., Kim, Y. L., Jun, A. S. & Wallace, D. C. (1996) Methods Enzymol. 264, 484-509. [DOI] [PubMed] [Google Scholar]
- 30.Trounce, I., Neill, S. & Wallace, D. C. (1994) Proc. Natl. Acad. Sci. USA 91, 8334-8338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakamura, Y., Leppert, M., O'Connell, P., Wolff, R., Holm, T., Culver, M., Martin, C., Fujimoto, E., Hoff, M., Kumlin, E., et al. (1987) Science 235, 1616-1622. [DOI] [PubMed] [Google Scholar]
- 32.Herrmann, P. C., Gillespie, J. W., Charboneau, L., Bichsel, V. E., Paweletz, C. P., Calvert, V. S., Kohn, E. C., Emmert-Buck, M. R., Liotta, L. A. & Petricoin, E. F., III (2003) Proteomics 3, 1801-1810. [DOI] [PubMed] [Google Scholar]
- 33.Krieg, R. C., Knuechel, R., Schiffmann, E., Liotta, L. A., Petricoin, E. F., III, & Herrmann, P. C. (2004) Proteomics 4, 2789-2795. [DOI] [PubMed] [Google Scholar]
- 34.Taylor, R. W., Barron, M. J., Borthwick, G. M., Gospel, A., Chinnery, P. F., Samuels, D. C., Taylor, G. A., Plusa, S. M., Needham, S. J., Greaves, L. C., et al. (2003) J. Clin. Invest. 112, 1351-1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Broker, S., Meunier, B., Rich, P., Gattermann, N. & Hofhaus, G. (1998) Eur. J. Biochem. 258, 132-138. [DOI] [PubMed] [Google Scholar]
- 36.Gattermann, N., Retzlaff, S., Wang, Y. L., Hofhaus, G., Heinisch, J., Aul, C. & Schneider, W. (1997) Blood 90, 4961-4972. [PubMed] [Google Scholar]
- 37.Mishmar, D., Ruiz-Pesini, E., Golik, P., Macaulay, V., Clark, A. G., Hosseini, S., Brandon, M., Easley, K., Chen, E., Brown, M. D., et al. (2003) Proc. Natl. Acad. Sci. USA 100, 171-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Holt, I. J., Harding, A. E., Petty, R. K. & Morgan-Hughes, J. A. (1990) Am. J. Hum. Genet. 46, 428-433. [PMC free article] [PubMed] [Google Scholar]
- 39.Mattiazzi, M., Vijayvergiya, C., Gajewski, C. D., DeVivo, D. C., Lenaz, G., Wiedmann, M. & Manfredi, G. (2004) Hum. Mol. Genet. 13, 869-879. [DOI] [PubMed] [Google Scholar]
- 40.Sakr, W. A. (February 20, 2004) Modern Pathol., 10.1038/modpathol.3800074.
- 41.Kokoszka, J. E., Waymire, K. G., Levy, S. E., Sligh, J. E., Cai, J., Jones, D. P., MacGregor, G. R. & Wallace, D. C. (2004) Nature 427, 461-465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Howell, A. N. & Sager, R. (1978) Proc. Natl. Acad. Sci. USA 75, 2358-2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wallace, D. C. (1981) Mol. Cell. Biol. 1, 697-710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Warburg, O. (1931) The Metabolism of Tumors (R. R. Smith, New York).
- 45.Pallotti, F., Baracca, A., Hernandez-Rosa, E., Walker, W. F., Solaini, G., Lenaz, G., Melzi D'Eril, G. V., DiMauro, S., Schon, E. A. & Davidson, M. M. (2004) Biochem. J. 384, 287-293. [DOI] [PMC free article] [PubMed] [Google Scholar]