

Abstract
The identification of epidermal growth factor receptor (EGFR) mutations represented a fundamental step forward in the treatment of advanced non-small cell lung cancer (NSCLC) as they define a subset of patients who benefit from the administration of specifically designed targeted therapies. The inhibition of mutant EGFR through EGFR-tyrosine kinase inhibitors (TKIs), either reversible, first-generation gefitinib and erlotinib, or irreversible, second-generation afatinib, has dramatically improved the prognosis of patients harboring this specific genetic alteration, leading to unexpected clinical benefit. Unfortunately, virtually all patients who initially respond to treatment develop acquired resistance to EGFR-TKIs within 9–14 months. The EGFR T790M secondary mutation has emerged as a cause of treatment failure in approximately 60% of resistant cases. To date, several compounds designed with the aim to overcome T790M-mediated resistance are under clinical investigation. The aim of this review is to discuss emerging data regarding the third-generation EGFR-TKI, osimertinib, for the treatment of EGFR T790M mutant advanced NSCLC.
Keywords: NSCLC, EGFR, tyrosine kinase inhibitors, resistance, osimertinib, T790M, liquid biopsy, brain metastasis
Introduction
With 1.6 million deaths expected annually, lung cancer is the most common cause of cancer-related death worldwide in both men and women. Currently, lung cancers are classified into non-small cell lung cancers (NSCLCs), which account for approximately 80% of cases, and small cell lung cancers representing the remaining 20%. Although in the last decade we witnessed an impressive advancement in the understanding the molecular mechanisms underlying the development and progression of NSCLC, the prognosis of patients with advanced disease is still disappointing, with 5-year survival rates of less than 5%, lower than many other leading cancer sites.1 In this scenario, the discovery of targetable genetic mutations that drive distinct subsets of NSCLC has successfully paved the way for personalized therapy in patients harboring specific actionable molecular alterations with astounding clinical results.
Epidermal growth factor receptor mutations in NSCLC
Activating mutations in the epidermal growth factor receptor (EGFR) gene occur in 10–15% of NSCLC cases in North American and European patients and in approximately 30–40% in East Asian patients.2–3 Of note, regardless of ethnicity, EGFR mutations are more frequently found in female patients with no history of smoking and in those with adenocarcinoma histology.4–6 However, the presence of EGFR mutations cannot be ruled out only on the basis of clinical characteristics.
It is noteworthy that EGFR mutations are typically non-overlapping with other oncogenic driver mutations found in NSCLC (e.g. KRAS, ALK, ROS1, MET, HER2, BRAF, NTRK1) which make EGFR-mutant (EGFRm) NSCLC a distinct disease entity.7
EGFR-activating mutations are included in a wide spectrum of genetic alterations, principally in-frame deletions, in-frame duplications/insertions, and point mutations.3,8 Altogether, frame deletions in exon 19 at the LeuArgGluAla sequence (E746-A750), and the exon 21-point mutation Leu858Arg (L858R), represent 85–90% of all EGFR mutations in NSCLC. Additional ‘uncommon’ mutations have been reported, including G719X in exon 18 (G719C, G719S, G719A), L861Q in exon 21, and S768I in exon 20. However, the predictive significance of these mutations to treatment with EGFR-TKI is still unclear, though data reported in literature so far seem to confirm a positive predictive role also for uncommon mutations.9 On the other hand, exon 20 frame insertions, along with de novo T790M point mutations in exon 20 are responsible for primary resistance to EGFR-TKIs.10–12 Clinical trials evaluating treatment-naïve patients with advanced NSCLC harboring sensitizing EGFR mutations treated with gefitinib, erlotinib or afatinib, showed a marked superiority in favor of EGFR-TKIs compared with platinum-based chemotherapy in terms of objective response rate (ORR), progression-free survival (PFS) and quality of life.13–15 Afatinib was also associated with a statistically significant improvement in overall survival (OS) in patients with the del19 EGFR mutation,16 though data from the LUX-lung 7 trial (a phase IIb trial comparing the activity of gefitinib and afatinib as upfront therapy for advanced EGFR-mutant NSCLC) showed that the differences in PFS, OS, time to treatment failure, and ORR with afatinib and gefitinib were largely unaffected by mutation type.17
Unfortunately, despite the initial response to treatment, virtually all patients develop acquired resistance (AR) to treatment, which occurs typically within 9–14 months. In order to overcome such resistance a number of third-generation EGFR-TKIs have been designed and are under clinical evaluation. The aim of this review is to discuss emerging data regarding the third-generation EGFR-TKI osimertinib against the EGFR T790M mutation in the treatment of patients with advanced NSCLC.
Mechanism of resistance to first and second-generation EGFR-TKIs
Despite the inhibition of the mutated form of EGFR being one of the most successful examples of targeted therapy in NSCLC, the prognosis of these patients still remains unfavorable because, in addition to primary or intrinsic resistance, almost all patients who initially benefited will develop AR to EGFR-TKIs. Several mechanisms of either intrinsic or AR to EGFR-TKIs have been described so far and many other are still unknown. Regarding pathogenesis, these mechanisms can be schematically divided in EGFR-dominant (secondary mutation) or EGFR-nondominant (bypass tracks with activation of other signaling pathways or phenotypic change). Overall, approximately 60% of AR cases are due to the secondary T790M mutation in exon 20 of the EGFR gene, followed by other EGFR mutations such as C797S, activation of alternative signaling pathways (such as MET or HER2 amplifications and secondary mutation of PI3KCA or BRAF), aberrance of the downstream pathways (KRAS mutations, and loss of PTEN), impairment of the EGFR-TKIs-mediated apoptosis (BCL2-like 11/BIM deletion polymorphism) and histological transformations.18–19
Primary resistance to EGFR-TKIs
Although many mechanisms are not completely understood, intrinsic resistance, defined as an immediate ineffectiveness of EGFR-TKIs, is often due the presence of a nonsensitive EGFR mutation. The most important EGFR mutations that generate TKI resistance are represented by exon 20 insertions or duplications, which account for 1–10% of the entire group of EGFR mutations. Most of these insertions occur between amino acids 767 and 775, and their preferential location is the C-helix (A767 to C775).20 This region is crucial in orienting the kinase into a state that controls both the ATP and the EGFR-TKI binding. The result is that the majority of exon 20 insertion mutations regulate the kinase domain conformation into an active position and reduce the affinity for EGFR-TKIs.21
Although the gatekeeper T790M mutation is recognized as the most common mechanism of AR, it has also been associated with primary resistance and in vitro studies have demonstrated transformational potential when T790M is concurrently expressed with an EGFR-TKI-sensitizing mutation.22 Regarding the allelic frequencies of this mutation in treatment-naïve tumors, with the conventional Sanger sequencing methods, this value has been reported as rather low. Interestingly, recent studies conducted using more sensitive techniques such as next-generation sequencing, locked nucleic acid polymerase chain reaction (PCR) and high-performance liquid chromatography, have reported higher frequency rates of T790M mutations in EGFR-mutant NSCLC pretreatment tumors (35%, 38% and 79% respectively).23–25
Another EGFR mutation that occurs in the extracellular domain, consisting of in-frame deletion of exons 2–7 (namely EGFR vIII), has been associated with EGFR-directed therapy failure. This form of resistance relies on structural changes in the EGFR protein, ultimately leading to constitutive activation of EGFR vIII.26
Intrinsic resistance may also be caused by concurrent molecular or genetic alterations that compromise the EGFR-TKI response in patients with sensitizing EGFR mutations. An example is represented by the pro-apoptotic Bcl-2 family member BIM, that is a crucial mediator of EGFR-TKI-induced apoptosis.27 In this regard, patients with BIM deletion polymorphisms or harboring low to intermediate levels of messenger RNA (mRNA) of BIM, have shown disappointing results when treated with EGFR-TKIs.28
Mechanism of acquired resistance toEGFR-TKIs
Secondary or AR typically occurs after prolonged treatment, and several molecular mechanisms have been discovered in around 60–70% of cases and fall into one of the following categories: secondary mutations in the EGFR gene, activation of alternative pathways and phenotypic transformation.
Insurgence of secondary mutations in the EGFR gene
The most common secondary mutation, detected in approximately 60% of the AR developed to erlotinib, gefitinib and afatinib, is the T790M mutation that occurs in exon 20 of the EGFR gene.29–31 Of note, this proportion could be underestimated as a higher prevalence of 68% has been shown using a locked nucleic acid (LNA)-PCR/sequencing assay.32 Originally reported in 2003 as a potential determinant of resistance to first-generation EGFR-TKIs in vitro,33 the mutation of threonine at residue 790 of EGFR had been increasingly identified in patients with NSCLC harboring EGFR-sensitizing mutations that progressed on first-generation EGFR-TKIs,34 and is currently considered the most common mechanism of AR to EGFR-TKI-based therapies. T790 is called ‘the gatekeeper residue’ because it is located in the ATP-binding pocket and constitutes a critical hydrogen bond with the EGFR-TKIs, thus being responsible for the drugs affinity to EGFR. At this level, resistance occurs because the substitution of the nonpolar, hydrophobic methionine 790 for hydrophilic threonine (T790M) results in a steric hindrance that interferes with TKI binding.35 Importantly, the acquisition of the T790M mutation leads to an increased affinity of EGFR for ATP, ultimately resulting into the displacement of ATP-competitive TKIs.35
Interestingly, even though the reason is still unclear, patients with disease progression due to a secondary EGFR T790M mutation, tend to have a more indolent natural history and longer post-progression survival as compared with patients with T790M-negative tumors.36 To date, two theories have been proposed in order to explain the development of these second mutations: subcloning and induced mutation/acquisition.37 In this respect, although this secondary mutation is rarely found in TKI-naïve tumors, resistant clones may be selected following prolonged exposure to TKIs and are detected in roughly 60% of EGFR-TKIs-treated patients.25,38 T790M mutation can also coexist with different EGFR mutations, such as L858R and D761Y and this combination leads to lung cancer cell survival.39
Regarding the relationship between the T790M mutation and the activity of the irreversible EGFR-TKI, afatinib, a preclinical study showed a certain grade of activity in EGFR T790M-positive NSCLC cell lines; however no evidence of survival benefit has been reported with afatinib after failure of platinum-doublet chemotherapy and a first-generation EGFR-TKI.40–41 In addition, the prevalence of T790M in NSCLC patients with AR to afatinib has never been properly studied. In a recent study, Wu and coworkers enrolled 42 patients with EGFR-mutated NSCLC (14 TKI-naïve and 28 pretreated with first-generation EGFR-TKIs) that presented with AR to afatinib and had suitable ‘post-afatinib’ tissue for molecular analysis. Intriguingly, the authors found acquired T790M in 47.6% of specimens, in line with data reported with first-generation EGFR-TKIs. Moreover, they showed that prevalence of T790M was also similar in the two groups of TKI-naïve and TKI-pretreated patients (50.0% and 46.4% respectively) and that clinical factors, including sex, age and smoking history, were not associated with a higher prevalence of T790M. This study did not report either second-site EGFR mutations besides T790M nor acquired C797S mutation that is responsible for the AR to third-generation EGFR-TKIs.31
Once it was established that T790M represents the major mechanism of AR to first- and second-generation EGFR-TKIs and that third-generation EGFR-TKIs with a specific activity against T790M such as osimertinib and rociletinib (CO1686) showed to be very effective also in this subgroup of patients, it was clear that re-biopsy at disease progression, whenever possible, is crucial in order to make the appropriate decision on management of these patients.42–43
Not surprisingly, several mechanisms of AR to third-generation EGFR-TKIs osimertinib and rociletinib, in vitro and in vivo, have recently been discovered. Niederst and colleagues analyzed cell-free DNA by next-generation sequencing (NGS), of 15 patients with resistance to osimertinib, and identified different genotypes before and after treatment.44 The most common was acquired C797S plus T790M mutation genotype (40%), followed by T790M mutation without the C797S mutation (33%) and loss of the T790M mutation in absence of the mutation C797S (27%).44 In these cases, proliferation of cancer cells is still EGFR-dependent but, under selective pressure of EGFR-TKIs, these cells may develop tertiary mutations in the EGFR gene, such as C797S.
Beyond T790M, rare EGFR resistance point mutations have been identified (<10% of patients), including T854A, D761Y, and L747S; however, the number of cases is extremely limited and the underlying mechanism is still unclear.45–46
Activation of alternative signaling pathways (e.g. MET, HER2, BRAF, PI3KA, IGFR1) and phenotypic transformation (epithelial to mesenchymal transition and small cell transformation) represent other common mechanism of AR to EGFR-TKIs but are not the subject of this review and so will not be discussed.
Liquid biopsy in NSCLC
The eligibility for treatment with osimertinib in advanced NSCLC requires tumor genotyping for EGFR and the presence of T790M resistance mutation. Until recently, molecular profiling of NSCLC was based only on tumor biopsy but the landmark for the detection of tumor genetic alterations is rapidly and excitingly changing. A new perspective for molecular tumor characterization is represented by the analysis of circulating tumor DNA (ctDNA) that provides a non-invasive alternative approach to tumor biopsy.
The use of liquid biopsy has multiple potential applications and advantages. It represents a fast and non-invasive method applicable for early diagnosis, assessment of prognosis, monitoring of tumor burden and identifying AR during the treatment,47 while repeating tissue biopsy to evaluate genomic tumor evolution at the time of disease progression or even during therapy is problematic, expensive and unsafe and may not be reflective of tumor heterogeneity.48
Several methods are available for the blood-based EGFR mutational status analysis which must be highly sensitive because of the low frequency of ctDNA in blood compared with germline cell-free DNA. These methods include: allele-specific PCR platforms like the Cobas® EGFR Mutation Test (Roche, Basel, Switzerland) and the Therascreen® EGFR amplification refractory mutation system (ARMS) assay (Qiagen, Hilden, Germany), digital assays including the droplet digital PCR (ddPCR), as well as the beads, emulsions, amplification and magnetics (BEAM)ing dPCR technique (Sysmex Inostics, Hamburg, Germany). Moreover, NGS is evolving to be applicable for ctDNA analysis (Amplicon-based targeted NGS as well as Captured-based Targeted NGS), offering efficiently large spectrum genotyping analysis starting from a limited amount of material.49 In recent years many studies have shown that ctDNA can be used to identify and quantify mutations in solid cancers. In 2009, Rosell and colleagues analyzed 164 samples of ctDNA extracted from the serum of patients, suffering from NSCLC with activating mutations of the EGFR determined on tumor tissue, yielding a sensitivity equal to 59.2%.15 Later, Liu and colleagues identified the same activating mutation in ctDNA extracted from plasma compared with that detected on the tissue sample by using the ARMS platform in 67.5% of the cases analyzed.50 In the last decade, several studies have shown that it is feasible to detect mutations in the plasma or serum of NSCLC patients with a sensitivity ranging between 46–80% and with a high level of specificity and concordance with tumor tissue analysis.50–56
A recent systematic review and meta-analysis including 25 studies on a total of 2605 patients showed, for a blood-based test, a sensitivity, specificity, and concordance rate of 0.61, 0.90, and 0.79, respectively. Serum testing was associated with a lower sensitivity (0.56 versus 0.65) but higher specificity (0.95 versus 0.85) and concordance (0.86 versus 0.74) than plasma. Furthermore, clinical data showed that EGFR mutations (exon 19 or 21) in blood were significantly associated with objective response [RR: 4.08; 95% confidence interval (CI) 2.48–6.70], PFS [hazard ratio (HR): 0.72; 95% CI 0.64–0.80], and OS (HR: 0.71; 95% CI 0.50–0.99). Collectively, these data demonstrate that blood can be a good surrogate when tumor tissue is absent or insufficient for testing EGFR mutations to guide EGFR-TKI treatment in patients with NSCLC.57
Recently Sacher and colleagues have analyzed by ddPCR (Bio-Rad/Molecular MD, California, USA) ctDNA of 120 patients at the time of diagnosis and 60 patients at the time of AR to an EGFR-TKI, demonstrating a sensitivity of plasma genotyping of 82% for EGFR exon 19 deletion, 74% for the L858R mutation, 77% for the T790M mutation, and 64% for the KRAS G12X mutation, compared with tumor biopsy molecular analysis.58 Another recent study focused on EGFR mutation-positive NSCLC patients with AR to EGFR-TKIs identified TKI-sensitizing and T790M mutations in plasma of 120 (46.2%) and 75 (28.8%) patients, respectively.59 T790M was detected in 56.7% of patients with plasma positive for TKI-sensitizing mutations. The concordance for mutation detection at the time of resistance by ddPCR in plasma compared with tumor tissue or malignant fluid specimens, evaluated on 41 patients with paired samples, was 78.0% for TKI-sensitizing mutations and 65.9% for T790M (identified especially in larger tumors and more heavily treated tumors).59
The ability of real-time based methods (Cobas, Therascreen, ddPCR and BEAMing dPCR) to detect EGFR mutations, including T790M, has been assessed using plasma samples collected in the phase I part of AURA trial.60 Results showed a sensitivity/specificity for EGFR-sensitizing mutations of 78–90%/100% and 90–100%/93–100% for nondroplet and droplet platforms, respectively. Digital platforms detected a higher percentage of T790M mutations, compared with nondigital platforms characterized by low sensitivity (29% for Therascreen and 41% for Cobas versus 71% of ddPCR and BEAMing dPCR) but high specificity (100% versus 83% for ddPCR and 67% for BEAMing dPCR). The Cobas® EGFR Mutation Test and BEAMing dPCR were used in a subsequent assessment on a different set of plasma samples independently from the AURA trial showing concordant results, with high sensitivity (73–81%) and low specificity (58–67%) for the detection of the T790M mutation. The T790M resistance mutation was more frequently detected in the plasma of patients with metastatic versus locally advanced disease as already reported.60 Genomic heterogeneity may explain the reduced specificity observed with plasma-based detection of T790M mutations versus tissue. The objective response rate (ORR) to osimertinib was 59% and 61% in patients positive for the T790M mutation in plasma and in tissue (both assessed using the Cobas® EGFR Mutation Test), respectively. Together, these results show that the Cobas® EGFR Mutation Test and BEAMing dPCR have the potential to identify patients harboring T790M mutations from plasma ctDNA.
More data about the clinical correlation between plasma genotyping and osimertinib have been recently reported by Oxnard and colleagues.61 Retrospective analysis of the AURA trial revealed a similar ORR and median PFS in patients with T790M-positive plasma (ORR, 63%; PFS, 9.7 months) or T790M-positive tumor (ORR, 62%; PFS, 9.7 months) with a sensitivity of ctDNA analysis for detection of T790M of 70%. Of 58 patients with T790M-negative tumors, T790M was detected in the plasma of 18 (31%). This study showed that tumor and plasma genotyping have complementary roles for T790M testing. For 30% of patients who usually test negative for T790M in plasma, a re-biopsy can be considered. The plasma genotyping can represent a first initial analysis; however, further investigation, in the presence of plasma T790M-negative assessment on tumor tissue, are warranted to identify patients who may benefit from anti-T790M agents.
CtDNA genotyping by the Therascreen® EGFR plasma PCR kit is approved for use with gefitinib in Europe and China, in patients with advanced NSCLC from whom a tumor biopsy sample is not available. In June 2016, The United States Food and Drug Administration (US FDA) approved the Cobas® EGFR Mutation Test (v2 blood-based companion diagnostic test) for eligibility for treatment with erlotinib. Furthermore, in Europe T790M mutation detection for osimertinib treatment eligibility can be performed using either a tissue-based or plasma-based test. Also, ctDNA sequencing could be used for patients with NSCLC that progressed during treatment with third-generation TKIs, for example for the detection of C797S EGFR mutation resistance.44,58,62 Several studies have shown that quantitative and qualitative EGFR mutational status monitoring by using ctDNA is feasible and permits detection of resistance mutations anticipating clinical progression.62–64 Sorensen and colleagues found that detection of resistance mutation in the plasma of patients with advanced NSCLC occurs very early (range, 15–344 days) before radiological evidence of progression during first-line erlotinib treatment and increases until progression of disease.64 Nevertheless, resistance monitoring cannot yet be used to change therapy before clinical evidence of resistance. No data are available on changing treatment early and outcomes in EGFR-mutant cancer patients.
Liquid biopsy is feasible and increases the number of patient eligible for target therapy but several issues are still open. More efforts are required to increase sensitivity, to avoid artifacts and reduce false positive, and finally translate the entire potential of ctDNA analysis in the clinical management of cancer patients.
Pharmacodynamics and pharmacokinetics
The most common sensitizing mutations of EGFR, including in-frame deletions of exon 19 and the point mutation L858R, have been demonstrated to reduce ATP affinity and increase the sensitivity of mutant receptors to competitive inhibitors.65 Consistently, gefitinib and erlotinib show a dramatic clinical benefit in patients with EGFR exon 19 or exon 21-mutated NSCLCs.66 The missense T790M variant in exon 20 of EGFR with the primary activating mutation of the EGFR allele results in a significantly increased affinity of mutant EGFR for ATP.35 In detail, pharmacokinetic analyses showed the Michaelis–Menten constant (Km) for ATP of respectively 8.4 ± 0.3 μM, 5.2 ± .02 μM and 148 ± 4 μM in the L858R/T790M mutant, wild-type and single L858R mutant receptor.35 As a consequence, the T790M mutation markedly reduces the potency of EGFR competitive inhibitors, representing the primary mechanism of T790M-mediated first-generation TKI resistance.
Osimertinib (TAGRISSO™, AZD9291; AstraZeneca, London, UK) is a mono-anilino-pyrimidine, orally available, irreversible, third-generation EGFR-TKI, which inhibits EGFR, via the C797 amino acid covalent bond, both sensitizing mutations (exon 19 deletion, L858R) and double mutants harboring T790M at a nine-fold lower concentration compared with wild-type EGFR. In vitro assays showed that osimertinib also possesses a certain grade of activity at clinically relevant concentration against ERBB2, ERBB3, ERBB4, BLK and ACK1. Consistently, preclinical models involving NSCLC cell lines and tumor xenografts showed that osimertinib exerts an impressive activity against those tumors harboring L858R, exon 19 deletions alone or in coexistence with T790M mutations. Of note, two of its metabolites AZ7550 and AZ5104, which are circulating at approximately 10% of the mother compound, retain a comparable inhibitory profile on EGFR mutations to osimertinib. AZ7550 showed also a similar potency to the original molecule while AZ5104 exhibits an eight-fold greater potency against exon 19 deletion and T790M mutation, and up to 15-fold higher against wild-type EGFR.30 Following a single oral administration, osimertinib exhibits a linear pharmacokinetics, the area under the plasma concentration-time curve (AUC), along with maximal plasma concentration (Cmax) and minimal concentration (Cmin) increases in a dose proportional manner over the 20–240 mg dose range. Osimertinib is given once daily, with a three-fold body accumulation after single administration, which results in steady-state exposure achieved in 22 days of dosing, reaching a mean volume of distribution of approximately 986 l. Importantly, the AUC of osimertinib derivative compounds AZ5104 and AZ7550 is approximately 10% of the exposure of osimertinib. Of note, pharmacokinetic exposure does not substantially differ between Asian and Western patients. Osimertinib has a mean half-life of 48 h and its primary degradation pathway involves oxidation (predominantly CYP3A) and dealkylation in vitro. Although osimertinib does not interact with CYP2C8, 1A2, 2A6, 2B6, 2C9, 2C19, 2D6 and 2E1, it is a competitive inhibitor of CYP3A. Besides, osimertinib has been demonstrated to be a substrate of P-glycoprotein and the ATP-binding cassette subfamily G member 2, but it is not a substrate of organic anion-transporting polypeptide proteins, OATP1B1 and OATP1B3 (Table 1) [AstraZeneca Pharmaceuticals LP 2015].30
Table 1.
Drug summary box.
| Name | AZD9291, osimertinib, Tagrisso™ |
|---|---|
| Chemical structure |
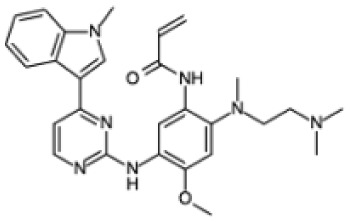
|
| Biochemical name | N-[2-[2-(dimethylamino)ethyl-methylamino]-4-methoxy-5-[[4-(1-methylindol-3-yl)pyrimidin-2- yl]amino]phenyl]prop-2-enamide |
| Pharmacodynamics | Irreversible, covalent bond and inhibition of sensitizing mutation (exon 19 deletion, L858R) and double mutants harboring T790M |
| Pharmacokinetics | Median time to Cmax = 6 h Mean volume of distribution Vss = 986 l Oral clearance = 14.2 l/h Mean half-life = 38 h Time to steady state: 22 days Elimination: 68% fecal and 14% urinary |
| Drug interaction | CYP3A inhibitors or inducers, substrates of CYP3A, BCRP and CYP1A2 |
| Adverse events | Any grade ⩾5%: diarrhea, rash, dry skin, nail toxicity, decreased appetite Select toxicities of special interest: ILD/pneumonitis, QTc interval prolongation, cardiomyopathy, hyperglycemia |
ILD, interstitial lung disease
Clinical efficacy of osimertinib in NSCLC
On the heels of promising preclinical data, the phase I/II AURA trial [ClinicalTrials.gov identifier: NCT01802632] was conducted in order to assess the safety and efficacy of osimertinib in patients with advanced EGFR-mutant (EGFRm) NSCLC who progressed on or following a first- or second-generation EGFR-TKI (Figure 1). A total of 253 patients were enrolled in this study. Of them, 33 were included in the dose-escalation portion, and 222 in the dose-expansion cohort that evaluated five doses ranging from 20–240 mg daily. All patients had received at least one EGFR-TKI prior to osimertinib, while 80% had also received prior cytotoxic chemotherapy. Overall, 138 of the 222 patients (62%) enrolled in the expansion cohorts had a documented T790M mutation whereas it was not detected in tumors from 62 patients in those cohorts (28%). For the remaining 22 patients, EGFR mutational status was unknown (10%). Overall, osimertinib showed a promising clinical activity with a an ORR of 51%. Of 239 patients evaluable for response, 123 (51%) experienced a partial response (PR, n = 122) or a complete response (CR, n = 1) and 78 (33%) had stable disease, while only 34 (14%) had progressive disease (PD). Importantly, in patients with a documented T790M mutation at central testing, the ORR was 61%, with a disease control rate (DCR) of 95%. On the other hand, the subgroup of patients who were negative for T790M experienced inferior ORR (21%) and DCR of (61%) compared with the T790M-positive counterpart. The median PFS was 8.2 months. Again, patients harboring a T790M mutation had a median PFS of 9.6 months, whereas those without the mutation had a lower median PFS of 2.8 months. No difference in ORR was observed between Asian and Western patients.42 In view of increasing toxicities observed at the 160 mg and 240 mg doses, and similar response rates reported across different osimertinib dosing schedules, 80 mg daily was the recommended dose for subsequent studies, including registration trials.
Figure 1.

Developmental program of osimertinib.
EMA, European Medicines Agency; US FDA, United States Food and Drug Administration.
These findings have been further enlarged by the preliminary results of the expansion cohort of the AURA trial and the phase II AURA2 trial (Figure 1). In the expansion cohort of AURA, which involved 201 patients with T790M-positive locally advanced or metastatic NSCLC, osimertinib was associated with an ORR assessed by an independent review committee (ICR) of 58% and a DCR of 92%.67 In the AURA2 trial, an open-label, single-arm phase II study, 210 patients with advanced NSCLC and harboring the T790M mutation as assessed by central testing confirmation (CobasTM EGFR mutation test) received osimertinib at the recommended dose of 80 mg. ORR assessed by ICR was 70% and DCR was 92%. The median duration of response was 11.4 months (95% CI 9.0–not calculable). Notably, data for 70 of these patients were censored at the time of data cutoff and the proportion of patients remaining in response was 76% and 59% at 6 and 9 months respectively, and 48% at 12 months. Median PFS as assessed by blinded independent central review was 9.9 months (95% CI 8.5–12.3) and the proportion of patients estimated to be progression-free at 6, 9 and 12 months was respectively 71%, 56% and 44%.68
More recently, updated results from the T790M-positive cohort of the AURA phase I trial, along with a pooled analysis of phase II studies (expansion of AURA trial and AURA2) were presented at the European Lung Cancer Conference (ELCC) 2016. Respectively, 63 and 411 patients received treatment within AURA phase I (only those who tested positive for T790M at central confirmation analysis) and AURA pooled phase II (expansion cohort of AURA + AURA2). In AURA phase I, investigator-assessed ORR was 71% (43/61; 95% CI 57–82), with a median duration of response (DoR) of 9.6 months (95% CI 7.7–15.6) and median PFS of 9.7 months (95% CI 8.3–13.6). On the other hand, in the pooled analysis of phase II studies, a blinded independent central review (BICR) confirmed an impressive ORR of 66% (262/397; 95% CI 61–71) with a median DoR approaching 12.5 months (95% CI 11.1–not calculable). The median PFS reached 11.0 months (95% CI 9.6–12.4) and the proportion of patients free of progression at 12 months was 47.5% (95% CI 42.4–52.5).69 Based on the above mentioned data, osimertinib was granted US FDA accelerated approval for T90M-positive patients who progress on a first or second-generation EGFR-TKI and European Medicines Agency (EMA) conditional authorization for patients with metastatic NSCLC who test positive for an EGFR T790M mutation regardless of prior EGFR-TKI treatment.
To further investigate the activity of osimertinib in EGFRm NSCLC, additional phase III studies have been designed with the aim to assess the role of osimertinib in different clinical settings (Figure 1). AURA3 is an open-label study in which 419 patients with locally advanced or metastatic NSCLC with an EGFR T790M mutation who had progressed to a previous EGFR-TKI were randomized in a 2:1 ratio to receive osimertinib or a platinum-based chemotherapy doublet as second-line therapy. The investigators reported a significantly better ORR with osimertinib (71%) compared with platinum-pemetrexed treatment (31%) (odds ratio, 5.39; 95% CI, 3.47–8.48, p < 0.001) The median DoR was 9.7 months with osimertinib and 4.1 months with platinum-pemetrexed. Accordingly, the duration of PFS according to a BICR was consistent with the investigator-assessed duration, and significantly favored osimertinib over platinum-pemetrexed treatment, with a median of 11.0 months versus 4.2 months (adjusted HR, 0.28; 95% CI, 0.20–0.38; p < 0.001).70 The efficacy outcomes are in line with those reported in the previous phase I/II studies. Due to a superior clinically meaningful outcome over platinum-based chemotherapy, osimertinib is now established as the standard of care for T790M-positive patients who progress on or following a first-line EGFR-TKI.
Whether osimertinib could provide a further clinical benefit as upfront therapy is still unknown. The ongoing phase III FLAURA trial will further characterize the potential of osimertinib 80 mg daily in the first-line EGFRm setting. This trial is a double-blind, randomized, phase III study aimed to assess the efficacy and safety of osimertinib compared with standard of care gefitinib or erlotinib in treatment-naïve patients with locally advanced or metastatic EGFRm NSCLC.71 This study is currently ongoing and results are eagerly awaited. However, though preliminary, updated data from the expansion cohort of the phase I/II AURA trial investigating the efficacy and safety of first-line osimertinib in treatment-naïve T790M positive advanced NSCLC patients, showed encouraging results in a first-line setting with a confirmed ORR of 77% and a median PFS of 19.3 months. Moreover, at 18 months 55% of patients were progression-free and the median duration of response had not yet been reached.72 Worthy of note, a ‘real life’ study of single agent osimertinib for metastatic EGFR T790M-positive NSCLC patients (ASTRIS), who have received prior EGFR-TKIs is currently ongoing worldwide. The primary outcome measures are OS and safety, and the estimated completion date is August 2019 [ClinicalTrials.gov identifier: NCT02474355].
Lastly, the ADAURA trial is a double-blind, placebo-controlled randomized study evaluating the efficacy and safety of osimertinib versus placebo as adjuvant treatment for patients with EGFRm stage Ib–IIIa NSCLC who underwent complete tumor resection followed or not by adjuvant chemotherapy [ClinicalTrials.gov identifier: NCT02511106] (Figure 1).
Safety and tolerability of osimertinib
First- and second-generation EGFR-TKIs have been tested on a large number of patients over the last decade, meanwhile most studies involving third-generation TKIs, including osimertinib are currently ongoing, and safety data are still immature. However, even though long-term follow up is required for a precise assessment on safety, osimertinib seems to have an acceptable toxicity profile and tolerability, better if indirectly compared with first- and second-generation EGFR-TKIs.
In the dose-escalation cohort of the phase I AURA trial, osimertinib displayed a satisfactory safety profile, and no dose limiting toxicities were observed at any dose level tested (ranging from 20–240 mg daily). The overall incidence of any grade of adverse event (AE) was 96%, with 32% of patients experiencing grade 3–5 AEs. AEs leading to dose reduction or drug withdrawal were observed in 7% and 6% of patients, respectively. Serious AEs considered to be treatment-related, as assessed by the site investigator, occurred in 6% of patients. Notably, no differences in the severity or frequency of AEs were observed between Asian and Western patients. The most common AEs were diarrhea (47%), rash (40%), nausea (22%), and decreased appetite (21%). Furthermore, 6 cases (2.4%) of pneumonitis-like AEs occurred, and resolved following osimertinib discontinuation. Additionally, 11 patients (4.3%) developed prolongation of the QTc interval while 6 patients (2.4%) experienced hyperglycemia during osimertinib treatment.42 The expansion cohort study and the AURA2 trial showed similar results in terms of safety and tolerability. In a pooled analysis of these studies the most common AEs of any grade with osimertinib 80 mg/daily were diarrhea (42%), rash (41%), dry skin (31%) followed by paronychia (25%). AEs requiring dose reduction or drug discontinuation manifested 4.4% and 5.6% of patients, respectively. A prolonged QTc interval (2.2%) and neutropenia (1.9%) were the most common event associated with dose reduction or discontinuation. Of note in both trials, four cases of deadly interstitial lung disease occurred that was considered eventually related to osimertinib by the investigator.69 More recently, osimertinib confirmed a satisfactory safety profile also in the AURA3 trial. In this study 273 out of 279 (98%) patients reported at least one AE. In the osimertinib arm, the most commonly reported AEs were diarrhea (41%), rash (34%), dry skin (23%), and paronychia (22%). AEs of grade ⩾3 occurred in 16 patients (6%) and included diarrhea (1%), anemia (1%), decreased appetite (1%), skin rash (1%) and interstitial lung disease (1%). Of note, interstitial lung disease-like AEs were recorded in 10 patients (4%) in the osimertinib group, with 9 events of grade 1 or 2 in severity and 1 death. Compared with platinum-pemetrexed chemotherapy, osimertinib was associated with a lower rate of AEs leading to permanent discontinuation (7% versus 10%).70
Efficacy of osimertinib in EGFR-mutant NSCLC brain metastases
Central nervous system (CNS) failure is a common event during treatment with EGFR-TKIs, probably reflecting the long survival benefit achieved with targeted therapies. In front of poor penetration rates of first-generation EGFR-TKIs across the blood–brain barrier (BBB), CNS response occurs in approximately 80% of patients with pre-existing brain metastases (BMs) treated with upfront gefitinib or erlotinib, suggesting a significant intracranial efficacy of these drugs. Unfortunately, after initial intracranial response 26–33% experience disease progression in CNS which associates with a very poor prognosis.73
Whether osimertinib exerts activity also against brain metastasis is still poorly addressed and data are lacking. In an early preclinical study involving an EGFRm BM xenograft mouse model, osimertinib distribution into the brain was 10-fold higher compared with gefitinib at clinically relevant doses, and resulted in a marked shrinkage of brain lesions.74 These data have been further corroborated by Ballard and colleagues who confirmed a greater penetration of osimertinib across the BBB than gefitinib, rociletinib (CO-1686), or afatinib, and at clinically relevant doses in a mouse model. Worthy of note, osimertinib was associated with a sustained tumor regression in an EGFRm PC9 mouse BM model; conversely the third-generation EGFR-TKI rociletinib, did not achieve tumor regression. Moreover, under positron emission tomography micro-dosing conditions, [11C]osimertinib showed a greater exposure in the cynomolgus monkey brain compared with both [11C]rociletinib and [11C]gefitinib.75 Interestingly, the authors also provided evidence of clinical activity of osimertinib against BMs in two patients (within the expansion cohort of AURA trial) with EGFRm and T790M-positive BMs who achieved an overall PR according to Response Evaluation Criteria In Solid Tumors (RECIST) 1.1, with noncomplete response-nonprogressive disease (SD) reported within the CNS.75 Consistent with these findings, our group has recently published the cases of two radiotherapy-naïve patients with EGFRm and T790M-positive pretreated advanced NSCLC and BMs who responded to osimertinib. There was one patient that had a solitary brain lesion at diagnosis and was treated with upfront erlotinib that led to a prolonged control of CNS disease and no need of cranial radiation. Upon documentation of T790M-positive disease progression (extra- and intracranial), brain radiotherapy was withheld in favor of osimertinib, which allowed a re-response in the brain and in extracranial sites, suggesting that osimertinib crosses the BBB and overcomes a T790M-mediated resistance in the CNS. On the other hand, the second patient developed CNS disease following T790M-mediated resistance to gefitinib. At the time of progression, the patient was switched to osimertinib from which they achieved an impressive intracranial response. Both cases confirmed the clinical activity of osimertinib in BMs from T790M-positive NSCLC. Overall, this report may suggest that delaying cranial radiation in favor of a third-generation EGFR-TKI osimertinib, might represent an excellent therapeutic alternative for radiotherapy-naïve patients progressing in the brain on a first- or second-generation EGFR-TKI, especially in view of the long life expectancy these patients usually encounter.76 More recently, pooled data from two phase II studies on CNS response to osimertinib in patients with T790M-positive advanced NSCLC has been presented at the International Association for the Study of Lung Cancer World Conference on Lung Cancer 2016. The authors reported an objective intracranial response rate (OIRR) of 54% and an impressive intracranial disease control rate (IDCR) of 92%. Of note, a CNS response was observed regardless of prior brain irradiation, and 82% of patients responded by the time of first assessment (within 6 weeks).77
Finally, it should be mentioned that an ongoing phase I study (BLOOM) [ClinicalTrials.gov identifier: NCT02228369] designed to assess the efficacy of osimertinib in EGFR-TKIs pretreated patients with BMs/leptomeningeal carcinomatosis from EGFRm NSCLC confirmed by positive cerebrospinal fluid (CSF) cytology. Preliminary results were presented at the 2016 American Society of Clinical Oncology (ASCO)meeting. A total of 20 patients were dosed with osimertinib 80 mg. Of 12 reaching a 12-week intracranial image assessment, 7 had radiological improvement, 2 had a stable disease and 3 were not evaluable. Importantly, of 12 patients reaching the 12-week neurological assessment, 6 were symptomatic, of which 3 had improvement in neurological symptoms, while 1 had no change and 2 were not evaluable. Notably, of 9 patients with pre-dose and cycle 2 day 1 CSF timepoint samples, 8 had a significant decrease in EGFRm DNA copy, which was >50% in 5 patients.78 Taken together, these data support the hypothesis that osimertinib can actually cross the BBB and be effective against CNS metastases from EGFRm NSCLC. Certainly these findings are still immature and should be interpreted with caution, however are very encouraging.
Regulatory affairs
In November 2015, the US FDA granted osimertinib approval for the treatment of patients with T790M-positive advanced NSCLC who had progressed on or following first- or second-generation EGFR-TKIs (gefitinib, erlotinib, afatinib) accompanied by the approval of a companion diagnostic test (Cobas® EGFR Mutation Test v2; Roche). Subsequently, osimertinib was registered with the same indication in Europe, Japan and South Korea. Of note, in Europe tissue- and blood-based EGFR mutation testing before treatment with osimertinib were allowed concurrently to the approval of the drug. Conversely, in the US the blood-based testing for EGFR mutations has been available for determining osimertinib eligibility since September 2016, but is not feasible in case tissue sampling.
Conclusion and future perspectives
EGFR-TKIs represent the mainstay of first-line therapy for patients with advanced NSCLC harboring sensitizing EGFR mutations. Nevertheless, resistance to treatment invariably occurs, generally within 1 year. Patients with disease progression after first-line TKIs have been traditionally switched to a platinum-doublet-based second-line chemotherapy, which unfortunately yields a disappointing ORR of 30%, considerably lower compared with 51% and 71% reported with osimertinib in phase I and II studies, respectively.79,80 Recently, the results of the phase III AURA3 trial have been published and confirmed an astounding clinical efficacy of osimertinib compared with platinum-doublet chemotherapy in terms of ORR and DCR (71% versus 31% and 93% versus 74%, respectively).2–3 Taken together, these data suggest the use of osimertinib upon documented T790M-mediated disease progression after therapy with EGFR-TKIs of the precedent generation, setting aside cytotoxic platinum-based chemotherapy as third-line treatment (Table 2).
Table 2.
Clinical efficacy of osimertinib in advanced NSCLC.
| Trial | Phase (n) | ORR | DCR | Median PFS | Remaining alive and progression-free |
|---|---|---|---|---|---|
| AURA67 | I (253) | Overall: 51% T790M-: 21% T790M+: 71% |
Overall: 84% T790M-: 61% T790M+: 93% |
Overall: 8.2 months T790M-: 2.8 months T790M+: 9.7 months |
12 months: 41% 18 months: 29% 24 months: 17% |
| Pooled AURA extension – AURA-269
|
II (441) | T790M+: 66% | T790M: 91% | T790M+: 11 months | 12 months: 48% 18 months: NA 24 months: NA |
| AURA 370 | III (279) | T790M+: 71% | T790M+: 93% | T790M+: 11 months | 6 months: 69% 12 months: 44% |
DCR, disease control rate; NSCLC, non-small cell lung cancer; ORR, objective response rate; PFS, progression-free survival; NA, not assessed.
Osimertinib is also being evaluated in a first-line setting within the phase III clinical trial FLAURA which compares osimertinib with gefitinib/erlotinib in EGFRm treatment-naïve advanced NSCLC. This study is expected to provide novel data that might elucidate whether osimertinib can gain a role as upfront therapy, as T790M-positive clones have been proven to coexist in untreated patients with an EGFR-sensitizing mutation. Finally, osimertinib is also being studied as part of combination therapy. In the phase Ib trial TATTON [ClinicalTrials.gov identifier: NCT02143466] osimertinib was administered in combination with either darvalumab (anti PD-L1), selumetinib (MEK inhibitor) or savoltinib (MET inhibitor). Preliminary results from the osimertinib plus darvalumab cohort showed an ORR of 67% (6/9) and 21% (3/14) in EGFR-TKI pretreated patients with T790M positive and T790M negative NSCLC, respectively, and 70% (7/10) in EGFRm treatment-naïve patients.79 However, the combination resulted in more toxicity, especially with immunotherapy, with interstitial lung disease and diarrhea occurring with higher incidence and grade in an osimertinib–darvalumab association then osimertinib or darvalumab alone.79 Interestingly, osimertinib is also being evaluated in combination with dasatinib for the treatment of patients with EGFRm NSCLC. Dasatinib is a potent, orally available Abelson murine leukemia viral oncogene homolog 1/Proto-Oncogene Tyrosine-Protein Kinase Src (ABL1/SRC) TKI, approved for the first-line treatment of chronic myeloid leukemia and in patients with imatinib-resistant disease and is being studied in patients with advanced solid tumors. Recently, researchers identified Cripto-1 overexpression as a mechanism of intrinsic resistance to EGFR-TKIs via activation of the SRC oncogene.80 On this basis, an open label, nonrandomized, phase I/II trial evaluating the combination of osimertinib and dasatinib in TKI-naïve, advanced, EGFRm NSCLC has been designed, and is currently recruiting participants [ClinicalTrials.gov identifier: NCT02954523]. Lastly, osimertinib is being studied in combination with bevacizumab for the treatment of TKI-naïve patients with EGFRm metastatic NSCLC in a phase I/II clinical trial [ClinicalTrials.gov identifier: NCT02803203]. The estimated primary completion date is June 2019.
Despite the impressive clinical results reported so far, patients with T790M-positive advanced NSCLC who initially respond to osimertinib constantly develop resistance to treatment, with preliminary data showing a PFS ranging from 9.7–11 months.42,69–70
In consideration of the clinical efficacy showed so far and the favorable safety profile, osimertinib is expected to become the standard treatment for patients with EGFRm and documented T790M mutation advanced NSCLC who progress on a first- or second-generation EGFR-TKI. Ongoing clinical trials will further clarify whether osimertinib can carve out a role in a first-line setting or in combination with other agents.
Along with osimertinib, several third-generation EGFR-TKIs are under clinical investigation. Rociletinib is a small mutant-selective, covalent inhibitor of both the activating EGFR mutations (exon 19 deletions and L858R) and the resistance mutation T790M. Despite initial promising results in terms of ORR (59%) and DCR (93%) among patients with centrally confirmed T790M-positive tumors in a phase I/II trial,43 the updated ‘confirmed’ response rates were extremely disappointing, reaching 34% at a 625 mg twice daily dose and 28% at a 500 mg twice daily dose in patients with T790M-positive NSCLC, with a median duration of response of 9 months for both doses (Clovis presentation at the JP Morgan Healthcare Conference, 13 January 2016). Following the achievement of these data, the developmental program of rociletinib has been recently discontinued.
EGF816 is another third-generation covalent EGFR inhibitor with potent inhibitory activity against the most common sensitizing del19 and L858R mutations and resistant T790M mutants.81 In preclinical models, EGF816 proved to be more efficacious than earlier generation EGFR inhibitors. Updated results of a phase I study conducted in advanced harboring T790M confirmed an ORR of 44% (56/127, with an additional 11 responders awaiting confirmation) with a DCR of 91%. Additionally, the Kaplan–Meier (KM) estimate of median duration of response (mDOR) was 9.2 months, while the median KM estimate of PFS was 9.2 months.82 Other third-generation EGFR-TKIs (e.g. PF-06747775, HM61713, ASP8273) are currently being evaluated in phase I/II trials, but final results of ORR and toxicities have not yet been published in peer-reviewed journals.
Certainly, the development of resistance to third-generation EGFR-TKIs represents a new challenge in the treatment of EGFR-addicted NSCLC. However, fourth generation TKIs that might overcome C797S-mediated resistance are already under preclinical evaluation and are expected to provide a new weapon against this deadly disease.
Footnotes
Funding: This work was supported by the Italian Association for Cancer Research (AIRC).
Conflict of interest statement: The authors declare that there is no conflict of interest.
Contributor Information
Biagio Ricciuti, Medical Oncology, Santa Maria della Misericordia Hospital, Azienda Ospedaliera di Perugia, via Dottori, 1 - 06156, Perugia, Italy.
Sara Baglivo, Medical Oncology, Santa Maria della Misericordia Hospital, Azienda Ospedaliera di Perugia, Perugia, Italy.
Luca Paglialunga, Medical Oncology, Santa Maria della Misericordia Hospital, Azienda Ospedaliera di Perugia, Perugia, Italy.
Andrea De Giglio, Medical Oncology, Santa Maria della Misericordia Hospital, Azienda Ospedaliera di Perugia, Perugia, Italy.
Guido Bellezza, Department of Experimental Medicine, Division of Pathology and Histology, University of Perugia Medical School, Perugia, Italy.
Rita Chiari, Medical Oncology, Santa Maria della Misericordia Hospital, Azienda Ospedaliera di Perugia, Perugia, Italy.
Lucio Crinò, Medical Oncology, Santa Maria della Misericordia Hospital, Azienda Ospedaliera di Perugia, Perugia, Italy.
Giulio Metro, Medical Oncology, Santa Maria della Misericordia Hospital, Azienda Ospedaliera di Perugia, Perugia, Italy.
References
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin 2015; 65: 5–29. [DOI] [PubMed] [Google Scholar]
- 2. Pao W, Chmielecki J. Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat Rev Cancer 2010; 10: 760–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sharma SV, Bell DW, Settleman J, et al. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer 2007; 7: 169–181. [DOI] [PubMed] [Google Scholar]
- 4. Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 2004; 350: 2129–2139. [DOI] [PubMed] [Google Scholar]
- 5. Pao W, Miller V, Zakowski M, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci USA 2004; 101: 13306–13311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Paez JG, Jänne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004; 304: 1497–1500. [DOI] [PubMed] [Google Scholar]
- 7. Califano R, Abidin A, Tariq NU, et al. Beyond EGFR and ALK inhibition: unravelling and exploiting novel genetic alterations in advanced non small-cell lung cancer. Cancer Treat Rev 2015; 41: 401–411. [DOI] [PubMed] [Google Scholar]
- 8. Murray S, Dahabreh IJ, Linardou H, et al. Somatic mutations of the tyrosine kinase domain of the epidermal growth factor receptor and tyrosine kinase inhibitor response to TKIs in non-small cell lung cancer: an analytical database. J Thorac Oncol 2008; 3: 832–839. [DOI] [PubMed] [Google Scholar]
- 9. Yang JC, Sequist LV, Geater SL, et al. Clinical activity of afatinib in patients with advanced non-small-cell lung cancer harbouring uncommon EGFR mutations: a combined post-hoc analysis of LUX-Lung 2, LUX-Lung 3, and LUX-Lung 6. Lancet Oncol 2015; 16: 830–838. [DOI] [PubMed] [Google Scholar]
- 10. Wu JY, Yu CJ, Chang YC, et al. Effectiveness of tyrosine kinase inhibitors on “uncommon” epidermal growth factor receptor mutations of unknown clinical significance in non-small cell lung cancer. Clin Cancer Res 2011; 17: 3812–3821. [DOI] [PubMed] [Google Scholar]
- 11. De Pas T, Toffalorio F, Manzotti M, et al. Activity of epidermal growth factor receptor-tyrosine kinase inhibitors in patients with non-small cell lung cancer harbouring rare epidermal growth factor receptor mutations. J Thorac Oncol 2011; 6: 1895–1901. [DOI] [PubMed] [Google Scholar]
- 12. Chiu CH, Yang CT, Shih JY, et al. Epidermal growth factor receptor tyrosine kinase inhibitor treatment response in advanced lung adenocarcinomas with G719X/L861Q/S768I mutations. J Thorac Oncol 2015; 10: 793–739. [DOI] [PubMed] [Google Scholar]
- 13. Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med 2009; 361: 947–957. [DOI] [PubMed] [Google Scholar]
- 14. Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med 2010; 362: 2380–2388. [DOI] [PubMed] [Google Scholar]
- 15. Rosell R, Moran T, Queralt C, et al. Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med 2009; 361: 958–967. [DOI] [PubMed] [Google Scholar]
- 16. Yang JC, Wu YL, Schuler M, et al. Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): analysis of overall survival data from two randomised, phase 3 trials. Lancet Oncol 2015; 16: 141–151. [DOI] [PubMed] [Google Scholar]
- 17. Park KS, Raffeld M, Moon YW, et al. CRIPTO1 expression in EGFR-mutant NSCLC elicits intrinsic EGFR-inhibitor resistance. J Clin Invest 2014; 124: 3003–3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yu HA, Arcila ME, Rekhtman N, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res 2013; 19: 2240–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Takezawa K, Pirazzoli V, Arcila ME, et al. HER2 amplification: a potential mechanism of acquired resistance to EGFR inhibition in EGFR-mutant lung cancers that lack the second-site EGFRT790M mutation. Cancer Discov 2012; 2: 922–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yasuda H, Kobayashi S, Costa DB. EGFR exon 20 insertion mutations in non-small-cell lung cancer: preclinical data and clinical implications. Lancet Oncol 2012; 13:e23–e31. [DOI] [PubMed] [Google Scholar]
- 21. Eck MJ, Yun CH. Structural and mechanistic underpinnings of the differential drug sensitivity of EGFR mutations in non-small cell lung cancer. Biochim Biophys Acta 2010; 1804: 559–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Godin-Heymann N, Bryant I, Rivera MN, et al. Oncogenic activity of epidermal growth factor receptor kinase mutant alleles is enhanced by the T790M drug resistance mutation. Cancer Res 2007; 67: 7319–7326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rosell R, Molina MA, Costa C, et al. Pretreatment EGFR T790M mutation and BRCA1 mRNA expression in erlotinib-treated advanced non-small-cell lung cancer patients with EGFR mutations. Clin Cancer Res 2011; 17: 1160–1168. [DOI] [PubMed] [Google Scholar]
- 24. Rosell R, Molina-Vila MA, Taron M, et al. EGFR compound mutants and survival on erlotinib in non-small cell lung cancer (NSCLC) patients (p) in the EURTAC study. J Clin Oncol 2012; 30: abstract 7522. [Google Scholar]
- 25. Fujita Y, Suda K, Kimura H, et al. Highly sensitive detection of EGFR T790M mutation using colony hybridization predicts favorable prognosis of patients with lung cancer harboring activating EGFR mutation. J Thorac Oncol 2012; 7: 1640–1644. [DOI] [PubMed] [Google Scholar]
- 26. Ji H, Zhao X, Yuza Y, et al. Epidermal growth factor receptor variant III mutations in lung tumorigenesis and sensitivity to tyrosine kinase inhibitors. Proc Natl Acad USA 2006; 103: 7817–7822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Faber AC, Corcoran RB, Ebi H, et al. BIM expression in treatment-naive cancers predicts responsiveness to kinase inhibitors. Cancer Discov 2011; 1: 352–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Murakami H, Nokihara H, Shimizu T, et al. 9LBA Antitumor activity of ASP8273, an irreversible mutant selective EGFR-TKI, in NSCLC patients with tumors harboring EGFR activating mutations and T790M resistance mutation. Eur J Cancer 2014; 50: 198. [Google Scholar]
- 29. Zhou W, Ercan D, Chen L, et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 2009; 462: 1070–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cross DA, Ashton SE, Ghiorghiu S, et al. AZD9291, an irreversible EGFR TKI, overcomes T790Mmediated resistance to EGFR inhibitors in lung cancer. Cancer Discov 2014; 4: 1046–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wu SG, Liu YN, Tsai MF, et al. The mechanism of acquired resistance to irreversible EGFR tyrosine kinase inhibitor-afatinib in lung adenocarcinoma patients. Oncotarget 2016; 7: 12404–12413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Arcila ME, Oxnard GR, Nafa K, et al. Rebiopsy of lung cancer patients with acquired resistance to EGFR inhibitors and enhanced detection of the T790M mutation using a locked nucleic acid-based assay. Clin Cancer Res 2011; 17: 1169–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Blencke S, Ullrich A, Daub H. Mutation of threonine 766 in the epidermal growth factor receptor reveals a hotspot for resistance formation against selective tyrosine kinase inhibitors. J Biol Chem 2003; 278: 15435–15440. [DOI] [PubMed] [Google Scholar]
- 34. Pao W, Miller VA, Politi KA, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med 2005; 2: e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yun CH, Mengwasser KE, Toms AV, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci USA 2008; 105: 2070–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hata A, Katakami N, Yoshioka H, et al. Rebiopsy of non-small cell lung cancer patients with acquired resistance to epidermal growth factor receptor-tyrosine kinase inhibitor: comparison between T790M mutation-positive and mutation-negative populations. Cancer 2013; 119: 4325–4332. [DOI] [PubMed] [Google Scholar]
- 37. Ma C, Wei S, Song Y. T790M and acquired resistance of EGFR TKI: a literature review of clinical reports. J Thorac Dis 2011; 3: 10–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Su KY, Chen HY, Li KC, et al. Pretreatment epidermal growth factor receptor (EGFR) T790M mutation predicts shorter EGFR tyrosine kinase inhibitor response duration in patients with non-small-cell lung cancer. J Clin Oncol 2012; 30: 433–440. [DOI] [PubMed] [Google Scholar]
- 39. Suda K, Onozato R, Yatabe Y, et al. EGFR T790M mutation: a double role in lung cancer cell survival? J Thorac Oncol 2009; 4: 1–4. [DOI] [PubMed] [Google Scholar]
- 40. Kim Y, Ko J, Cui Z, et al. The EGFR T790M mutation in acquired resistance to an irreversible second-generation EGFR inhibitor. Mol Cancer Ther 2012; 11: 784–791. [DOI] [PubMed] [Google Scholar]
- 41. Miller VA, Hirsh V, Cadranel J, et al. Afatinib versus placebo for patients with advanced, metastatic non-small-cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUX-Lung 1): a phase 2b/3 randomised trial. Lancet Onco 2012; 13: 528–538. [DOI] [PubMed] [Google Scholar]
- 42. Janne PA, Yang JC, Kim DW, et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N Engl J Med 2015; 372: 1689–1699. [DOI] [PubMed] [Google Scholar]
- 43. Sequist LV, Soria JC, Goldman JW, et al. Rociletinib in EGFR-mutated non-small-cell lung cancer. N Engl J Med 2015; 372: 1700–1709. [DOI] [PubMed] [Google Scholar]
- 44. Niederst MJ, Hu H, Mulvey HE, et al. The allelic context of the C797S mutation acquired upon treatment with third-generation EGFR inhibitors impacts sensitivity to subsequent treatment strategies. Clin Cancer Res 2015; 21: 3924–3933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Costa DB, Halmos B, Kumar A, et al. BIM mediates EGFR tyrosine kinase inhibitor-induced apoptosis in lung cancers with oncogenic EGFR mutations. PLoS Med 2007; 4: 1669–1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Balak MN, Gong Y, Riely GJ, et al. Novel D761Y and common secondary T790M mutations in epidermal growth factor receptor-mutant lung adenocarcinomas with acquired resistance to kinase inhibitors. Clin Cancer Res 2006; 12: 6494–6501. [DOI] [PubMed] [Google Scholar]
- 47. Diaz LA, Jr, Bardelli A. Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol 2014; 32: 579–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Weber B, Meldgaard P, Hager H, et al. Detection of EGFR mutations in plasma and biopsies from non-small cell lung cancer patients by allele-specific PCR assays. BMC Cancer 2014; 14: 294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Oxnard GR, Paweletz CP, Sholl LM. Genomic analysis of plasma cell-free DNA in patients with cancer. JAMA Oncol. Epub ahead of print 2016. DOI: 10.1001/jamaoncol.2016.2835 [DOI] [PubMed] [Google Scholar]
- 50. Liu Y, Liu B, Li XY, et al. A comparison of ARMS and direct sequencing for EGFR mutation analysis and tyrosine kinase inhibitors treatment prediction in body fluid samples of non-small-cell lung cancer patients. J Exp Clin Cancer Res 2011; 30: 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Goto K, Ichinose Y, Ohe Y, et al. Epidermal growth factor receptor mutation status in circulating free DNA in serum: from IPASS, a phase III study of gefitinib or carboplatin/paclitaxel in non-small cell lung cancer. J Thorac Oncol 2012; 7: 115–121. [DOI] [PubMed] [Google Scholar]
- 52. Douillard JY, Ostoros G, Cobo M, et al. Gefitinib treatment in EGFR mutated Caucasian NSCLC: circulating-free tumor DNA as a surrogate for determination of EGFR status. J Thorac Oncol 2014; 9: 1345–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mitsudomi T, Morita S, Yatabe Y, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol 2010; 11: 121–128. [DOI] [PubMed] [Google Scholar]
- 54. Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol 2012; 13: 239–246. [DOI] [PubMed] [Google Scholar]
- 55. Reck M, Hagiwara K, Han B, et al. ctDNA determination of EGFR mutation status in European and Japanese patients with advanced NSCLC: The ASSESS Study. J Thorac Oncol 2016; 11(10): 1682–1689. [DOI] [PubMed] [Google Scholar]
- 56. Luo J, Shen L, Zheng D. Diagnostic value of circulating free DNA for the detection of EGFR mutation status in NSCLC: a systematic review and meta-analysis. Sci Rep 2014; 4: 6269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mao C, Yuan JQ, Yang ZY, et al. Blood as a substitute for tumor tissue in detecting EGFR mutations for guiding EGFR TKIs treatment of non-small cell lung cancer: a systematic review and meta-analysis. Medicine (Baltimore) 2015; 94: e775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sacher AG, Paweletz C, Dahlberg SE, et al. Prospective validation of rapid plasma genotyping for the detection of EGFR and KRAS mutations in advanced lung cancer. JAMA Oncol 2016; 2: 1014–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Takahama T, Sakai K, Takeda M, et al. Detection of the T790M mutation of EGFR in plasma of advanced non-small cell lung cancer patients with acquired resistance to tyrosine kinase inhibitors (West Japan oncology group 8014LTR study). Oncotarget 2016; 7: 58492–58499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sun JM, Karlovich C, Wen W, et al. Serial monitoring of EGFR mutations in plasma and evaluation of EGFR mutation status in matched tissue and plasma from NSCLC patients treated with CO-1686. Mol Cancer Ther 2013; 12(Suppl. 11): abstract. B25. [Google Scholar]
- 61. Oxnard GR, Thress KS, Alden RS, et al. Association between plasma genotyping and outcomes of treatment with osimertinib (AZD9291) in advanced non-small-cell lung cancer. J Clin Oncol 2016; 34: 3375–3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Thress KS, Brant R, Carr TH, et al. EGFR mutation detection in ctDNA from NSCLC patient plasma: a cross-platform comparison of leading technologies to support the clinical development of AZD9291. Lung Cancer 2015; 90: 509–515. [DOI] [PubMed] [Google Scholar]
- 63. Oxnard GR, Paweletz CP, Kuang Y, et al. Noninvasive detection of response and resistance in EGFR-mutant lung cancer using quantitative next-generation genotyping of cell-free plasma DNA. Clin Cancer Res 2014; 20: 1698–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sorensen BS, Wu L, Wei W, et al. Monitoring of epidermal growth factor receptor tyrosine kinase inhibitor-sensitizing and resistance mutations in the plasma DNA of patients with advanced non-small cell lung cancer during treatment with erlotinib. Cancer 2014; 120: 3896–3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Carey KD, Garton AJ, Romero MS, et al. Kinetic analysis of epidermal growth factor receptor somatic mutant proteins shows increased sensitivity to the epidermal growth factor receptor tyrosine kinase inhibitor, erlotinib. Cancer Res 2006; 66: 8163–81671. [DOI] [PubMed] [Google Scholar]
- 66. Gazdar AF. Activating and resistance mutations of EGFR in non-small-cell lung cancer: role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene 2009; 28(Suppl. 1): S24–S31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Yang JC, Ahn M, Ramalingam SS, et al. AZD9291 in pre-treated T790M positive advanced NSCLC: AURA study phase II extension cohort. J Thorac Oncol 2015; 10(Suppl. 2): S319. [Google Scholar]
- 68. Goss G, Tsai CM, Shepherd FA, et al. Osimertinib for pretreated EGFR Thr790Met-positive advanced non-small-cell lung cancer (AURA2): a multicentre, open-label, single-arm, phase 2 study. Lancet Oncol 2016; 17: 1643–1652. [DOI] [PubMed] [Google Scholar]
- 69. Yang JC, Ramalingam SS, Jänne PA, et al. Osimertinib (AZD9291) in pre-treated patients with T790M-positive advanced NSCLC: updated phase I and pooled phase II results. J Thorac Oncol 2016; 11(Suppl. 4): S57–S166. [Google Scholar]
- 70. Mok TS, Wu YL, Ahn MJ, et al. Osimertinib or platinum-pemetrexed in EGFR T790M-positive lung cancer. N Engl J Med 2016; 376: 629–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ramalingam S, Rukazenkov Y, Thomas K, et al. A randomized, phase III study (FLAURA) of AZD9291, a novel EGFR-TKI, versus gefitinib or erlotinib in treatment-naïve patients with advanced non-small cell lung cancer and an EGFR-TKI-sensitizing mutation. J Clin Oncol 2015; 33(Suppl. 33): abstract TPS8102. [Google Scholar]
- 72. Ramalingam S, Yang J, Lee CH, et al. Osimertinib (AZD9291) as first-line treatment for EGFR mutation-positive advanced NSCLC: updated efficacy and safety results from two phase I expansion cohorts. J Thorac Oncol 2016; 11(Suppl. l4): S152. [Google Scholar]
- 73. Metro G, Chiari R, Ricciuti B, et al. Pharmacotherapeutic options for treating brain metastases in non-small cell lung cancer. Expert Opin Pharmacother 2016; 16: 2601–2613. [DOI] [PubMed] [Google Scholar]
- 74. Remon J, Planchard D. AZD9291 in EGFR-mutant advanced non-small-cell lung cancer patients. Future Oncol 2015; 11: 3069–3081. [DOI] [PubMed] [Google Scholar]
- 75. Ballard P, Yates JW, Yang Z, et al. Preclinical comparison of osimertinib with other EGFR-TKIs in EGFR-mutant NSCLC brain metastases models, and early evidence of clinical brain metastases activity. Clin Cancer Res 2016; 22: 5130–5140. [DOI] [PubMed] [Google Scholar]
- 76. Ricciuti B, Chiari R, Chiarini P, et al. Osimertinib (AZD9291) and CNS response in two radiotherapy-naïve patients with EGFR-mutant and T790M-positive advanced non-small cell lung cancer. Clin Drug Investig 2016; 36: 683–686. [DOI] [PubMed] [Google Scholar]
- 77. Goss G, Shepherd FA, Ahn MJ, et al. CNS response to osimertinib in patients with T790M-positive advanced NSCLC: pooled data from two phase II trials. J Thorac Oncol 2016; 12(Suppl. 1S): S440–S441. [DOI] [PubMed] [Google Scholar]
- 78. Yang J, Kim DW, Kim SW, et al. Osimertinib activity in patients (pts) with leptomeningeal (LM) disease from non-small cell lung cancer (NSCLC): updated results from BLOOM, a phase I study. J Clin Oncol 2016; 34(Suppl. 34): abstract 9002. [Google Scholar]
- 79. Ahn MJ, Yang J, Yu H, et al. Osimertinib combined with durvalumab in EGFR-mutant non-small cell lung cancer: results from the TATTON phase Ib trial. J Thorac Oncol 2016; 11(Suppl. S115): 4. [Google Scholar]
- 80. Park KS, Raffeld M, Moon YW, et al. CRIPTO1 expression in EGFR-mutant NSCLC elicits intrinsic EGFR-inhibitor resistance. J Clin Invest 2014; 124: 3003–3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Jia Y, Juarez J, Li J, et al. EGF816 exerts anticancer effects in non-small cell lung cancer by irreversibly and selectively targeting primary and acquired activating mutations in the EGF receptor. Cancer Res 2016; 76: 1591–1602. [DOI] [PubMed] [Google Scholar]
- 82. Tan D, Yang JC, Leighl NB, et al. Updated results of a phase 1 study of EGF816, a third-generation, mutant-selective EGFR tyrosine kinase inhibitor (TKI), in advanced non-small cell lung cancer (NSCLC) harboring T790M. J Clin Oncol 2016; 34(Suppl. 34): abstract 9044. [Google Scholar]