

Abstract
Many bacteria, including the model bacterium Escherichia coli can survive for years within spent media, following resource exhaustion. We carried out evolutionary experiments, followed by whole genome sequencing of hundreds of evolved clones to study the dynamics by which E. coli adapts during the first 4 months of survival under resource exhaustion. Our results reveal that bacteria evolving under resource exhaustion are subject to intense selection, manifesting in rapid mutation accumulation, enrichment in functional mutation categories and extremely convergent adaptation. In the most striking example of convergent adaptation, we found that across five independent populations adaptation to conditions of resource exhaustion occurs through mutations to the three same specific positions of the RNA polymerase core enzyme. Mutations to these three sites are strongly antagonistically pleiotropic, in that they sharply reduce exponential growth rates in fresh media. Such antagonistically pleiotropic mutations, combined with the accumulation of additional mutations, severely reduce the ability of bacteria surviving under resource exhaustion to grow exponentially in fresh media. We further demonstrate that the three positions at which these resource exhaustion mutations occur are conserved for the ancestral E. coli allele, across bacterial phyla, with the exception of nonculturable bacteria that carry the resource exhaustion allele at one of these positions, at very high frequencies. Finally, our results demonstrate that adaptation to resource exhaustion is not limited by mutational input and that bacteria are able to rapidly adapt under resource exhaustion in a temporally precise manner through allele frequency fluctuations.
Keywords: experimental evolution, adaptation, resource exhaustion, convergent evolution, antagonistic pleiotropy, mutators
Introduction
Elucidating the selective pressures to which bacteria adapt within natural environments and characterizing the dynamics with which such natural adaptation occurs is important from the evolutionary, ecological as well as the medical perspectives. Within their environments, bacteria likely often experience periods of boom, in which they freely grow, followed by periods of bust in which they no longer have the resources needed for growth and must maintain some viability until they again face growth-enabling conditions.
Fitting with this need for long-term maintenance, Eshcerichia coli and many other nonsporulating bacteria show a remarkable ability to maintain themselves in spent media without any external nutrient replenishment, for prolonged periods of time. Following exponential growth, when such bacteria exhaust their growth-resources, they will enter a short stationary phase followed by a rapid death phase. However, this death phase does not lead to complete loss of viability within the bacterial population. Rather, a small subset of cells is able to maintain viability and enter a growth-phase known as long-term stationary phase (LTSP; Zambrano et al., 1993; Finkel and Kolter, 1999; Finkel, 2006). During LTSP some cells manage to replicate, by recycling the remains of their deceased brethren. E. coli cells can be maintained in LTSP for a large number of years, without any replenishment of nutrients. Past studies have demonstrated the emergence of novel phenotypes with time within LTSP populations as well as fluctuations in phenotype frequencies (Zambrano et al., 1993; Finkel and Kolter, 1999; Finkel, 2006). Specific alleles were identified as conferring an advantage under conditions of LTSP, most notably within the gene encoding RpoS, the master regulator of the stress response, but also within other genes (Zambrano et al., 1993; Finkel and Kolter, 1999; Finkel, 2006). Despite many years of research into LTSP, the dynamics of adaptation under this fascinating stage of growth remain ill characterized.
Due to their immense complexity, it is extremely difficult to study the dynamics of adaptation within natural environments. An approach that has proven very useful in the study of adaption is experimental evolution, or the study of adaptation as it occurs under more controlled selective regimens in the lab (reviewed in Barrick and Lenski, 2013; Kassen, 2014). Normally, such experiments are designed to maintain selective pressures as well as growth constant or semiconstant (fig. 1A and B;Barrick and Lenski, 2013; Kassen, 2014). Microbial evolutionary experiments have revealed several important insights into the dynamics of adaptation under conditions of continuous or semicontinuous growth. For example, it has been demonstrated that adaptation tends to be quite convergent across independent populations exposed to a similar selective regimen (Conrad et al., 2010; Tenaillon et al., 2012; Avrani and Lindell, 2015; Tenaillon et al., 2016). Such convergence manifests at various levels including in the extent of fitness gains with time, in phenotypic changes and in the specific genes and pathways that are involved in adaptation. It has also been shown that bacterial adaptation often involves soft, rather than hard sweeps, resulting from clonal interference between alleles (Barrick and Lenski, 2009; Maddamsetti et al., 2015). Such clonal interference is not expected to occur in small populations with limited mutational input (Fogle et al., 2008). Adaptation can sometimes involve mutations demonstrating antagonistic pleiotropy, meaning that a mutation adaptive under the selective regimen studied may be deleterious under other conditions (Cooper and Lenski, 2000; Kvitek and Sherlock, 2013). Finally, it was demonstrated that during adaptation mutator strains often emerge that increase the rate of mutation within the adapting population (Barrick and Lenski, 2009; Barrick et al., 2009; Wielgoss et al., 2013; Tenaillon et al., 2016). Whether or not these characteristics of the adaptive process also hold under conditions of severe growth-limitation, following exhaustion of resources is unknown.
Fig. 1.

Experimental evolutionary setups. (A) Continuous culture—Through a constant inflow of nutrients and outflow of random cells and waste, bacteria are allowed to grow continuously, while maintaining a nearly constant population size (B) Serial dilution—semi-continuous growth—bacteria are transferred into fresh media at set intervals, allowing for a consistent number of replications between dilution cycles. (C) LTSP evolutionary experiments—bacteria are only initially provided with resources needed for growth. Shortly after they spend their resources they enter a phase of accelerated death, followed by LTSP. Panels A and B of this figure were adapted from (Barrick and Lenski, 2013).
Here, we carried out a different type of evolutionary experiment, which we refer to as an LTSP evolutionary experiment (fig. 1C). In this experiment five independent populations inoculated from the same E. coli genotype are maintained under LTSP for prolonged periods, following a short initial phases of growth. Almost 250 clones regrown from six time points, spanning the first four months of LTSP, were sequenced and high frequency mutations present within the LTSP populations were identified. The resulting data were used to characterize the dynamics of adaptation under LTSP.
Results
LTSP Evolutionary Experiment and Sequencing of Evolved Clones
Five populations were initiated by inoculating ∼5 × 106 cells per ml of the E. coli lab strain K12 MG1655 into 400 ml of Luria Broth (LB) inside 2 liter Erlenmeyer flasks. The flasks were placed into an incubator set to 37 °C and shaken at 225 rpm. One ml samples were drawn at first daily, then weekly, then monthly and finally at longer intervals over the duration of the experiment so far. No new resources were added to the cultures with time and only water was added to counter dehydration of the cultures. Samples were used to quantify the number of viable cells present with time (fig. 2). Bacteria from each sample were frozen for future sequencing and additional analyses.
Fig. 2.

Mean number of viable cells with time in five LTSP populations maintained in 400 ml LB culture, in 2 liter flasks. Error bars represent the standard deviation across the five populations. Number of viable cells was estimated by counting colony-forming units (CFU), following regrowth on LB agar plates. 9–11 clones from each of the five populations were sequenced, from three times points (red dots). 9–11 clones from each of three of the populations were sequenced from an additional three time points (blue dots).
As can be seen in figure 2, bacteria grew to ∼6.8 × 109 cells per ml over the first 24 hours of the experiment and then started dying rapidly over the following 24 hours. By day 11, viability declined almost 300-fold to ∼2.3 × 107 cells per ml. Viability continued decreasing following day 11, but the rate of loss of viability reduced substantially and over the next year viability only decreased around 20-fold. Given these data we consider day 11 as a time point that represents a switch between death phase and LTSP.
Next, we obtained whole genome sequences to allow us to identify mutations arising during LTSP. To obtain DNA for sequencing for a certain population at a certain time point, we streaked out bacteria from the sample extracted from that population at that time point. We then isolated individual colonies and extracted DNA for sequencing from these clones. Such clones must represent individual cells that have survived so far in the experiment and that maintained their ability to regrow. 9–11 clones from each of the five populations were sequenced from the day 11, day 64, and day 127 time points. In addition, we also sequenced 9–11 clones from populations 1, 2, and 4 from the day 22, day 32, and day 42 time points (fig. 2, supplementary table S1, Supplementary Material online). The ancestral clones used to initiate the five populations were also fully sequenced. In total, we obtained sufficient sequence coverage for 242 evolved clones and for the five ancestral genotypes (that all turned out to be genetically identical, as expected).
In order to call mutations occurring in the evolved clones, we aligned the sequences of each clone as well as of the ancestors used to initiate the five populations to the E. coli K12 MG1655 reference genome. We then used the breseq pipeline (Deatherage and Barrick, 2014) to call mutations as variants appearing in a clone, but not within its ancestor. The numbers of mutations identified per clone are given in supplementary table S1, Supplementary Material online. A detailed list of all mutations observed is provided in supplementary table S2, Supplementary Material online. The raw sequence data has been deposited in the Sequence Read Archive (SRA), under accession number: PRJNA380864.
The Majority of Cells Surviving to Enter LTSP Carry a Genetic Modification
As seen in figure 2, prior to entering LTSP, bacteria experience a phase of rapid death in which their numbers diminish ∼300-fold. We wished to examine whether cells surviving this rapid death phase to reach day 11 tend to survive because they carry a genetic modification that contributes to their survival, or whether the subset of surviving cells constitutes a random sample of the population that entered death phase. Prior to entry into death phase each of our five populations contained an average of 400 × 6.8 × 109 cells (fig. 2). 41 generations of growth are required for a single ancestral cell to grow to such a number. In the absence of any selection, based on a mutation rate of 0.001 mutations per genome per generation estimated for E. coli (Lee et al., 2012), and based on 41 generations of growth, we would expect cells within our population to carry an average of 0.041 mutations per genome. Based on this expected average number of mutations, we carried out 1000 independent computer simulations to estimate the proportion of cells that would be expected to carry at least one mutation, prior to entry into death phase (see Materials and Methods). Across these 1,000 simulations the percentage of cells carrying one or more mutations ranged between 3.5% and 4.6%. This means that if cells surviving to day 11 constitute a random sample of those cells that entered death phase, we would expect the vast majority of them not to carry any mutations. However, we found a very different result in our actual LTSP experiments. Namely, we find that 93.5% of cells surviving to day 11 carry at least one mutation. Even more striking, the vast majority of mutations found at day 11, across all populations occur within a single gene complex (The RNA-polymerase core enzyme, see below), strongly indicating they carry an adaptive function. Combined, our results imply that the cells that survive to day 11 are not a random subset of those cells that entered death phase. Rather, the majority of cells surviving to day 11 likely do so because they carry an adaptive mutation contributing to their survival.
Rapid Accumulation of Mutations in Both Mutator and NonMutator Clones
Across all five populations examined, mutations accumulate with time spent under LTSP (fig. 3). In three of the five populations, mutator strains, carrying a mutation to a mismatch repair gene emerged during LTSP (table 1). As expected, mutator clones tend to carry a significantly higher number of mutations than nonmutator clones, extracted at the same time points (P ≪ 0.0001, for all comparisons, according to a Mann–Whitney test). Yet, even nonmutator clones accumulate on average eight mutations per clone by day 127 of our experiments (fig. 3). For comparison sake, we plotted the accumulation of mutations with time in nonmutator clones from our experiments, against mutation accumulation with time estimated from Richard Lenski’s long-term evolutionary experiment (LTEE [Tenaillon et al., 2016], fig. 3). Within the LTEE bacteria have been serially diluted daily into fresh media, guaranteeing ∼6.6 generations of growth per day (Lenski et al., 1991). According to this comparison, it has taken the LTEE clones 2000 generations, or nearly 300 serial dilutions to accumulate an average number of mutations, that is similar to that our LTSP clones have accumulated following only 127 days under LTSP.
Fig. 3.
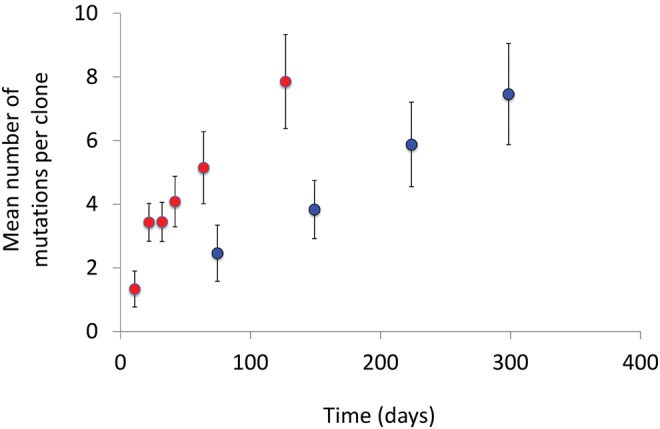
Mean number of mutations per clone increases with time spent under LTSP. Depicted is the average number of mutations observed per nonmutator LTSP clone across all sampled populations (Red dots). For comparison sake we also depict the number of mutations accumulated as a function of time within clones sequenced from Richard Lenski’s long-term serial dilution evolutionary experiment (Tenaillon et al., 2016, blue dots). Error bars represent the standard deviation across clones.
Table 1.
Observed Mutator Mutations.
| Population | Mutator Mutation | Mutator First Observed at |
|---|---|---|
| 2 | MutS T300K | Day 22 |
| 3 | Frame-shift mutation within mutS gene | Day 64 (the Day 22, 32, and 42 time points were not yet sequenced for this population) |
| 4 | 87 bp deletion within mutL gene | Day 32 |
Strong Signal of Positive Selection Affecting the Mutations Observed within LTSP Clones
Under neutrality we would expect to observe all mutation types at certain relative frequencies that can be calculated based on the distribution of site types within the E. coli genome. However, the mutations we observed within LTSP clones are significantly enriched for classes of mutations that are more likely to carry a functional effect. Such an enrichment of potentially functional mutations is widely considered to be a signal of positive selection affecting mutations (Graur and Wen-Hsiung, 2000; Ostrow et al., 2014; Tenaillon et al., 2016). Mutations occurring within protein-coding genes can be classified into nonsynonymous, if they change both the DNA and amino acid sequence of the gene, or synonymous, if they change only the DNA, but not the amino acid sequence. Because they affect the amino acid sequence of a gene, nonsynonymous mutations are more likely to be functional. We find within our LTSP clones a significant enrichment in nonsynonymous mutations, relative to neutral expectations (supplementary fig. S1A, Supplementary Material online, P ≪ 0.0001 for nonmutator clones and P = 0.008 for mutator clones, according to a χ2 test [df = 1]). Furthermore, when examining intergenic sequences, it is possible to classify them into those that likely contain a gene promoter and those that likely don’t (see Materials and Methods). We expect that mutations within intergenic regions that more likely contain a promoter will be more likely functional than those appearing in intergenic regions that are less likely to contain a promoter. We find a significant enrichment in LTSP mutations within likely promoter-containing intergenic regions, compared with intergenic regions less likely to contain a promoter (supplementary fig. S1B, Supplementary Material online, P ≪ 0.0001 for nonmutator and P = 0.016 for mutator clones, according to a χ2 test [df = 1]). Combined, these results suggest that there is a significant enrichment of adaptive mutations within our LTSP clones and that this enrichment is stronger for nonmutator compared with mutator clones.
Extremely Convergent Adaptation via Mutations to the RNA-Polymerase Core Enzyme
Not only do most cells surviving to day 11 carry a mutation. Almost all cells surviving to LTSP carry a mutation within the exact same gene complex, the RNA-polymerase core enzyme. ∼67% of day 11 clones and ∼90% of all clones, extracted from the six time points carried a replacement (i.e., amino acid sequence altering) mutation in one of the three genes (rpoA, rpoB, and rpoC) encoding the core RNA polymerase (fig. 4A). Given that the E. coli K12 MG1655 genome carries over 4,000 genes, this is a very clear signal of convergent adaptation.
Fig. 4.

(A) Very high frequency of mutations within the core RNA-polymerase enzyme. Depicted is the mean percentage of sequenced clones carrying a mutation within the core RNA polymerase enzyme across all five examined populations. Error bars represent the standard deviation across the five populations. (B) The distribution of RNA polymerase mutation types within one of the five populations studied (similar patterns were also observed within other populations).
Even more striking, 93.5% of the 217 clones carrying a mutation within the core RNA-polymerase carried a mutation in one of only three specific sites within the RpoB and RpoC protein sequences: RpoB 1272, RpoC 334, and RpoC 428. A total of five different mutations were found in these sites across all clones: RpoB E1272G, RpoB E1272V, RpoC K334Q, RpoC K334T, and RpoC T428S. Mutations at each of these three sites were observed at high frequencies across each of the five independently sampled populations (fig. 4B). The rpoB and rpoC genes are relatively large genes and while they are essential, the target size for possible viable mutations within these genes is demonstrably large. For example various mutations, at various sites within rpoB confer resistance to the antibiotic rifampicin (Wrande et al., 2008; Katz and Hershberg, 2013; Field and Hershberg, 2015) and mutations within rpoB and rpoC were also shown to be involved in adaptation to high temperatures (Tenaillon et al., 2012) and to growth in minimal media (Conrad et al., 2010). The above mentioned rifampicin resistance mutations and high temperature and minimal media adaptive mutations all occur at positions different than the three observed in our experiments. Even within the experiments described here we also observed mutations at additional sites of the RNA polymerase core enzyme. Additionally, rpoB and rpoC have been demonstrated to be useful as markers for metagenomic studies, due to their substantial levels of sequence variation (Lan et al., 2016). The fact that we found mutations segregating at very high frequency across three particular positions within rpoB and rpoC, across five independent LTSP populations, therefore demonstrates a striking level of convergence in adaptation. Such high adaptive convergence within an evolutionary experiment was to our knowledge never before described.
LTSP Adaptive Mutations within the Three Most Frequently Mutated Sites of the RNA Polymerase Core Enzyme Display Sharp Antagonistic Pleiotropy
Clones carrying a mutation to position 428 of RpoC, position 334 of rpoC or position 1272 of RpoB that are otherwise entirely identical to the ancestral strain grow exponentially at a rate that is ∼17 to ∼21% slower than that of the ancestral strain (supplementary fig. S2, Supplementary Material online). Since their high frequency across LTSP populations suggests these mutations likely confer a very strong advantage under LTSP, this appears to be a very striking example of antagonistic pleiotropy.
To examine whether passenger mutations accumulated during LTSP may further reduce growth, once resources are available, we also characterized the exponential growth rates of three clones that all carried a mutation at position 1272 of RpoB, but varied in the total number of mutations they suffered (4, 18, and 290 for the three clones, respectively). We found that the exponential growth rate of the two RpoB mutants that carried the smaller number of mutations was somewhat lower than that of the RpoB mutant that did not carry any additional mutations (an exponential growth rate reduction of ∼26% to ∼29% relative the ancestor, supplementary fig. S2, Supplementary Material online). At the same time, the exponential growth rate of the clone that carried the largest number of mutations was much lower (a reduction of ∼76% relative the ancestral growth rate, supplementary fig. S2, Supplementary Material online). Combined, these results demonstrate that both antagonistic pleiotropy and mutation accumulation can significantly reduce the ability of bacteria that survive under resource exhaustion to grow, once resources again become available.
The LTSP Adaptive RpoC 428S Allele Segregates at High Frequencies among Unculturable Bacteria
Fitting with their harmful effect on exponential growth, we find that the most frequent LTSP adaptive core RNA polymerase alleles we observe in our experiment are very rarely present within fully sequenced genomes. Indeed, the E. coli ancestral E allele at RpoB position 1272 and the ancestral K allele at RpoC position 334 are universally conserved across over 3,600 fully sequenced bacterial genomes we examined. The E. coli ancestral T allele at RpoC position 428 is conserved in 99.3% of fully sequenced genomes. Interestingly, the remaining 13 genomes all carry the S allele, which is the mutation we identified as present at high frequency under LTSP. The 13 genomes carrying this LTSP adaptive allele belong to bacteria that are either nonculturable, or extreme thermophiles (supplementary table S3, Supplementary Material online).
We further sought to explore the frequency with which LTSP adaptive mutations at these three sites of the RNA-polymerase core enzyme segregate within natural environments. Towards this end we extracted 17,162 sequences of the RpoB gene, and 16,085 sequences of RpoC, including the sites of interest, from a large collection of human and environmental microbiomes (see Materials and Methods). We found that, fitting with the results obtained for completed sequences, the ancestral RpoC position 334 K and RpoB position 1272 E alleles are almost universally conserved across all bacterial phyla (supplementary fig. S3, Supplementary Material online). Very rare instances in which a different allele is found at these positions may reflect sequencing errors. The ancestral RpoC position 428 T allele is also largely extremely conserved within most phyla. However, we do find a high frequency of the LTSP adaptive 428 S allele within some phyla (fig. 5). Nonculturable bacteria from the Candidatus Saccharibacteria phylum (Hugenholtz et al., 2001) carry the S allele in a nearly universal manner. The S allele is also frequent among sequences that could not be classified according to phyla, because they did not match well any known phyla. It is tempting to predict that many of these unclassifiable sequences may also belong to nonculturable bacteria. After all, to classify metagenomic RpoB and RpoC sequences according to their phyla, we aligned them to all available fully sequenced genome sequences of the same genes (see Materials and Methods). Nonculturable bacteria are far more difficult to sequence and are therefore less likely to be included among the fully sequenced bacteria we used. We also find a relatively high frequency (∼10%) of the S allele among the Chloroflexi phylum, which is known to contain some hard to culture bacteria (Yamada and Sekiguchi, 2009). Combined, these results suggest that the LTSP adaptive RpoC 428 S allele may be highly abundant, specifically among nonculturable bacteria.
Fig. 5.

High frequency of LTSP adaptive RpoC position 428 S allele among nonculturable bacteria within natural microbiomes. Depicted is the relative frequency of the E. coli ancestral T allele (Red), the LTSP adaptives allele (black) and all other alleles (white) within RpoC sequences extracted from a large collection of metagenomics samples (see Materials and Methods). Only phyla for which at least 100 RpoC sequences could be analyzed are depicted.
Adaptation through Temporally Precise Fluctuations in Allele Frequencies within Genes Regulating Fatty Acid Metabolism
In a second striking pattern of convergence, we found that at day 22, within all sampled populations 73% or more of clones carry a mutation within the fadR gene combined with a second mutation within either the atoS or atoC genes (supplementary fig. S4, Supplementary Material online). The fadR gene encodes a major regulator of fatty-acid metabolism whereas atoS and atoC together encode a two-component system that regulates the expression of enzymes involved in short-chain fatty acid catabolism (Lioliou et al., 2005). These mutations do not continue to segregate at notable frequencies following day 22, and are also not seen at day 11 (supplementary fig. S4, Supplementary Material online). Thus, the LTSP populations experience a temporally precise sharp increase, and subsequent decrease in the frequency of clones carrying mutations at two genes involved in fatty acid metabolism. This implies that these populations have the ability to adapt rapidly and in a precise temporal manner through fluctuations in allele frequencies.
Prevalent Clonal Interference under LTSP
Given the relatively small population sizes under LTSP and that LTSP bacteria are assumingly not growing freely, it was perhaps expected that the number of mutations available for adaptation would be rather limited, predicting a pattern of hard, rather than soft sweeps. However, we observe a clear pattern of clonal interference within our LTSP populations (fig. 6, and supplementary figs. S5–S8, Supplementary Material online). Such clonal interference, combined with the relatively rapid mutation accumulation we observe (fig. 3), and the ability of populations to adapt in a precise temporal manner through fluctuations in allele frequencies, indicates that mutational input within LTSP populations may be surprisingly high.
Fig. 6.

Clear pattern of clonal interference observed within LTSP populations. Presented is a Muller diagram depicting the relative frequencies of different haplotypes segregating within LTSP population 1. The X-axis indicates the sampling times (not to scale). Only mutations appearing in 30% or more of population 1 clones, in at least one time point, were used to generate the plot. Gene names appearing in bold represent genes that are mutated in at least three of the five LTSP populations. Similar patterns of clonal interference were observed for the other four populations as well (supplementary figs. S5–S8, Supplementary Material online). *The mutation in rpoC is identical in both drawn lineages. There is no way to tell with complete certainty whether this mutation occurred within this population once or twice independently. up = upstream.
Discussion
When first one hears that E. coli can survive for years on end in spent media, it is quite tempting to imagine that survival under these conditions will be achieved by maintaining a high level of dormancy. Yet, past studies have already provided evidence that E. coli cells do multiply under LTSP (reviewed in Finkel, 2006). Still, it is easy to imagine that following death phase, bacterial populations may be quite small and that cellular replication may be quite slow. Thus, it may seem quite likely that adaptation under LTSP would be limited by low mutational input. Our results demonstrate that this is not actually the case. Nonmutator clones of E. coli accumulate as many mutations in 127 days spent under LTSP, as do E. coli cells passed through ∼300 daily serial dilutions (allowing for 2,000 generations of growth) in Richard Lenski’s long-term evolutionary experiment (LTEE Tenaillon et al., 2016). Many LTSP clones incur a mutation within a mismatch repair gene, further increasing the numbers of mutations they accumulate. As a result of what seems to be a substantial mutational input into adaptation under LTSP, adaptation is characterized by soft, rather than hard sweeps.
It is quite likely that LTSP clones undergo a lower number of generations in 127 days within spent media, than clones sequenced from the LTEE following 300 days of serial transfer. That both types of clones accumulate a similar number of mutations therefore likely reflects very strong selection exerted on cells surviving under LTSP, requiring that they accumulate mutations allowing them to survive. Such strong selection is also demonstrated by the enrichment observed in functional categories of mutations, relative neutral expectations. Perhaps most indicative of the strong selection exerted on cells surviving under LTSP is the extreme convergence we observe in the presence of certain mutations, at high frequencies, across sampled populations. We find that across all populations almost all clones carry a mutation within the core enzyme of RNA polymerase. Within each population several different mutations are found within RNA polymerase and across all populations the same three sites of the enzyme carry mutations at high frequencies. This is a very strong case of convergent evolution that undoubtedly represents strong selection in favor of these mutations under LTSP. That these mutations within the RNA polymerase already appear at very high frequency at day 11 of our experiment likely reflects that this adaptation provides a very basic advantage in surviving death phase and entering LTSP. Further studies will need to be taken to understand the exact nature of the adaptive effect of these mutations and to elucidate exactly what these mutations do to the functionality of RNA polymerase and whether and how they may alter global transcriptional patterns.
In a second striking example of temporal convergence, we observe that across all populations sampled at day 22 there is a very high frequency of mutations within genes regulating fatty acid metabolism. Specifically, across populations, 73% or more of clones carry a mutation within the fadR gene together with a second mutation in either atoC or atoS. Strikingly, these mutations are not seen at the previous, day 11 sampling point and at following time points these mutations nearly disappear from all sampled populations. This indicates that the LTSP populations are able to adapt to the conditions they face through fairly rapid and temporally precise fluctuations in allele frequencies. Such a pronounced temporal pattern of convergence is interesting for a number of reasons. First, this convergence indicates that at least early on in LTSP, independent bacterial populations seem to experience similar temporal selective pressures. Second, these results also shed light on the physiological conditions faced by the LTSP populations. The convergent increase in the frequency of mutations to fatty acid metabolism genes may indicate that around day 22 there is a sudden and temporary increase in the availability of fatty acids for consumption, perhaps due to the breakdown of cells that have perished during death phase. Finally, this striking pattern of temporal convergence indicates that E. coli populations under prolonged resource exhaustion carry sufficient genetic variation to allow them to rapidly adapt to changing conditions, through fluctuations in allele frequencies, and not only by regulatory changes possible within any one individual cell. This again suggests that mutational input is not limiting and reflects the great genetic flexibility bacterial populations have in adapting, even under conditions of resource exhaustion.
Interestingly, while the fatty acid metabolism mutants are present at high frequencies only within a specific timeframe, RNA polymerase core enzyme mutants remain at very high frequencies across all sampled time points. This may indicate that they carry an important role across the first 4 months of LTSP.
Adapting to survival under LTSP seems to incur a pronounced cost for growth, once resources are again available. We find that clones carrying a mutation in one of the three most frequently mutated RNA polymerase sites grow exponentially ∼20% slower, in fresh media, than the ancestral wildtype strain. This is a striking example of antagonistic pleiotropy, by which a mutation that is adaptive under one condition (resource exhaustion) is deleterious under another (growth in fresh rich media). Additional mutations accumulated by LTSP clones, that are not necessarily adaptive under resource exhaustion, also reduce exponential growth rates. We observe that the mutator clone carrying the highest number of mutations in our experiment exponentially grows ∼76% slower than the ancestral wildtype, within fresh media.
Past studies into LTSP, that did not utilize the sequencing of whole genomes, have suggested that adaptation to LTSP often involves changes to the function of the master regulator of the stress response, RpoS (Zambrano et al., 1993; Farrell and Finkel, 2003; Finkel, 2006). It was therefore somewhat surprising that in our sequencing-based, whole-genome identification of alleles contributing to adaptation to LTSP, we did not observe any mutations within the rpoS gene. This surprising result may have several explanations. The first possible explanation is that the involvement of RpoS in adaptation to LTSP may often be indirect. Many studies demonstrating the involvement of RpoS did so by examining a phenotype associated with RpoS function, rather than by identifying specific mutations within the rpoS gene or promoter (Zambrano et al., 1993; Farrell and Finkel, 2003). Since the function of RpoS is tightly regulated, and since RpoS itself regulates a very large number of genes (Battesti et al., 2011), it is quite possible that there are many mutations outside of the rpoS gene that can affect RpoS function. It is therefore possible that while RpoS function often contributes to adaptation under resource exhaustion, actual mutations within rpoS are much more rarely involved. (Although, it is important to note that some such mutations within the rpoS gene have been documented [Zambrano et al., 1993; Finkel, 2006]).
Variation in the genetic background of the E. coli strains used in different studies may also affect the identity of genes involved in adaptation to resource exhaustion. However, this does not appear to be very likely to explain why we did not observe rpoS mutations, since our study used an E. coli K12 strain, very similar to the one used in previous studies that did report the involvement of changes in RpoS function (Zambrano et al., 1993; Farrell and Finkel, 2003).
Finally, it possible we did not observe any rpoS mutations due to variation in the experimental conditions used between our study and previous studies. While most past studies examined adaptation to LTSP as it occurs in much smaller volumes (≤5 ml) in test tubes, we examined adaptation to LTSP in large volumes (400 ml) within flasks. Past studies have demonstrated that growth volume and receptacle, as well as media type can affect survival dynamics under LTSP (Kram and Finkel, 2014; Kram and Finkel, 2015). Growth volume, receptacle and media type may greatly affect the conditions bacteria face prior to entering death phase, during death phase and subsequently in LTSP. Such variation in conditions may lead to variation in the identity of the mutations that are most adaptive under each growth regimen. In addition to affecting the conditions bacteria are exposed to, growth volume can also affect the size of the population prior to entry into death phase and likely also subsequently during LTSP. This in turn should affect the amount of genetic variation that will be available for adaptation, as larger populations will accumulate more mutations and therefore contain more genetic variation. This in turn may enable larger populations to adapt in a more convergent manner, through mutations that are more adaptive, since they are less limited by the availability of such adaptive mutations. In contrast smaller populations can be more limited by mutational input and may therefore often not acquire the most adaptive mutations, leading them to adapt in a less convergent manner, sometimes via less adaptive mutations. This in turn may lead to variation between populations of different sizes in the identity of the mutations that contribute to their adaptation to resource exhaustion. More studies will be needed to determine whether, how and why growth volume affects the dynamics of adaptation to conditions of resource exhaustion.
We found that mutations within the RNA polymerase core enzyme are strongly involved in adaptation to conditions of resource exhaustion. In this, we joined a number of previous studies that have demonstrated the involvement of RNA polymerase core enzyme mutations in adaptation to growth in minimal media (Conrad et al., 2010) and to adaptation to growth at high temperatures (Tenaillon et al., 2012). It is therefore becoming more and more evident that the RNA polymerase core enzyme acts as a hub of adaptation to a variety of conditions. It will be interesting to further study the role of RNA polymerase in adaptation and to elucidate how this role affects the diversification of this important gene complex. It will also be interesting to determine whether the RNA polymerase core enzyme is unique in its involvement in adaptation to a large variety of conditions, or whether in general, regulatory hubs tend to more often be involved in adaptation.
The patterns of clonal interference we observe in our experiments may arise from various genotypes competing with each other for a similar niche, or from various genotypes specializing for specific subniches. The exhausted media the LTSP populations inhabit during the first 4 months of our experiment likely includes a variety of resources. Additionally, cells surviving under LTSP likely also face a variety of challenges. It is possible that soft sweeps are generated because each competing genotype is similarly adapted to live on the variety of resources available and face the variety of challenges imposed, or because different genotypes are specialized to survive on particular sets of resources and/or better face particular challenges.
Irrespective of the causes of the observed soft sweeps, such soft sweeps mean that we generally do not observe any genotypes that fix across an entire population. Most clones sequenced following 4 months under LTSP carry an antagonistically pleiotropic mutation within RNA polymerase and a substantial number of additional mutations that may further reduce their ability to grow exponentially. However even at the last time point of our experiment (day 127) we did find some clones that do not carry an RNA polymerase mutation and that carry a very small number of mutations overall (supplementary tables S1 and S2, Supplementary Material online). It is therefore possible that once a 4-month-old LTSP population is provided with resources to grow, such clones will allow it to quickly reconstitute the ancestral ability to grow and the ancestral genetic makeup. Thus, it is tempting to hypothesize that cycles of boom and bust may be associated with fluctuations in allele frequencies. Under such a model, the mutations we observe within our LTSP populations, at the three most frequently mutated sites of RNA polymerase will be present only within natural populations that are currently experiencing a period of bust. Once the same bacterial populations receive conditions that allow them to grow, these alleles will rapidly be reduced to very low frequencies. In metagenomic analyses DNA from abundant bacteria will be substantially over-represented relative DNA from cells that are currently experiencing a period of bust. We would therefore expect to observe within microbiome data only those alleles that are found within more abundant strains that are likely currently capable of growth. Fitting with this, within natural microbiomes, we almost only observe the ancestral RNA polymerase alleles and almost never observe the frequently occurring LTSP mutations that strongly reduce exponential growth. The exception seems to be bacteria that cannot be lab-cultured, which carry a specific LTSP RNA polymerase allele (RpoC 428S) at extremely high frequencies. Such nonculturable bacteria likely grow very differently from other bacteria. Indeed, it is interesting that we may have stumbled across an allele that is a marker of sorts of unculturability. Future studies will be required to determine whether within natural environments bacteria experiencing periods of resource exhaustion, tend to more frequently carry LTSP alleles.
Materials and Methods
LTSP Evolutionary Experiments
To initiate our five LTSP evolutionary experiments, single colonies of E. coli K12 MG1655 were grown over night. Five colonies were used to inoculate test tubes with 4 ml of fresh medium. Each culture was then grown until it reached an OD of 1 and then ∼2 × 109 cells (1 ml) were used to inoculate 400 ml of Luria Broth (LB) in a 2 liter polycarbonate breathing flask. This procedure was used in order to both start with a similar number of cells in the five populations and start with isogenic populations by reducing the generations passed since each population was a single cell. The five resulting flasks were placed in an incubator set at 37 °C, with shaking at 225 rpm.
No new nutrients or resources were added to the cultures with time, except for sterile water that was added to compensate for evaporation every 10–15 days, according to the weight lost by each flask during that time period.
Sampling LTSP Populations and Estimating Viability
Initially, every day, then every week, then every month and following that at longer intervals (fig. 2) one ml of each culture was sampled. Dilutions were plated using a robotic plater to evaluate viability through live counts. Samples were frozen in 50% glycerol in a −80°C freezer for future analysis.
Sequencing of LTSP Clones
Frozen cultures of the desired populations and time points were thawed and dilutions were plated and grown over night. The thawing efficiency was ∼50% (data not shown). Ten colonies from each culture were used to inoculate 4 ml of medium in a test tube and were grown until they reached an OD of ∼1. Again, this was done to reduce the time these colonies can evolve and accumulate mutations. 1 ml of the culture was centrifuged at 10,000 g for 5 min and the pellet was used for DNA extraction. The remainder of each culture was then archived by freezing in 50% of glycerol in a −80°C freezer.
DNA was extracted using the Qiagen DNeasy Blood & Tissue Kit. Library preparation followed the protocol outlined in (Baym et al., 2015). Sequencing was carried out at the Technion Genome Center using an Illumina HiSeq 2500 machine. Clones extracted from time points 11, 22, 32, 42, and 64 of population 1 were sequenced using a single-end 50 bp reads. All remaining clones were sequenced using paired end 150 bp reads. The ancestral clones used to initiate the five populations were sequenced as well.
Calling of Mutations
In order to call mutations, the reads obtained for each LTSP clone or ancestral strain were aligned to the E. coli K12 MG1655 reference genome (accession NC_000913). LTSP clone mutations were then recorded if they appear within an LTSP clone’s genome, but not within the ancestral genome. Alignment and mutation calling were carried out using the breseq platform, which allows for the identification of point mutations, short insertions and deletions, larger deletions, and the creation of new junctions (Deatherage and Barrick, 2014).
Exponential Growth Rate Estimation
In order to calculate the exponential growth rate of a specific LTSP clone, single colonies were used to inoculate test tubes with 4 ml of medium. These cultures were grown over night and in the morning they were diluted to an OD of ∼0.2. Then the cultures were allowed to grow for an additional hour and diluted again to an OD of ∼0.2 and transferred to a 96 well plate. This plate was incubated and OD was read by the plate reader every 10 min. The exponential growth rate was calculated from the slope of the growth at the exponential phase. We used the maximum rate looking at seven sequential time points, where R2 > 0.99. For each clone we tested five colonies with two technical replicates.
Mutation Simulations
In order to estimate the proportion of cells expected to carry at least a single mutation within a population with a mean number of mutations of 0.041 per genome, in the absence of selection, we carried out 1,000 simulations. Since mutation is a Poisson process (Luria and Delbruck, 1943), we assumed that the number of mutations that will be present in each genome within the population will be Poisson distributed around the 0.041 mean. In each simulation we therefore drew 10,000 numbers (representing 10,000 cells) from a Poisson distribution with a mean of 0.041. Drawing of numbers was done using the R package’s rpois function. For each simulation we then calculated the proportion of the 10,000 cells to carry at least a single mutation. We found that across our 1,000 simulations, between 3.5% and 4.6% of cells carried at least one mutation.
Calculating Proportion of NonSynonymous Versus Synonymous Mutations Expected Under Neutrality
The DNA sequences of all protein coding genes in E. coli strain MG1655 were downloaded from the NCBI database. For each position of each gene, we examined the likelihood a mutation at that position would lead to a nonsynonymous or a synonymous change. For example, any change to the third codon position of a 4-fold degenerate codon would be synonymous so mutations at such a position would be 100% likely to be synonymous. In contrast mutations to the third codon position of a 2-fold-degenerate codon will be synonymous for a third of possible mutations and nonsynonymous for two thirds of possible mutations. In such a manner, we could add up the likelihood of a mutation being synonymous or nonsynonymous across the entirety of E coli K12. MG1655’s protein coding genes.
Calculating Proportion of Mutations Expected Under Neutrality to Fall Within Intergenic Regions that are More Likely to Contain Promoters
The intergenic regions of the E. coli K12 MG1655 genome were classified as more likely to contain a promoter, if the genes surrounding them had a unidirectional orientation relative each other (→→ or ←←), or a divergent orientation (←→). Intergenic regions were classified as less likely to contain a promoter if the genes surrounding them had a convergent orientation relative each other (→←). Based on this classification we could calculate what proportion of base pairs within the genome fall in each of these categories. This proportion is considered by us to be the proportion of mutations that would be expected to fall within each category under neutrality.
Quantifying the Frequency of LTSP RNA Polymerase Adaptive Alleles among Fully Sequenced Bacterial Genomes
All 4004 fully sequenced (nondraft) bacterial genomes were downloaded from the NCBI database on January 2016. The RpoB and RpoC genes of each genome were identified using gene annotations provided by the NCBI, allowing us to extract each gene’s protein sequence. A total of 3606 RpoB and 3788 RpoC sequences were extracted. Each of these protein sequences was aligned against its matching E. coli K12 MG1655 reference sequence, using the EMBOSS package’s Needle program (Rice et al., 2000) and the identity of amino acids at the positions of interest was determined.
Quantifying the Frequency of LTSP RNA Polymerase Adaptive Alleles Within Metagenomic Data Sets
Metagenomic data sets used in our analysis were extracted the JGI database (Nordberg et al., 2014) between March and June of 2016. For environmental samples, we used only “Permanent draft” and “Finished” metagenomic projects, whereas for Human samples we also used “draft” projects, as the vast majority of Human microbiome projects carried this status. The Metagenomic sequences extracted from the resulting 191 environmental and 874 Human projects were searched using BLAST (Altschul et al., 1990) for RpoB and RpoC sequences (using the E. coli K12 MG1655 sequences as a query. For this BLAST search, we used an E-value cutoff of 1. Each obtained sequence was further inspected to ensure that it indeed represents part of a RpoB or RpoC sequence. To this end the COG and KO classifications of each sequence provided by the JGI were used. After filtering unreliable sequences, the remaining sequences were aligned against the reference gene sequences from E. coli K12 MG1655 using the EMBOSS needle program (Rice et al., 2000) and the identity of amino acids at the positions of interest was determined.
To phylogenetically classify the metagenomic RpoB and RpoC sequences extracted from the JGI database each sequence was compared using FASTA (Pearson and Lipman, 1988) against RpoB or RpoC sequences extracted from all fully sequenced bacterial genomes available in the NCBI database. Phylogeny was then determined according to the phylogeny of the best fully sequenced match obtained by a given metagenomic sequence. The match threshold utilized was 70% sequence identity between the query and the reference, with at least 2/3 of the query length aligned to the reference. Sequences that did not match any fully sequenced genome according to this threshold were considered unclassified.
Supplementary Material
Supplementary data are available at Molecular Biology and Evolution online.
Supplementary Material
Acknowledgments
This work was supported by an Ali Kaufman postdoctoral fellowship (to S.A.), by a BSF grant (No. 2013463, to R.H.) by a Yigal Allon Fellowship awarded by the Israeli Council for Higher Education (to R.H.), by the Rappaport Family Institute for Research in the Medical Sciences (to R.H.), and by the Robert J. Shillman Career Advancement Chair (to R.H.). The described research was carried out in the Rachel & Menachem Mendelovitch Evolutionary Process of Mutation & Natural Selection Research Laboratory.
References
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ.. 1990. Basic local alignment search tool. J Mol Biol. 215:403–410. [DOI] [PubMed] [Google Scholar]
- Avrani S, Lindell D.. 2015. Convergent evolution toward an improved growth rate and a reduced resistance range in Prochlorococcus strains resistant to phage. Proc Natl Acad Sci U S A. 112:E2191–E2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrick JE, Lenski RE.. 2009. Genome-wide mutational diversity in an evolving population of Escherichia coli. Cold Spring Harb Sym. 74:119–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrick JE, Lenski RE.. 2013. Genome dynamics during experimental evolution. Nat Rev Genet. 14:827–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrick JE, Yu DS, Yoon SH, Jeong H, Oh TK, Schneider D, Lenski RE, Kim JF.. 2009. Genome evolution and adaptation in a long-term experiment with Escherichia coli. Nature 461:1243–1247. [DOI] [PubMed] [Google Scholar]
- Battesti A, Majdalani N, Gottesman S.. 2011. The RpoS-mediated general stress response in Escherichia coli. Annu Rev Microbiol. 65:189–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baym M, Kryazhimskiy S, Lieberman TD, Chung H, Desai MM, Kishony R.. 2015. Inexpensive multiplexed library preparation for megabase-sized genomes. PLoS One 10:e0128036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad TM, Frazier M, Joyce AR, Cho BK, Knight EM, Lewis NE, Landick R, Palsson BO.. 2010. RNA polymerase mutants found through adaptive evolution reprogram Escherichia coli for optimal growth in minimal media. Proc Natl Acad Sci U S A. 107:20500–20505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper VS, Lenski RE.. 2000. The population genetics of ecological specialization in evolving Escherichia coli populations. Nature 407:736–739. [DOI] [PubMed] [Google Scholar]
- Deatherage DE, Barrick JE.. 2014. Identification of mutations in laboratory-evolved microbes from next-generation sequencing data using breseq. Method Mol Biol. 1151:165–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell MJ, Finkel SE.. 2003. The growth advantage in stationary-phase phenotype conferred by rpoS mutations is dependent on the pH and nutrient environment. J Bacteriol. 185:7044–7052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field W, Hershberg R.. 2015. Alarmingly high segregation frequencies of quinolone resistance alleles within human and animal microbiomes are not explained by direct clinical antibiotic exposure. Genome Biol Evol .7:1743–1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel SE. 2006. Long-term survival during stationary phase: evolution and the GASP phenotype. Nature Rev Microbiol 4:113–120. [DOI] [PubMed] [Google Scholar]
- Finkel SE, Kolter R.. 1999. Evolution of microbial diversity during prolonged starvation. Proc Natl Acad Sci U S A. 96:4023–4027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogle CA, Nagle JL, Desai MM.. 2008. Clonal interference, multiple mutations and adaptation in large asexual populations. Genetics 180:2163–2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graur D, Wen-Hsiung L.. 2000. Fundementals of Molecular Evolution, 2nd edn.Sunderland (MA: ): Sinauer Associates Inc. [Google Scholar]
- Hugenholtz P, Tyson GW, Webb RI, Wagner AM, Blackall LL.. 2001. Investigation of candidate division TM7, a recently recognized major lineage of the domain Bacteria with no known pure-culture representatives. Appl Environ Microb. 67:411–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassen R. 2014. Experimental evolution and the nature of biodiversity. Greenwood Village (CO: ): Roberts and Company. [Google Scholar]
- Katz S, Hershberg R.. 2013. Elevated mutagenesis does not explain the increased frequency of antibiotic resistant mutants in starved aging colonies. PLoS Genet. 9:e1003968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kram KE, Finkel SE.. 2014. Culture volume and vessel affect long-term survival, mutation frequency, and oxidative stress of Escherichia coli. Appl Environ Microb. 80:1732–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kram KE, Finkel SE.. 2015. Rich medium composition affects Escherichia coli survival, glycation, and mutation frequency during long-term batch culture. Appl Environ Microb. 81:4442–4450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvitek DJ, Sherlock G.. 2013. Whole genome, whole population sequencing reveals that loss of signaling networks is the major adaptive strategy in a constant environment. PLoS Genet. 9:e1003972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan Y, Rosen G, Hershberg R.. 2016. Marker genes that are less conserved in their sequences are useful for predicting genome-wide similarity levels between closely related prokaryotic strains. Microbiome 4:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Popodi E, Tang H, Foster PL.. 2012. Rate and molecular spectrum of spontaneous mutations in the bacterium Escherichia coli as determined by whole-genome sequencing. Proc Natl Acad Sci U S A. 109:E2774–E2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenski RE, Rose MR, Simpson SC, Tadler SC.. 1991. Long-term experimental evolution in Escherichia coli. I. Adaptation and divergence during 2000 generations. Am Nat. 138:1315–1341. [Google Scholar]
- Lioliou EE, Mimitou EP, Grigoroudis AI, Panagiotidis CH, Panagiotidis CA, Kyriakidis DA.. 2005. Phosphorylation activity of the response regulator of the two-component signal transduction system AtoS-AtoC in E. coli. Biochim Biophys Acta 1725:257–268. [DOI] [PubMed] [Google Scholar]
- Luria SE, Delbruck M.. 1943. Mutations of bacteria from virus sensitivity to virus resistance. Genetics 28:491–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddamsetti R, Lenski RE, Barrick JE.. 2015. Adaptation, clonal interference, and frequency-dependent interactions in a long-term evolution experiment with Escherichia coli. Genetics 200:619–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordberg H, Cantor M, Dusheyko S, Hua S, Poliakov A, Shabalov I, Smirnova T, Grigoriev IV, Dubchak I.. 2014. The genome portal of the Department of Energy Joint Genome Institute: 2014 updates. Nucleic Acids Res. 42:D26–D31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrow SL, Barshir R, DeGregori J, Yeger-Lotem E, Hershberg R.. 2014. Cancer evolution is associated with pervasive positive selection on globally expressed genes. PLoS Genet. 10:e1004239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson WR, Lipman DJ.. 1988. Improved tools for biological sequence comparison. Proc Natl Acad Sci U S A. 85:2444–2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice P, Longden I, Bleasby A.. 2000. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 16:276–277. [DOI] [PubMed] [Google Scholar]
- Tenaillon O, Barrick JE, Ribeck N, Deatherage DE, Blanchard JL, Dasgupta A, Wu GC, Wielgoss S, Cruveiller S, Medigue C, et al. 2016. Tempo and mode of genome evolution in a 50,000-generation experiment. Nature 536:165–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenaillon O, Rodriguez-Verdugo A, Gaut RL, McDonald P, Bennett AF, Long AD, Gaut BS.. 2012. The molecular diversity of adaptive convergence. Science 335:457–461. [DOI] [PubMed] [Google Scholar]
- Wielgoss S, Barrick JE, Tenaillon O, Wiser MJ, Dittmar WJ, Cruveiller S, Chane-Woon-Ming B, Medigue C, Lenski RE, Schneider D.. 2013. Mutation rate dynamics in a bacterial population reflect tension between adaptation and genetic load. Proc Natl Acad Sci U S A. 110:222–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrande M, Roth JR, Hughes D.. 2008. Accumulation of mutants in “aging” bacterial colonies is due to growth under selection, not stress-induced mutagenesis. Proc Natl Acad Sci U S A. 105:11863–11868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T, Sekiguchi Y.. 2009. Cultivation of uncultured chloroflexi subphyla: significance and ecophysiology of formerly uncultured chloroflexi ‘subphylum’ with natural and biotechnological relevance. Microbes Environ. 24:205–216. [DOI] [PubMed] [Google Scholar]
- Zambrano MM, Siegele DA, Almiron M, Tormo A, Kolter R.. 1993. Microbial competition: Escherichia coli mutants that take over stationary phase cultures. Science 259: 1757–1760. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.