

Abstract
Alcoholic liver disease (ALD) is a leading cause of liver related morbidity and mortality worldwide. ALD encompasses a spectrum of disorders including asymptomatic steatosis, steatohepatitis, fibrosis, cirrhosis and its related complications, and the acute on chronic state of alcoholic hepatitis. While multidisciplinary efforts continue to be aimed at curbing progression of this spectrum of disorders, there is an urgent need to focus our efforts on effective therapeutic interventions for alcoholic hepatitis (AH), the most severe form of ALD. AH is characterized by an abrupt development of jaundice and complications related to liver insufficiency and portal hypertension in patients with heavy alcohol intake. The mortality of patients with severe AH is very high (20–50% at 3 months). The current therapeutic regimens are limited. The development of new therapies requires translational studies in human samples and suitable animal models that reproduce clinical and histological features of human AH. This review article summarizes the clinical syndrome, pre-clinical translational tools, and pathogenesis of AH at a molecular and cellular level, with the aim to identify new targets of potential therapeutic intervention.
1. Introduction
Alcoholic Liver Disease is the leading cause of liver-related morbidity and mortality worldwide and is a major cause of death among adults with prolonged alcohol abuse1. According to WHO, 3.3 million deaths occur worldwide every year due to the harmful use of alcohol, representing 5.9% of all deaths. Alcohol is the third leading preventable cause of death in the US accounting for about 88,000 deaths per year2,3. This review highlights the natural history, pathogenesis, molecular and cellular targets, as well as the current and new therapies being investigated for Alcoholic Hepatitis.
Alcoholic liver disease encompasses different stages of liver disease as a consequence of susceptibility factors and duration of alcohol consumption; listed from least to most severe, are steatosis, alcoholic steatohepatitis (ASH), progressive fibrosis, end stage cirrhosis, decompensated cirrhosis, and superimposed hepatocellular carcinoma (HCC).
Alcoholic hepatitis is an acute on chronic condition, diagnosed clinically by new-onset jaundice and/or ascites in the setting of ongoing alcohol abuse and underlying ALD. Severe forms of AH have very high short term mortality and represent one of the deadliest diseases in clinical hepatology, with a mortality rate of 30–50% at 3 months4.
The true incidence of AH is not well known; population based studies estimate approximately 4.5 hospitalizations for AH per 100,000 persons per year5. Patients with ALD can present with acute episodes of jaundice and liver decompensation from other reasons, such as sepsis, biliary obstruction, diffuse HCC, drug-induced liver injury and ischemic hepatitis. All the etiologies stated above present with a similar clinical picture and there is a lack of biomarkers or other laboratory tests to distinguish these acute entities. Where diagnosis is unclear, transjugular liver biopsies in patients hospitalized for acute hepatitis with underlying ALD to confirm the existence of AH is important.
2. Environmental and Genetic Risk Factors
There is a positive correlation between cumulative alcohol intake and degree of liver fibrosis; however extensive variability in the histological response to alcohol abuse exists in individuals. At similar levels of ethanol consumption, some patients only develop fatty liver or macrovesicular steatosis, while others progress to fibrosis and cirrhosis. Several risk factors for the susceptibility of ALD have been identified including sex, obesity, drinking patterns, dietary factors, non-sex-linked genetic factors, and cigarette smoking1,6,7(Figure 1).
Figure 1.
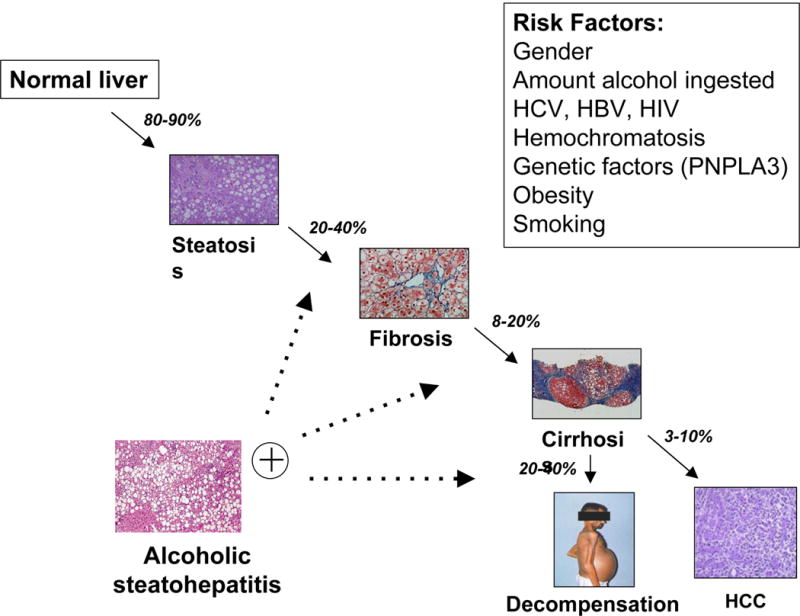
Natural progression along the spectrum of ALD, from steatosis, to the inflammatory state of steatohepatitis, to progressive fibrosis and cirrhosis, and finally, to decompensated cirrhosis and HCC. Exacerbations of AH occur at many of the later stages of disease. Predisposing risk factors to accelerated progression are listed.
Epidemiological studies suggest that several genetic factors influence the severity of steatosis and oxidative stress, and that the cytokine milieu, the magnitude of the immune response, and the severity of fibrosis also modulate an individual’s propensity to progress to advanced ALD. The genetic factors that influence an individual’s susceptibility to develop advanced ALD are largely unknown. Variations in genes encoding antioxidant enzymes, cytokines, other inflammatory mediators, and alcohol-metabolizing enzymes seem to play a role7.
Owing to its fibrogenic potential, variations in the rate of generation of acetaldehyde could explain the differences in the susceptibility of individuals to ALD after abusive alcohol consumption. Although polymorphisms in the genes encoding the main alcohol-metabolizing enzymes such as ADH, ALDH and CYP2E1 are accepted to be involved in an individual’s susceptibility to alcoholism, their role in the progression of ALD remains controversial8.
Also, recent studies indicate that variations in the patatin-like phospholipase domain-containing protein 3 (PNPLA3) strongly influence the development of cirrhosis in alcoholic Caucasians and Mexicans. PNPLA3 polymorphisms can be considered as the only confirmed and replicated genetic risk factor for ALD. However deletion of the PNPLA3 gene did not affect obesity-associated fatty liver or liver enzyme elevation in animal studies9–11.
Polymorphisms in genes that encode pro-inflammatory cytokines like TNF, IRAK-M, interleukin-1β, IL-1 receptor antagonists, IL-2, IL-6 and IL-10, involved in the pathogenesis of ALD, have also been examined12. Moreover, studies have also investigated the role of genetic variation in factors involved in lipopolysaccharide induced intracellular pathways, including CD14 and toll-like receptor 4 (TLR4), as potential risk factors for ALD13.
Despite the large number of studies that have assessed the role of genetic variation in susceptibility to ALD, a large-scale, well-designed, genome-wide association study of factors linked to the development of ALD remains to be performed. Consequently, a genetic test capable of identifying which patients are susceptible to advanced ALD is yet to be developed. Such a test could be useful in clinical settings, as it would help to identify the main genetic determinants of ALD, which could potentially assist in the development of future therapies14.
3. Current treatment in management of alcoholic hepatitis
Patients that develop severe AH usually require hospitalization for initial management, a summary of current interventions is listed in Table 1. Primary prevention is aimed at alcohol abstinence; active management of alcohol use disorders is critical to achieving continued abstinence. For the successful management of these patients, a multidisciplinary team composed of hepatologists, psychologists, psychiatrists and social workers is highly recommended15. Significant protein calorie malnutrition is a common finding in alcoholics, as are deficiencies in a number of vitamins and trace minerals, including vitamin A, vitamin D, thiamine, folate, pyridoxine, and zinc1,16, 17. Nutritional support improves liver function and short-term follow-up studies suggest that improved nutrition might improve survival times and histological findings in patients with AH18. Most patients improve spontaneously with abstinence and supportive care. Medical treatment is considered for patients who present with a very severe clinical picture or continue to deteriorate (Figure 2).
Table 1.
Current therapeutic interventions for management of alcoholic hepatitis.
| Intervention Class | Examples, Indications and Limitations |
|---|---|
| Nutritional support | Address protein calorie malnutrition (High calorie meals and supplements), and vitamin deficiency (Vitamin A, Vitamin D, Thiamine, Folate) through supplementation. |
| Alcohol Abstinence | Most important component of therapy, often a multidisciplinary team recommended for successful management. |
| Corticosteroids | Prednisolone: first line therapy for severe AH (indicated with DF > 32 or HE) AASLD/EASL recommendations: 40mg/day for 4 weeks Assess response after a week: Lille score> 0.45, discontinue therapy Rule out infection and GI bleeding before initiating therapy |
| Phosphodiesterase inhibitor: | Pentoxifylline: inhibits TNF Second line agent. Not an effective rescue therapy agent. |
| Anti-TNF agents | Infliximab/Etanercept Large studies showed increased adverse effects: Infections, mortality Currently not recommended for AH |
| N-acetylcysteine | Replenishes Glutathione in damaged hepatocytes and prevents cell death. Used in Acute Hepatitis from other causes. Studies on Prednisolone and NAC combination therapy have shown reduction in 1-month mortality rate, HRS incidence, and Infection incidence, but no effect on improving 6-month mortality. |
DF: Maddrey’s discriminant function, HE: Hepatic encephalopathy, TNF: Tumor necrosis factor, HRS: Hepatorenal Syndrome
Figure 2.
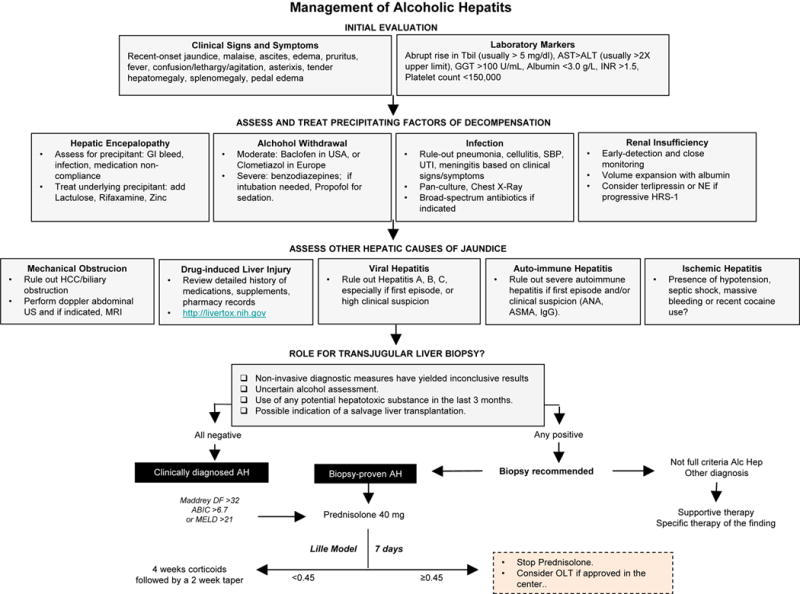
Clinical evaluation for a patient with high suspicion of AH involves ruling out precipitating factors of decompensated liver disease and confounding illnesses. Role of transjugular liver biopsy is dependent on the care provider’s confidence in the diagnosis. Initial treatment requires ongoing surveillance for improvement.
Several clinical scoring models have been developed to help predict outcomes of patients with AH and to guide therapy, including the Maddrey Discriminant Function (DF); Glasgow Alcoholic Hepatitis Score (GAHS); Mayo End-stage Liver Disease (MELD); Age, Bilirubin, INR, Creatinine (ABIC); MELD-Na, UK End-stage Liver Disease (UKELD), and three scores of corticosteroid response at 1 week: an Early Change in Bilirubin Levels (ECBL), a 25% fall in bilirubin, and the Lille score. The MELD, DF, GAHS, ABIC and scores of corticosteroid response prove to be valid in an independent cohort of biopsy-proven alcoholic hepatitis19. Combining features of various scoring models, for example, from the DF, ABIC, MELD and Lille, may prove to be a better prognosis indicator20.
Corticosteroids have been used in the treatment of AH for more than 40 years. Prednisolone is widely considered the first line therapy for severe AH. Both the AASLD and EASL practice guidelines recommend the use of corticosteroids (i.e. prednisolone 40 mg daily for 4 weeks) for patients with severe AH, defined by Maddrey’s discriminant function >32 or the presence of hepatic encephalopathy 1,21. Clinical trials conducted, to determine efficacy of steroid administration, so far suffer from heterogeneity and a high risk of bias. A recent meta-analysis from individual data however showed improved survival in patients with a high DF. In this study patients were categorized as complete responders, partial responders, or null responders and was able to predict the 6-month survival of each group using 2 new cut-offs of the Lille score 22. In another study, the response to prednisolone was assessed based on the change in bilirubin after one week of therapy and quantified using the Lille score. The Lille score was calculated after 7 days of initiation of therapy and it was determined that corticosteroids can be discontinued in non-responders, defined by a Lille score >0.4520. A Lille score greater than 0.45 predicts a 6-month survival rate of less than 25% 23. Clinical practice guidelines recommend stopping corticosteroids after one week in those with an unfavorable Lille score, as the risks of continued therapy likely outweigh the benefits. The contraindications for the use of corticosteroids are not well defined. When considering treatment with corticosteroids, patients require careful monitoring for evidence of present or developing infections and/or active GI bleeding.
Pentoxifylline is a phosphodiesterase inhibitor that inhibits the synthesis of tumor necrosis factor, which is increased in patients with AH. In practice, pentoxifylline is typically reserved as a second-line agent for patients with contraindications to corticosteroid therapy. A large trial, STOPAH, comparing prednisolone and pentoxifylline is underway and should prove to be a definitive study for assessing the efficacy of these drugs for AH24. Current consensus regarding Pentoxifylline is that it is not effective rescue therapy in patients who do not respond to corticosteroids25.
Infliximab and etanercept, anti-TNF agents, were also investigated as potential therapies for patients with AH. Rationale for their use was similar to pentoxifylline; TNF-α was implicated as a key culprit in the potentiation of hepatocyte inflammation. The use of these agents should theoretically benefit patients hospitalized with AH. However translational studies did not support the hypothesis26,27. Other larger studies resulted in adverse side effects such as increased rates of infection and increased mortality. Presently anti-TNF-α agents are not recommended for treatment of AH28.
N-acetylcysteine is known to replenish glutathione in damaged hepatocytes and prevent cell death in ALD. A recent randomized trial showed that the combination of N-acetylcysteine with prednisolone showed a clear trend to improve survival, by reducing 1-month mortality (8% vs. 24%) and reduce incidence of hepatorenal syndrome and infection, although the study was underpowered to reach statistical significance. In addition, it was found to have no effect on six-month survival29. The favorable safety profile of N-acetylcysteine makes it a potential option, in combination with corticosteroids, for patients with severe disease.
4. Role of transplantation in Alcoholic Hepatitis
ALD is the second most common indication for liver transplantation (LT) for chronic liver disease after HCV cirrhosis. Despite this, it is estimated that as many as 95% of patients with end-stage liver disease related to alcohol are never formally evaluated for candidacy for liver transplantation.
An important issue that is still unresolved is the role of LT in patients with alcoholic hepatitis, who are generally excluded from transplant. Patients with severe AH who do not respond to medical are unlikely to survive the “mandatory” 6-month abstinence period as their risk of mortality is quite high30,31. However post-transplant outcomes appear to be good for highly selected patients with severe AH unresponsive to medical therapy with low rates of alcohol relapse32,33. Salvage liver transplantation in highly selected patients has been shown to improve survival significantly, but is not available in the vast majority of transplant centers32. The availability of living donor transplantation and extended criteria donor liver transplantation are likely to heighten the debate on this issue.
5. Translation research in ALD: integrating animal and human studies
While animal models serve as the cornerstone of many research studies, their use in exploring potential novel targets for the treatment of alcoholic liver disease, particularly alcoholic hepatitis, is limited. Many rodent models produce some features of chronic alcoholic liver disease, but it is difficult to mimic acute-on-chronic liver injury as seen in patients with alcoholic hepatitis, though improved models are emerging.
While many models exist to mimic mild to moderate levels of hepatocyte damage, a better model of alcoholic hepatitis, is the chronic-plus-binge feeding model. As the name implies, it models the chronic drinking behaviors with intermittent binges seen as in patients presenting with alcoholic hepatitis. This model causes hepatocyte damage, disruption of mitochondrial function, and oxidative stress, resulting in moderate rises in AST and ALT, but in addition to mimicking steatosis, liver injury, and early fibrogenic response, it was also able to demonstrate hepatic neutrophil infiltration as seen in early stages of alcoholic hepatitis34, though the hepatocellular damage and inflammation were noted to be transient. Given the prevalence of obesity, rodent models have been developed to encompass both non-alcoholic steatohepatitis and alcoholic liver disease. The hybrid model with high-fat and high cholesterol plus chronic and binge ethanol feeding involves a high-fat and high cholesterol diet comprising 40% of the caloric intake, while chronic intragastric ethanol feeding comprised 60% of the caloric intake of these mice, and was supplemented by weekly binges of ethanol. This model represents moderate to severe alcoholic hepatitis, causing significant liver inflammation, by reproducing chronic alcoholic steatohepatitis characterized by balloon cell degeneration, macrophage activation and infiltration, and progression of liver fibrosis. It closely mimics alcoholic hepatitis histologically, with findings of neutrophil infiltration, but also clinically represents this acute entity as mice develop splenomegaly, hypoalbuminemia, and hyperbilirubinemia35. Models to show advanced fibrosis and cholestasis, as observed in alcoholic hepatitis, do not yet exist. “Second-hit” or “Multiple-hit” models exist in which use chronic ethanol feeding to induce hepatic susceptibility to other agents including nutritional modification or pharmacologic agents to achieve the acute on chronic disease state have shown some promising pathologic similarities to human alcoholic hepatitis including coagulative necrosis and inflammation 36, but often as a consequence of the second agent used, and thus calls into question their clinical relevance.
The traditional approach of identifying molecular drivers in animal models and translating this work to human disease poses many challenges. In addition, the the challenges of finding appropriate models to mimic histologic findings in human alcoholic liver disease and incorporate co-morbidities faced by patients with alcoholic liver disease and those presenting with alcoholic hepatitis, remains difficult to do.
A systematic rational approach to designing translational studies to examine alcoholic hepatitis begins by first determining the phenotype of the patient, disease severity and comorbidities that contribute to the acute disease state, with careful consideration given to related overlapping syndromes. Disease severity and patient prognosis can be computed using a variety of scoring systems as discussed previously. The second step involves obtaining anthropometrical, clinical, and histological data, though liver biopsy, and collection of ancillary biospecimens including suprahepatic and peripheral blood, PMNs, and stool for genomic, proteomic, and metabolomic studies. The third step involves the correlation of gene and protein expression in biospecimens obtained with clinical or histological features observed in patients to identify potential molecular drivers of disease. Finally, the fourth step involves testing hypothetical relations in in vitro and in vivo models in carefully designed animal studies, with the eventual goal in mind of identifying druggable molecular targets to reverse or abort progression of this disease (Figure 3).
Figure 3.
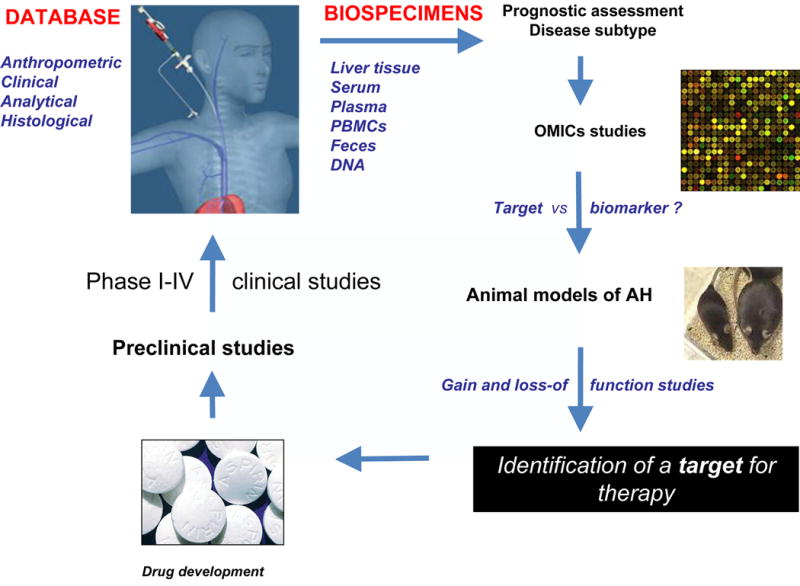
Systematic approach to designing translational studies to evaluate important molecular markers and potential therapeutic targets in AH.
6. Molecular pathogenesis of Alcoholic Liver Disease and potential targets for intervention
An understanding of the spectrum of disease states comprising ALD and the pathogenic mechanisms at work is imperative to our understanding of the development of alcoholic hepatitis as an acute on chronic state, and provides ample opportunity to halt progression in early disease stages. Table 2 summarizes some of the key molecular and cellular markers that are potential new targets for therapeutic intervention. Chronic alcohol ingestion leads to steatosis, an asymptomatic state reversible on cessation of alcohol consumption. Prolonged alcohol consumption can lead to alcoholic steatohepatitis, and inflammatory stated marked my neutrophil infiltration and hepatocellular damage. Histological evaluation demonstrates fat accumulation, hepatocyte ballooning, neutrophil infiltration, and even early signs of pericellular and sinusoidal fibrosis. Fibrosis can then progress to cirrhosis complicated by poor synthetic function portal hypertension and its associated complications. Alcoholic hepatitis, as previously mentioned, is an acute on chronic disease state, in which the majority of patients have underlying advanced fibrosis, and in addition to a marked inflammatory response, histologically, canalicular and lobular bilirubinostasis may be present37. The interplay of molecular and cellular markers contributing to disease is complex (Figure 4) and is discussed in detail below.
Table 2.
Potential novel therapeutic targets for intervention.
| Pathologic Process | Potential Therapeutic Targets | Mechanism of Action |
|---|---|---|
| Steatosis Decrease in fatty acid oxidation |
CYP2E1 and PPAR-α | Ethanol upregulates CYP2E1 production resulting in hepatic oxidative stress, which in turn prevents up-regulation of PPAR-α, a nuclear hormone receptor, that regulates transcription of many genes involved in free fatty acid transport and oxidation. |
| Steatosis Increase in fatty acid uptake |
SREBP-1c | Acetaldehyde upregulates SREBP-1c, a master transcription factor that upregulates the expression of genes encoding lipogenic enzymes, resulting in increased fatty acid synthesis. |
| CB1, CB2 | Endocannabimoids are also responsible for up-regulating SREBP-1c leading to increased fatty acid synthesis, but also mediate alcoholic liver injury through signaling through either of two cannabinoid receptors, CB1 and CB2. Signaling through CB1 worsens alcoholic liver injury while signaling through the CB2 confers a protective role in the development of steatosis. | |
| SIRT1 | SIRT1, a NAD(+)-dependent class III protein deacetylase, also inhibits SREBP-1c activation decreasing fatty acid synthesis. SIRT1 agonists could be potentially be used to curtail the effects of ethanol on steatosis. | |
| Adiponectin, AMPK | AMPK, a serine-threonine kinase, inactivates the rate-limiting enzyme ACC in fatty acid synthesis. Inactivation of ACC leads to a reduction in the levels of malonyl Co-A, a precursor in fatty acid synthesis and an inhibitor of the rate-limiting enzyme CPT1, in fatty acid oxidation, resulting in less fatty acid synthesis and increased oxidation. While ethanol inhibits this pathway, adiponectin can confer protection by modulating this pathway. | |
| Steatosis Autophagy |
mTOR | Chronic alcohol consumption inhibits autophagy, leading to fat accumulation and. Inhibition of the mTOR signaling pathway inhibits progression to steatosis. |
|
| ||
| Steatohepatitis Bacterial translocation and the inflammatory cascade |
LPS, TLR | Ethanol increases gut permeability resulting in translocation of bacterial products, such as LPS, into portal circulation, resulting in activation of Kupffer cells through TLR4 leading to production and release of pro-inflammatory cytokines. TLR antagonists have been proposed as potential therapeutic agents. |
| IL-6, IL-10, STAT3 | Kupffer cells secrete anti-inflammatory cytokines, IL-6 and IL-10, which bind STAT3 to curb inflammation and slow progression of liver injury. | |
| Steatohepatitis Neutrophillic infiltration |
IL-1, osteopontin, CXCL4, CXCL5, CXCL6 | IL-1, osteopontin, CXCL4, CXCL5, and CXCL6 are chemokines that attract and activate macrophages. |
|
| ||
| Fibrosis Promotion of fibrosis |
ERK1, ERK2, phosphinositide 3 kinase-Akt and JNK | ROS production stimulates collagen production though stimulation of pro-fibrogenic signaling pathways in HSCs including ERK1, ERK2, phosphinositide 3 kinase-Akt and JNK. |
| Fibrosis Inhibition of collagen degradation |
metalloproteinases | ROS produced with heavy alcohol consumotion propagate collagen accumulation by preventing collagen degradation, through direct inhibition of metalloproteinases which degrade collagen. |
|
| ||
| Apoptosis and massive cell death | Caspase | Caspase inhibitors are known to inhibit apoptosis. |
|
| ||
| Innate immune response Neutrophil recruitment |
IL-17, IL-8, CXCL1, ostepontin | IL-17 induces neutrophil recruitment by stimulating HSCs to secrete IL-8 and CXCL1. The modification of these chemokines may mediate neutrophil infiltration and attenuate alcoholic hepatitis. Osteopontin, an extracellular matrix protein, also contributes to neutrophillic recruitment, and is markedly upregulated in alcoholic hepatitis, and is an attractive target in considering new therapeutic agents. |
|
| ||
| Adaptive immune response T cells |
Circulating T cells | Patients with alcoholic hepatitis have been found to have circulating T cells with antibodies to free-radical adducts |
|
| ||
| Intestinal dysbiosis | LCFA | Genomic and metabolomic analyses of intestinal bacteria revealed low levels of lactobacilli and reduced production of LCFA in those with alcoholic liver injury. Supplementation with LCFA restored eubiosis, intestinal barrier function, and reduced liver injury in mice. |
|
| ||
| Hepatic regeneration | The mechanism remains to be elucidated, but severe alcoholic hepatitis is marked by a failure of liver progenitor cells to progress past massive proliferation to maturation into mature hepatocytes. Agents to promote hepatic regeneration are being explored. | |
CYP2E1, cytochrome P450 2E1, PPAR-α: proliferator-activated receptor-α, SREBP-1c: sterol regulatory element-binding protein-1c, CB1: cannabinoid receptor 1, CB2: cannabinoid receptor 2, SIRT1: sirtuin 1, AMPK: AMP-activated protein kinase, mTOR: mammalian target of rapamycin, LPS: lipoplysaccharide, TLR: toll-like receptor, IL-6: interleukin-6, IL-10: interleukin-10, IL-1: interleukin-1, IL-17: interleukin-17, IL-8: interleukin-8, LCFA: long-chain fatty acids
Figure 4.
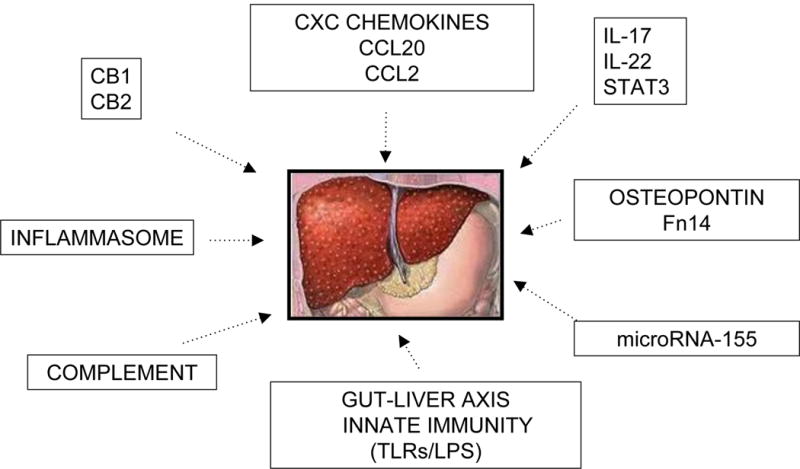
Potential molecular and cellular targets for therapy for AH identified in translational studies including human samples.
Steatosis as a result of decreased fatty acid oxidation
Steatosis evolves from the intrahepatic accumulation of fats, mainly triglycerides, phospholipids, and cholesterol esters, which is aggravated by excessive alcohol intake through disruption of fatty acid oxidation, and increase of fatty acid and triglyceride synthesis and uptake. Alcohol intake increases the NADH/NAD+ ratio in hepatocytes, which disrupts mitochondrial β-oxidation of fatty acids resulting in accumulation and steatosis38. Excessive alcohol consumption is also responsible for decreasing β-oxidation of fatty acids through it’s metabolite, acetaldehyde, which directly inhibits the DNA-binding ability and transcriptional activation ability of peroxisome proliferator-activated receptor-α (PPAR-α)38, a nuclear hormone receptor that regulates transcription of many genes involved in free fatty acid transport and oxidation39,40. Ethanol can also indirectly inhibit the function of PPAR-α through various mechanisms. The first is by up-regulating cytochrome P450 2E1 (CYP2E1) and hepatic oxidative stress, which in turn inhibits oxidation of fatty acids by preventing up-regulation of PPAR-α as demonstrated in wild-type mice as compared to CYP2E1 knock-out mice41. The second and third include down regulation of adiponectin, and zinc, both of which have been demonstrated to down-regulate PPAR-α42,43.
Steatosis as a result of increased fatty acid uptake and synthesis
It is well known that alcohol consumption increases hepatic influx of free fatty acids from adipose tissue and chylomicrons from the intestinal mucosa38. Fatty acid synthesis is increased through up-regulation of lipogenic enzymes. This is mainly accomplished directly through up-regulation of sterol regulatory element-binding protein-1c (SREBP-1c), a master transcription factor that up-regulates the expression of genes encoding lipogenic enzymes, or indirectly through inhibition of factors that inhibit SREBP-1c. Acetaldehyde promotes transcription of SREBP-1c, which in turn up-regulates expression of lipogenic enzyme genes, contributing to increased fatty acid synthesis44. SREBP-1c is also up-regulated through multiple other processes including ethanol-induced hepatocyte ER stress45, production and binding of adenosine to A1 receptors46, and through endocannnabinoids47. Endocannabimoids mediate alcoholic liver injury through signaling through either of two cannabinoid receptors, cannabinoid receptor 1 (CB1) and cannabinoid receptor 2 (CB2)48. Mouse studies have demonstrated that signaling through the CB1 cannabinoid receptor worsens alcoholic liver injury while signaling through the CB2 confers a protective role in the development of steatosis and alcoholic liver injury47. This suggests that CB1 receptor antagonists and CB2 receptor agonists could serve as potential therapeutic agents in preventing steatosis and alcoholic liver disease. CB1 receptor antagonists have their limitations, due to their neuropsychiatric side effects, however, peripherally acting CB1 antagonists are currently being explored48. SREBP-1c is upregulated indirectly though down-regulation of various factors that reduce its expression, such as ethanol-mediated AMP-activated protein kinase (AMPK) inhibition49. Studies in rat hepatoma cells demonstrated that activation of SREBP-1c by ethanol is also mediated by mammalian sirtuin 1 (SIRT1), a NAD(+)-dependent class III protein deacetylase; ethanol exposure induced SREBP-1c lysine acetylation and SREBP-1c transcriptional activity, which was reversed by adding a SIRT1, suggesting that ethanol must have an inhibitory effect on SIRT1, inhibiting its inhibitory effect on SREBP-1c50. This finding suggests that SIRT1 agonists could be potentially be used to curtail the effects of ethanol on steatosis. To extend this further, it has been shown that adiponectin confers protection against alcoholic fatty liver via modulation of complex hepatic signaling pathways through central regulatory system, SIRT1-AMPK axis 51.
Direct effects of ethanol on enzymes involved in fatty acid metabolism
In addition to regulating transcription factors involved in the regulation of genes involved in fatty acid metabolism, ethanol can also directly affect the activities of these enzymes through it’s inhibition of AMPK, a serine-threonine kinase. This was demonstrated through animal models of concomitant obesity and alcohol consumption, in which these mice demonstrated defective adiponectin-AMPK signaling even in the presence of increased adiponectin downstream of p-AMPK, suggesting a role for AMPK in steatosis52. AMPK functions to inactivate the rate-limiting enzyme acetyl-CoA carboxylase (ACC) in fatty acid synthesis. In turn, inactivation of ACC leads to a reduction in the levels of malonyl Co-A, a precursor in fatty acid synthesis, and an inhibitor of the rate-limiting enzyme carnitine palmitoyltransferase 1 (CPT1), in fatty acid oxidation, resulting in less fatty acid synthesis and increased oxidation53.
In addition, AMPK directly phosphorylates and inhibits SREBP activity in hepatocytes, thereby attenuating steatosis. In this manner, AMPK inhibits fatty acid synthesis but promotes fatty acid oxidation via the inactivation of ACC enzyme activity54. Alcohol consumption inhibits AMPK activity in the liver, leading to increased activity of ACC and decreased activity of CPT1 contributing to hepatic fatty acid accumulation and progression to steatosis49.
6. Cellular pathogenesis of alcoholic liver disease
The role of autophagy in steatosis
Autophagy removes lipid droplets from hepatocytes55. The role of alcohol intake on the autophagic process depends greatly on the chronicity or acuity of intake. Chronic alcohol consumption inhibits autophagy, and thus leads to fat accumulation and steatosis56,57. Mouse studies have demonstrated that acute ethanol consumption conversely activates autophagy through the production of reactive oxygen species (ROS) and inhibition of the mammalian target of rapamycin (mTOR) signaling pathway, inhibiting progression to steatosis58.
The role of the innate immune system in development of steatohepatitis
The innate immune system plays a large role in propagation of inflammation in steatohepatitis and the development of fibrosis. Excessive alcohol intake results in the mass production of ROS, which react with nucleic acids, fatty acids, proteins, and structural cell components to form adducts. These adducts are known to be potent activators of the innate immune system59.
The contribution of bacterial-immune system interplay has received more attention in many gastrointestinal disorders. Kupffer cells, the livers native phagocytes, serve as the first line of innate immune system defense. It is known that alcohol increases gut permeability resulting in translocation of bacterial products such as LPS into portal circulation. This results in activation of Kupffer cells through TLR4 signaling through MyD88-dependent and independent (TRIF/IRF-3) pathways60 leading to production and release of pro-inflammatory cytokines, including TNF-α 61–63. TLR antagonists have been proposed as potential therapeutic agents for the management of alcoholic liver disease. Kupffer cells are also activated through binding of complement proteins, C3 and C5, induced through ethanol activation of the complement cascade64,65.
While the innate inflammatory response is generally believed to propagate liver injury, Kupffer cells also have mechanisms in place to slow down and halt progression of inflammation through secretion of anti-inflammatory cytokines including IL-6 and IL-10. Through activation of STAT3, these cytokines curb inflammation and slow progression of liver injury67, 68.
Neutrophil infiltration is cytokine-mediated, through IL-17; IL-17 also stimulates hepatic stellate cells (HSCs) to produce IL-8 and CXCL1, these chemokines in turn, recruit more neutrophils, completing the cycle. Additional mediators that assist with neutrophil recruitment include IL-1, osteopontin, CXCL4, CXCL5, and CXCL6; these also activate macrophages during liver injury68,69.
The role of the innate immune system in development of fibrosis
Hepatic fibrosis is characterized by the excessive accumulation of collagen and other extracellular matrix proteins. The extensive hepatocellular damage that occurs at this stage of liver injury, results in the production of cytokines, neuroendocrine factors, and angiogenic factors, leading to activations HSCs. Portal and bone marrow-derived fibroblasts also contribute to fibrosis70,71.
The ethanol metabolite, acetaldehyde plays an important role in the instigation and maintenance of fibrosis, through HSC activation and maintenance of the activated phenotype. Through its rapid reaction with cellular components to form adducts, acetaldehyde and its adducts, malondialdehyde, 4-hydroxynonenal, and malondialdehyde-acetaldehyde, act upon HSCs to keep them in the perpetually activated “on” state72.
In a similar manner, ROS can cause activation of HSCs. ROS also directly stimulate fibrosis through stimulating production of collagen though stimulation of pro-fibrogenic signaling pathways in HSCs including ERK1, ERK2, phosphinositide 3 kinase-Akt and JNK73. ROS also propagate collagen accumulation by preventing collagen degradation, first through direct inhibition of metalloproteinases which degrade collagen, and the secondly, through upregulation of the tissue inhibitor of metalloproteinases73.
Extrinsic propagators of fibrosis include transluminal translocation of bacterial LPS. LPS activates the TLR4 on HSCs directly inducing HSC activation74, it activates TLR4 signaling pathways on hepatic sinusoidal endothelial cells promoting angiogenesis and subsequent fibrosis75. Finally, TLR4 indirectly stimulates fibrosis through activation of Kupffer cells, which in exchange, release ROS and other pro-fibrogenic cytokines, causing activation of HSCs76,77.
7. Cellular targets for therapeutic management of alcoholic hepatitis
Cell death via apoptosis
Massive hepatocyte cell death is a prominent feature of alcoholic hepatitis, and as previously discussed, apoptosis is a prominent feature of many of the preceding stages of alcoholic liver disease. Since caspase inhibitors are known to inhibit apoptosis, animal studies have been done in models of chronic liver injury from viral hepatitis secondary to hepatitis C infection, and non-alcoholic steatohepatitis, and caspase inhibitors have shown promising results in ameliorating liver injury and impeding progression to fibrosis78–80. It is reasonable to think such an approach would work in alcoholic liver disease, in particular in alcoholic hepatitis.
Role of Innate Immune System
Studies from other models of liver disease suggest that following activation, neutrophils undergo transmigration into the liver parenchyma where they destroy damaged hepatocytes through the release of ROS and proteases, supporting their prominent role in ALD81. IL-17 is increased in patients with alcoholic hepatitis and directly induces neutrophil recruitment, but also indirectly promotes neutrophil recruitment by stimulating HSCs to secrete IL-8 and CXCL182,83. This suggests that the modification of these chemokines, or their precursors or activators, may mediate neutrophil infiltration and perhaps attenuate alcoholic hepatitis. Translational studies have examined the role of the CXCL family of chemokines, and found elevated levels correlate with severity of disease, degree of portal hypertension, and patient survival84,85. Given these promising findings, therapeutic agents that target CXCL chemokines may be considered in the treatment of AH. Osteopontin is an extracellular matrix protein that is markedly upregulated in alcoholic hepatitis, similar to other CXCL chemokines86. Agents that inhibit osteopontin, therefore, are also attractive in considering new therapeutic agents. The redundancy of chemokines and their receptors makes the development of targeted therapeutics challenging.
Instigators of inflammation are also thought to play an important role. Sources of inflammatory mediators can be classified as sterile, originating from intracellular sources, or microbiological, from bacterial translocation in the gut. Damage-associated molecular patterns (DAMPs) are intracellular molecules released by dying cells that trigger the innate immune system87. Among the DAMPs, high mobility group box-1 (HMGB-1) has been implicated in the development of alcoholic steatohepatitis88, and likely also has a role in alcoholic hepatitis. Gut-derived bacterial products belong to the class of pathogen-associated molecular patterns (PAMPs). These PAMPs circulate trough the portal circulation and induce an inflammatory response through activation of HSCs and Kupffer cells88,89. Inhibition of gut leakage could be a potential target for therapy aimed at preventing the initiation of the innate immune response in alcoholic hepatitis.
Role of the Adaptive Immune System
While the role of the innate immune system has been widely explored, the role of the adaptive immune system in hepatocellular injury and propagation of alcoholic hepatitis leaves many questions unanswered. It is well-known that the adaptive immune system responds to oxidative stress and peroxidation adducts, but it’s role in hepatocellular damage and inflammation in alcoholic hepatitis remains unknown. As previously described, increased alcohol consumption generates ROS through multiple mechanisms and leads to adduct formation; protein adducts have altered conformation and function, and are relatively immunogenic. Patients with alcoholic hepatitis have been found to have circulating T cells with antibodies to these adducts, enforcing that the adaptive immune response likely plays a large, yet undiscovered role in AH 90–93.
Targeting dysbiosis
Alterations in the gut microbiome has unique implications on the development of alcoholic hepatitis, this was first suggested in the intragastric mouse feeding model in which elevated serum ethanol levels were maintained, treated mouse populations were observed to have both microbial translocation and dysbiosis94. In studies involving patients with chronic alcoholic liver disease, administration of probiotics appeared to improve liver function in this patient group, further supporting that the intestinal bacterial milieu is of great importance95. Work examining the applicability of probiotics in patients with alcoholic hepatitis is still underway. Other studies which included genomic and metabolomic analyses of intestinal bacteria revealed low levels of lactobacilli and reduced production of saturated long chain fatty acids (LCFA). In this model, supplementation with LCFA restored eubiosis, intestinal barrier function, and reduced liver injury in mice96, suggesting a role for potential supplementation of LCFA in this patient group.
The role of hepatocyte proliferation and regeneration
Hepatic regeneration in the healthy liver results from expansion of the remaining healthy hepatocytes. In the diseased state, in which hepatocyte proliferation is inhibited, pluripotent liver progenitor cells, also referred to as oval cells, or ductal hepatocytes, proliferate and differentiate to repopulate hepatocytes or biliary epithelial cells99. In the rodent model, alcohol attenuates regeneration of hepatocytes following partial surgical hepatectomy100, so though human studies lack, it is reasonable to hypothesize that alcohol not only causes hepatocellular injury and death, but also prevents regeneration. While histologically, the presence of bilirubinostasis and severe fibrosis are associated with a poorer prognosis in alcoholic hepatitis, the presence of proliferating hepatocytes is associated with better prognosis101. In addition, intense neutrophilic infiltrate was also associated with better prognosis99, suggesting that cytokines released by neutrophils likely play a role in hepatic regeneration following cessation of alcohol, and that resolving inflammation may actually have a beneficial, rather than detrimental role in alcoholic liver disease, contributing to hepatic regeneration. Severe alcoholic hepatitis is marked by a failure of liver progenitor cells to progress past massive proliferation to maturation into mature hepatocytes102, the mechanism for this remains to be elucidated. Potential therapeutic agents to promote hepatic regeneration are being explored.
8. Conclusion
ALD is a leading cause of liver related morbidity and mortality, encompassing a spectrum of disorders ranging from asymptomatic steatosis, steatohepatitis, fibrosis, cirrhosis and its related complications, as well as the the acute on chronic state of AH. While multidisciplinary efforts continue to be aimed at curbing progression of this spectrum of disorders, there is an urgent need for effective therapeutic interventions for AH given it’s high mortality rate and limitations of current treatment regimens. Corticosteroids and pentoxyfylline, though used extensively, offer only a modest survival benefit. Anti-TNF agents, including infliximab and etanercept, did not prove to be safe or effective, due to their increased rates of infection and increased mortality observed in larger studies. Adjunctive therapies, such as N-acetylcysteine, which is known to replenish glutathione in damaged hepatocytes and prevent cell death in ALD, was also found to have no effect on six-month survival. Liver transplantation continues to be limited for patients with ALD, and though risk of associated complications is not higher among this group, there remains a hesitancy and concern among providers, and it is estimated that as many as 95% of patients with end-stage liver disease related to alcohol are never formally evaluated for candidacy for LT. This drives our need to find better therapeutic targets for ALD with focus on AH. While our understanding of the molecular and cellular mechanisms of disease stems from animal models, there remains a dire need for translational research in this field to aid in bridging our gaps in understanding. Recent work in discovering the molecular and cellular basis of disease progression in ALD has helped uncover potential new targets for therapeutic intervention. In early stages of ALD, particularly in steatosis, many molecular targets involved in modifying transcription of genes responsible in fatty acid synthesis and accumulation have been explored as potential targets of therapeutic intervention including SREBP-1c, and the cannabinoid receptors CB1, and CB2. Similar pathways, including activation of the receptor PPAR-α have been explored in their role in fatty acid oxidation as it relates to progression to steatosis. The role of various inflammatory pathways in progression to steatohepatitis have been explored as potential targets including inhibition of translocation of bacterial products, such as LPS, which are known to trigger an inflammatory response, inhibition of TLR signaling pathways, and release of pro-inflammatory cytokines including IL-6, IL-10. Inhibition of profibrogenic pathways in HSCs, including ERK1, ERK2, phosphinositide 3 kinase-Akt and JNK, have also been explored and are attractive candidates for therapeutic intervention to prevent progression to fibrosis. Much work remains to elucidate the role of the cellular responses of the innate and adaptive immune systems in AH in uncovering potential targets for intervention. The role of IL-17 in inducing neutrophil recruitment by stimulating HSCs to secrete IL-8 and CXCL1 has been explored, with the thought that modification of these chemokines may mediate neutrophil infiltration and attenuate alcoholic hepatitis. The role of osteopontin has also been explored as it contributes to neutrophillic recruitment in alcoholic hepatitis, but much work remains ahead. Similarly, the mechanisms have not yet been elucidated, but it is well known that the adaptive immune response plays an important role in progression to AH, as antibodies to adducts generated through free radical oxidation have been found on circulating T cells, though potential targets for therapeutic intervention remain to be discovered.
Acknowledgments
Funding sources: This work was supported by the National Institute on Alcohol Abuse and Alcoholism (NIAAA)(1U01AA021908-01) and P30 DK34987.
Abbreviations
- AASLD
American association for the Study of Liver Disease
- ALD
alcoholic liver disease
- HCC
hepatocellular carcinoma
- ASH
alcoholic steatohepatitis
- AH
alcoholic hepatitis
- ADH
alcohol dehydrogenase
- ALDH
acetaldehyde dehydrogenase
- CYP2E1
Cytochrome P450 2E1
- EASL
European Association for the Study of the Liver
- ER
endoplasmic reticulum
- IRAK-M
Interleukin-1 receptor-associated kinase
- MELD
model for end-stage liver disease
- DF
Maddrey Discriminant Function
- GAHS
Glasgow Alcoholic Hepatitis Score
- ABIC
Age, Bilirubin, INR, Creatinine
- UKELD
UK End-stage Liver Disease
- IL
interleukin
- LPS
lipopolysaccharide
- LT
liver transplant
- TLR
toll-like receptor
- SREBP-1c
sterol regulatory element-binding protein 1c
- TLR4
toll-like receptor 4
- TNF
tumor necrosis factor
- AMPK
AMP-activated protein kinase
- PNPLA3
patatin-like phospholipase domain-containing protein 3
- PPAR
peroxisome proliferator-activated receptor
- ROS
ROS
- HSC
hepatic stellate cell
- NK
natural killer
- IFN
interferon
- SAMe
S-adenosylmethionine
- NIAA
National Institute of Alcohol Abuse and Alcoholism
- PMN
Polymorphonuclear leukocytes
- WHO
World Health Organization
References
- 1.O’Shea RS, et al. Alcoholic liver disease. Hepatology. 2010;51:307–328. doi: 10.1002/hep.23258. [DOI] [PubMed] [Google Scholar]
- 2.World Health Organization. Alcohol Fact Sheet WHO Media Center Fact Sheets. 2015 < http://www.who.int/mediacentre/factsheets/fs349/en/>.
- 3.NIAAA. Alcohol Facts and Statistics. < http://www.niaaa.nih.gov/alcohol-health/overview-alcohol-consumption/alcohol-facts-and-statistics>.
- 4.Mathurin P, Bataller R. Trends in the management and burden of alcoholic liver disease. J Hepatol. 2015;62(1 Suppl):S38–46. doi: 10.1016/j.jhep.2015.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sandahl TD, Jepsen P, Thomsen KL, Vilstrup H. Incidence and mortality of alcoholic hepatitis in Denmark 1999–2008: a nationwide population based cohort study. J Hepatol. 2011;54:760–4. doi: 10.1016/j.jhep.2010.07.016. [DOI] [PubMed] [Google Scholar]
- 6.Tsukamoto H, et al. “Second hit” models of alcoholic liver disease. Semin Liver Dis. 2009;29:178–187. doi: 10.1055/s-0029-1214373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wilfred de Alwis NM, Day CP. Genetics of alcoholic liver disease and nonalcoholic fatty liver disease. Semin Liver Dis. 2007;27:44–54. doi: 10.1055/s-2006-960170. [DOI] [PubMed] [Google Scholar]
- 8.Zintzaras E, et al. Do alcohol-metabolizing enzyme gene polymorphisms increase the risk of alcoholism and alcoholic liver disease? Hepatology. 2006;43:352–61. doi: 10.1002/hep.21023. [DOI] [PubMed] [Google Scholar]
- 9.Tian C, et al. Variant in PNPLA3 is associated with alcoholic liver disease. Nat Genet. 2010;42:21–3. doi: 10.1038/ng.488. [DOI] [PubMed] [Google Scholar]
- 10.Stickel F, Hampe J. Genetic determinants of alcoholic liver disease. Gut. 2012;61:150–9. doi: 10.1136/gutjnl-2011-301239. [DOI] [PubMed] [Google Scholar]
- 11.Trépo E, et al. Common polymorphism in the PNPLA3/adiponutrin gene confers higher risk of cirrhosis and liver damage in alcoholic liver disease. J Hepatol. 2011;55:906–12. doi: 10.1016/j.jhep.2011.01.028. [DOI] [PubMed] [Google Scholar]
- 12.Marcos M, et al. Tumor necrosis factor polymorphisms and alcoholic liver disease: a HuGE review and meta-analysis. Am J Epidemiol. 2009;170:948–56. doi: 10.1093/aje/kwp236. [DOI] [PubMed] [Google Scholar]
- 13.Zeng T, et al. Association between CD14-159C>T polymorphisms and the risk for alcoholic liver disease: a meta-analysis. Eur J Gastroenterol Hepatol. 25:1183–9. doi: 10.1097/MEG.0b013e3283612ff1. 201. [DOI] [PubMed] [Google Scholar]
- 14.Altamirano J, Bataller R. Alcoholic liver disease: pathogenesis and new targets for therapy. Nat Rev Gastroenterol Hepatol. 2011;8:491–501. doi: 10.1038/nrgastro.2011.134. [DOI] [PubMed] [Google Scholar]
- 15.Casanova J, Bataller R. Alcoholic hepatitis: Prognosis and treatment. Gastroenterol Hepatol. 2014;37:262–8. doi: 10.1016/j.gastrohep.2014.02.001. Epub 2014 Mar 20. [DOI] [PubMed] [Google Scholar]
- 16.Singal AK, et al. Nutritional status of patients with alcoholic cirrhosis undergoing liver transplantation: time trends and impact on survival. Transpl Int. 2013;26:788–94. doi: 10.1111/tri.12123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rambaldi A, et al. Anabolic-androgenic steroids for alcoholic liver disease: a Cochrane review. Am J Gastroenterol. 2002;97:1674–81. doi: 10.1111/j.1572-0241.2002.05826.x. [DOI] [PubMed] [Google Scholar]
- 18.Cabré E, et al. Short- and long-term outcome of severe alcohol-induced hepatitis treated with steroids or enteral nutrition: a multicenter randomized trial. Hepatology. 2000;32:36–42. doi: 10.1053/jhep.2000.8627. [DOI] [PubMed] [Google Scholar]
- 19.Papastergiou V, et al. Nine scoring models for short-term mortality in alcoholic hepatitis: cross-validation in a biopsy-proven cohort. Aliment Pharmacol Ther. 2014;39:721–32. doi: 10.1111/apt.12654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Louvet A, et al. Combining Data From Liver Disease Scoring Systems Better Predicts Outcomes of Patients With Alcoholic Hepatitis. Gastroenterology. 2015;149:398–406.e8. doi: 10.1053/j.gastro.2015.04.044. Epub 2015 Apr 29. [DOI] [PubMed] [Google Scholar]
- 21.European Association for the Study of Liver. EASL clinical practical guidelines: management of alcoholic liver disease. J Hepatol. 2012;57:399–420. doi: 10.1016/j.jhep.2012.04.004. [DOI] [PubMed] [Google Scholar]
- 22.Mathurin P, et al. Corticosteroids improve short-term survival in patients with severe alcoholic hepatitis: meta-analysis of individual patient data. Gut. 2011;60:255–60. doi: 10.1136/gut.2010.224097. [DOI] [PubMed] [Google Scholar]
- 23.Louvet A, et al. The Lille model: a new tool for therapeutic strategy in patients with severe alcoholic hepatitis treated with steroids. Hepatology. 2007;45:1348–54. doi: 10.1002/hep.21607. [DOI] [PubMed] [Google Scholar]
- 24.Forrest E, et al. Steroids or pentoxifylline for alcoholic hepatitis (STOPAH): study protocol for a randomised controlled trial. Trials. 2013;14:262. doi: 10.1186/1745-6215-14-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Whitfield K, et al. Pentoxifylline for alcoholic hepatitis. Cochrane Database Syst Rev. 2009;4 doi: 10.1002/14651858.CD007339.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Colmenero J, Bataller R, Sancho-Bru P, et al. Hepatic expression of candidate genes in patients with alcoholic hepatitis: correlation with disease severity. Gastroenterology. 2007;132:687–97. doi: 10.1053/j.gastro.2006.12.036. [DOI] [PubMed] [Google Scholar]
- 27.Dominguez M, Miquel R, Colmenero J, et al. Hepatic expression of CXC chemokines predicts portal hypertension and survival in patients with alcoholic hepatitis. Gastroenterology. 2009;136:1639–50. doi: 10.1053/j.gastro.2009.01.056. [DOI] [PubMed] [Google Scholar]
- 28.Boetticher NC, Peine CJ, Kwo P, Abrams GA, Patel T, Aqel B, et al. A randomized, double-blinded, placebo-controlled multicenter trial of etanercept in the treatment of alcoholic hepatitis. Gastroenterology. 2008;135:1953–60. doi: 10.1053/j.gastro.2008.08.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nguyen-Khac E, Thevenot T, Piquet MA, et al. Glucocorticoids plus N-acetylcysteine in severe alcoholic hepatitis. N Engl J Med. 2011;365:1781–9. doi: 10.1056/NEJMoa1101214. [DOI] [PubMed] [Google Scholar]
- 30.Stickel F, Seitz HK. Alcoholic steatohepatitis. Best Pract Res Clin Gastroenterol. 2010;24:683–93. doi: 10.1016/j.bpg.2010.07.003. [DOI] [PubMed] [Google Scholar]
- 31.Dureja P, Lucey MR. The place of liver transplantation in the treatment of severe alcoholic hepatitis. J Hepatol. 2010;52:759–64. doi: 10.1016/j.jhep.2009.12.021. [DOI] [PubMed] [Google Scholar]
- 32.Mathurin P, Moreno C, Samuel D, Dumortier J, Salleron J, Durand F, et al. Early liver transplantation for severe alcoholic hepatitis. N Engl J Med. 2011;365:1790–800. doi: 10.1056/NEJMoa1105703. [DOI] [PubMed] [Google Scholar]
- 33.Burra P, Senzolo M, Adam R, et al. Liver transplantation for alcoholic liver disease in Europe: a study from the ELTR (European Liver Transplant Registry) Am J Transplant. 2010;10:138–48. doi: 10.1111/j.1600-6143.2009.02869.x. [DOI] [PubMed] [Google Scholar]
- 34.Bertola A, Mathews S, Ki SH, Wang H, Gao B. Mouse model of chronic and binge ethanol feeding (the NIAAA model) Nat Protoc. 2013;8:627–37. doi: 10.1038/nprot.2013.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lazaro R, Wu R, Lee S, Zhu NL, Chen CL, French SW, et al. Osteopontin deficiency does not prevent but promotes alcoholic neutrophilic hepatitis in mice. Hepatology. 2015;61:129–40. doi: 10.1002/hep.27383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsukamoto H, Machida K, Dynnyk A, Mkrtchyan H. “Second hit” models of alcoholic liver disease. Semin Liver Dis. 2009;29:178–87. doi: 10.1055/s-0029-1214373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Altamirano J, Bataller R. Alcoholic liver disease: pathogenesis and new targets for therapy. Nat Rev Gastroenterol Hepatol. 2011;8:491–501. doi: 10.1038/nrgastro.2011.134. [DOI] [PubMed] [Google Scholar]
- 38.Baraona E, Lieber CS. Effects of ethanol on lipid metabolism. J Lipid Res. 1979;20:289–315. [PubMed] [Google Scholar]
- 39.Yu S, Rao S, Reddy JK. Peroxisome proliferator-activated receptors, fatty acid oxidation, steatohepatitis and hepatocarcinogenesis. Curr Mol Med. 2003;3:561–72. doi: 10.2174/1566524033479537. [DOI] [PubMed] [Google Scholar]
- 40.Wagner M, Zollner G, Trauner M. Nuclear receptors in liver disease. Hepatology. 2011;53:1023–34. doi: 10.1002/hep.24148. [DOI] [PubMed] [Google Scholar]
- 41.Lu Y, Zhuge J, Wang X, et al. Cytochrome P450 2E1 contributes to ethanol-induced fatty liver in mice. Hepatology. 2008;47:1483–94. doi: 10.1002/hep.22222. [DOI] [PubMed] [Google Scholar]
- 42.You M, Considine RV, Leone TC, et al. Role of adiponectin in the protective action of dietary saturated fat against alcoholic fatty liver in mice. Hepatology. 2005;42:568–77. doi: 10.1002/hep.20821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kang X, Zhong W, Liu J, et al. Zinc supplementation reverses alcohol-induced steatosis in mice through reactivating hepatocyte nuclear factor-4alpha and peroxisome proliferator-activated receptor-alpha. Hepatology. 2009;50:1241–50. doi: 10.1002/hep.23090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.You M, Fischer M, Deeg MA, et al. Ethanol induces fatty acid synthesis pathways by activation of sterol regulatory element-binding protein (SREBP) J Biol Chem. 2002;277:29342–7. doi: 10.1074/jbc.M202411200. [DOI] [PubMed] [Google Scholar]
- 45.Esfandiari F, Medici V, Wong DH, et al. Epigenetic regulation of hepatic endoplasmic reticulum stress pathways in the ethanol-fed cystathionine beta synthase-deficient mouse. Hepatology. 2010;51:932–41. doi: 10.1002/hep.23382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Peng Z, Borea PA, Varani K, et al. Adenosine signaling contributes to ethanol-induced fatty liver in mice. J Clin Invest. 2009;119:582–94. doi: 10.1172/JCI37409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jeong WI, Osei-Hyiaman D, Park O, et al. Paracrine activation of hepatic CB1 receptors by stellate cell-derived endocannabinoids mediates alcoholic fatty liver. Cell Metab. 2008;7:227–35. doi: 10.1016/j.cmet.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 48.Tam J, Liu J, Mukhopadhyay B, et al. Endocannabinoids in liver disease. Hepatology. 2011;53:346–55. doi: 10.1002/hep.24077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.You M, Matsumoto M, Pacold CM, et al. The role of AMP-activated protein kinase in the action of ethanol in the liver. Gastroenterology. 2004;127:1798–808. doi: 10.1053/j.gastro.2004.09.049. [DOI] [PubMed] [Google Scholar]
- 50.You M, Liang X, Ajmo JM, et al. Involvement of mammalian sirtuin 1 in the action of ethanol in the liver. Am J Physiol Gastrointest Liver Physiol. 2008;294:G892–8. doi: 10.1152/ajpgi.00575.2007. [DOI] [PubMed] [Google Scholar]
- 51.You MRC. Adiponectin: a key adipokine in alcoholic fatty liver. Exp Biol Med (Maywood) 2009;2009:850–859. doi: 10.3181/0902-MR-61. [DOI] [PubMed] [Google Scholar]
- 52.Xu J, Lai KK, Verlinsky A, Lugea A, French SW, Cooper MP, et al. Synergistic steatohepatitis by moderate obesity and alcohol in mice despite increased adiponectin and p-AMPK. J Hepatol. 2011;55:673–82. doi: 10.1016/j.jhep.2010.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Viollet BGB, Leclerc J, Hébrard S, Lantier L, Mounier R, Andreelli F, Foretz M. AMP-activated protein kinase in the regulation of hepatic energy metabolism: from physiology to therapeutic perspectives. Acta Physiol (Oxf) 2009;196:81–98. doi: 10.1111/j.1748-1716.2009.01970.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li Y, Xu S, Mihaylova MM, et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 2011;13:376–88. doi: 10.1016/j.cmet.2011.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Czaja MJ. Functions of autophagy in hepatic and pancreatic physiology and disease. Gastroenterology. 2011;140:1895–908. doi: 10.1053/j.gastro.2011.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Donohue TM., Jr Autophagy and ethanol-induced liver injury. World J Gastroenterol. 2009;15:1178–85. doi: 10.3748/wjg.15.1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu D, Wang X, Zhou R, et al. CYP2E1 enhances ethanol-induced lipid accumulation but impairs autophagy in HepG2 E47 cells. Biochem Biophys Res Commun. 2010;402:116–22. doi: 10.1016/j.bbrc.2010.09.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ding WX, Li M, Chen X, et al. Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology. 2010;139:1740–52. doi: 10.1053/j.gastro.2010.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chedid A, Arain S, et al. The immunology of fibrogenesis in alcoholic liver disease. Arch Pathol Lab Med. 2004;128:1230–8. doi: 10.5858/2004-128-1230-TIOFIA. [DOI] [PubMed] [Google Scholar]
- 60.Zhao XJ, Dong Q, Bindas J, et al. TRIF and IRF-3 binding to the TNF promoter results in macrophage TNF dysregulation and steatosis induced by chronic ethanol. J Immunol. 2008;181:3049–56. doi: 10.4049/jimmunol.181.5.3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Petrasek J, Mandrekar P, Szabo G. Toll-like receptors in the pathogenesis of alcoholic liver disease. Gastroenterol Res Pract. 2010;2010 doi: 10.1155/2010/710381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yin M, Wheeler MD, Kono H, Bradford BU, Gallucci RM, Luster MI, et al. Essential role of tumor necrosis factor alpha in alcohol-induced liver injury in mice. Gastroenterology. 1999;117:942–52. doi: 10.1016/s0016-5085(99)70354-9. [DOI] [PubMed] [Google Scholar]
- 63.Hritz I, Mandrekar P, Velayudham A, et al. The critical role of toll-like receptor (TLR) 4 in alcoholic liver disease is independent of the common TLR adapter MyD88. Hepatology. 2008;48:1224–31. doi: 10.1002/hep.22470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pritchard MT, McMullen MR, Stavitsky AB, et al. Differential contributions of C3, C5, and decay-accelerating factor to ethanol-induced fatty liver in mice. Gastroenterology. 2007;132:1117–26. doi: 10.1053/j.gastro.2007.01.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Roychowdhury S, McMullen MR, Pritchard MT, et al. An early complement-dependent and TLR-4-independent phase in the pathogenesis of ethanol-induced liver injury in mice. Hepatology. 2009;49:1326–34. doi: 10.1002/hep.22776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Horiguchi N, Wang L, Mukhopadhyay P, et al. Cell type-dependent pro- and anti-inflammatory role of signal transducer and activator of transcription 3 in alcoholic liver injury. Gastroenterology. 2008;134:1148–58. doi: 10.1053/j.gastro.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Miller AMWH, Bertola A, et al. Inflammation-associated IL-6/STAT3 activation ameliorates alcoholic and nonalcoholic fatty liver diseases in IL-10 deficient mice. Hepatology. 2011;54:846–856. doi: 10.1002/hep.24517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mandrekar P, Ambade A, Lim A, Szabo G, Catalano D. An essential role for monocyte chemoattractant protein-1 in alcoholic liver injury: regulation of proinflammatory cytokines and hepatic steatosis in mice. Hepatology. 2011;54:2185–97. doi: 10.1002/hep.24599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Petrasek J, Bala S, Csak T, Lippai D, Kodys K, Menashy V, et al. IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J Clin Invest. 2012;122:3476–89. doi: 10.1172/JCI60777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–18. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655–69. doi: 10.1053/j.gastro.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Setshedi M, Wands JR, Monte SM. Acetaldehyde adducts in alcoholic liver disease. Oxid Med Cell Longev. 2010;3:178–85. doi: 10.4161/oxim.3.3.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bataller R, Schwabe RF, Choi YH, et al. NADPH oxidase signal transduces angiotensin II in hepatic stellate cells and is critical in hepatic fibrosis. J Clin Invest. 2003;112:1383–94. doi: 10.1172/JCI18212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, et al. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med. 2007;13:1324–32. doi: 10.1038/nm1663. [DOI] [PubMed] [Google Scholar]
- 75.Jagavelu K, Routray C, Shergill U, et al. Endothelial cell toll-like receptor 4 regulates fibrosis-associated angiogenesis in the liver. Hepatology. 2010;52:590–601. doi: 10.1002/hep.23739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Purohit V, Brenner DA. Mechanisms of alcohol-induced hepatic fibrosis: a summary of the Ron Thurman Symposium. Hepatology. 2006;43:872–8. doi: 10.1002/hep.21107. [DOI] [PubMed] [Google Scholar]
- 77.Cubero FJ, Urtasun R, Nieto N. Alcohol and liver fibrosis. Semin Liver Dis. 2009;29:211–21. doi: 10.1055/s-0029-1214376. [DOI] [PubMed] [Google Scholar]
- 78.Pockros PJ, Schiff ER, Shiffman ML, et al. Oral IDN-6556, an antiapoptotic caspase inhibitor, may lower aminotransferase activity in patients with chronic hepatitis C. Hepatology. 2007;46:324–9. doi: 10.1002/hep.21664. [DOI] [PubMed] [Google Scholar]
- 79.Shiffman ML, Pockros P, McHutchison JG, et al. Clinical trial: the efficacy and safety of oral PF-03491390, a pancaspase inhibitor - a randomized placebo-controlled study in patients with chronic hepatitis C. Aliment Pharmacol Ther. 2010;31:969–78. doi: 10.1111/j.1365-2036.2010.04264.x. [DOI] [PubMed] [Google Scholar]
- 80.Ratziu V, Chojkiet M, Sheikh M, et al. Safety, tolerability and preliminary activity of GS-9450, a selective caspase inhibitor, in patients with non-alcoholic steatohepatitis. J Hepatol. 2010;52:S38. [Google Scholar]
- 81.Ramaiah SK, Jaeschke H. Hepatic neutrophil infiltration in the pathogenesis of alcohol-induced liver injury. Toxicol Mech Methods. 2007;17:431–40. doi: 10.1080/00952990701407702. [DOI] [PubMed] [Google Scholar]
- 82.Maltby J, Wright S, Bird G, et al. Chemokine levels in human liver homogenates: associations between GRO alpha and histopathological evidence of alcoholic hepatitis. Hepatology. 1996;24:1156–60. doi: 10.1053/jhep.1996.v24.pm0008903391. [DOI] [PubMed] [Google Scholar]
- 83.Lemmers A, Moreno C, Gustot T, et al. The interleukin-17 pathway is involved in human alcoholic liver disease. Hepatology. 2009;49:646–57. doi: 10.1002/hep.22680. [DOI] [PubMed] [Google Scholar]
- 84.Colmenero J, Bataller R, Sancho-Bru P, et al. Hepatic expression of candidate genes in patients with alcoholic hepatitis: correlation with disease severity. Gastroenterology. 2007;132:687–97. doi: 10.1053/j.gastro.2006.12.036. [DOI] [PubMed] [Google Scholar]
- 85.Dominguez M, Miquel R, Colmenero J, et al. Hepatic expression of CXC chemokines predicts portal hypertension and survival in patients with alcoholic hepatitis. Gastroenterology. 2009;136:1639–50. doi: 10.1053/j.gastro.2009.01.056. [DOI] [PubMed] [Google Scholar]
- 86.Seth D, Gorrell MD, Cordoba S, et al. Intrahepatic gene expression in human alcoholic hepatitis. J Hepatol. 2006;45:306–20. doi: 10.1016/j.jhep.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 87.Kubes P, Mehal WZ. Sterile inflammation in the liver. Gastroenterology. 2012;143:1158–72. doi: 10.1053/j.gastro.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 88.Ge X, Antoine DJ, Lu Y, et al. High mobility group box-1 (HMGB1) participates in the pathogenesis of alcoholic liver disease (ALD) J Biol Chem. 2014;289:22672–91. doi: 10.1074/jbc.M114.552141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Szabo G, Bala S, Petrasek J, Gattu A. Gut-liver axis and sensing microbes. Dig Dis. 2010;28:737–44. doi: 10.1159/000324281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Albano EVM. Immuno mechanisms in alcoholic liver disease. Genes Nutr. 2010;5:141–147. doi: 10.1007/s12263-009-0151-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mottaran E, Stewart SF, Rolla R, et al. Lipid peroxidation contributes to immune reactions associated with alcoholic liver disease. Free Radic Biol Med. 2002;32:38–45. doi: 10.1016/s0891-5849(01)00757-2. [DOI] [PubMed] [Google Scholar]
- 92.Thiele GM, Freeman TL, Klassen LW. Immunologic mechanisms of alcoholic liver injury. Semin Liver Dis. 2004;24:273–87. doi: 10.1055/s-2004-832940. [DOI] [PubMed] [Google Scholar]
- 93.Thiele GM, Duryee MJ, Willis MS, et al. Autoimmune hepatitis induced by syngeneic liver cytosolic proteins biotransformed by alcohol metabolites. Alcohol Clin Exp Res. 2010;34:2126–36. doi: 10.1111/j.1530-0277.2010.01309.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yan AW, Fouts DE, Brandl J, Starkel P, Torralba M, Schott E, et al. Enteric dysbiosis associated with a mouse model of alcoholic liver disease. Hepatology. 2011;53:96–105. doi: 10.1002/hep.24018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.ProKirpich IA, Solovieva NV, Leikhter SN, Shidakova NA, Lebedeva OV, Sidorov PI, et al. Probiotics restore bowel flora and improve liver enzymes in human alcohol-induced liver injury: a pilot study. Alcohol. 2008;42:675–82. doi: 10.1016/j.alcohol.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chen P, Torralba M, Tan J, Embree M, Zengler K, Starkel P, et al. Supplementation of Saturated Long-Chain Fatty Acids Maintains Intestinal Eubiosis and Reduces Ethanol-induced Liver Injury in Mice. Gastroenterology. 2015;148:203–214. doi: 10.1053/j.gastro.2014.09.014. e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Michalopoulos GK. Liver regeneration. J Cell Physiol. 2007;213:286–300. doi: 10.1002/jcp.21172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Saso K, Moehren G, Higashi K, et al. Differential inhibition of epidermal growth factor signaling pathways in rat hepatocytes by long-term ethanol treatment. Gastroenterology. 1997;112:2073–88. doi: 10.1053/gast.1997.v112.pm9178701. [DOI] [PubMed] [Google Scholar]
- 99.Altamirano J, Miquel R, Katoonizadeh A, Abraldes JG, Duarte-Rojo A, Louvet A, et al. A histologic scoring system for prognosis of patients with alcoholic hepatitis. Gastroenterology. 2014;146:1231–9. e1–6. doi: 10.1053/j.gastro.2014.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Dubuquoy L, Louvet A, Lassailly G, Truant S, Boleslawski E, Artru F, et al. Progenitor cell expansion and impaired hepatocyte regeneration in explanted livers from alcoholic hepatitis. Gut. 2015 doi: 10.1136/gutjnl-2014-308410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lanthier N, Rubbia-Brandt L, Lin-Marq N, Clement S, Frossard JL, Goossens N, et al. Hepatic cell proliferation plays a pivotal role in the prognosis of alcoholic hepatitis. J Hepatol. 2015 doi: 10.1016/j.jhep.2015.04.003. [DOI] [PubMed] [Google Scholar]
- 102.Sancho-Bru P, Altamirano J, Rodrigo-Torres D, et al. Liver progenitor cell markers correlate with liver damage and predict short-term mortality in patients with alcoholic hepatitis. Hepatology. 2012;55:1931–41. doi: 10.1002/hep.25614. [DOI] [PubMed] [Google Scholar]