

Abstract
Tumor, calor, rubor, and dolor describe four cardinal signs of inflammation. The fifth—functio laesa, or loss of function—was promulgated by Rudolf Virchow, who, in the 19th century, also noted an intricate link between inflammation and cancer. However, the role of cancer inflammation in driving loss of therapeutic efficacy has only recently been fully appreciated, as a result of molecular and immunohistochemical approaches applied in clinical medicine and the availability of novel agents for modulating inflammation. This review focuses on clinical evidence from solid malignancies that have shaped our view of how the immune system regulates cancer development, progression, and response to treatment. Understanding the multifaceted relationship between inflammation and patient outcomes has the potential to advance prognostic tools and uncover therapeutic opportunities for improving clinical outcomes.
INTRODUCTION
Since Virchow’s early observations in the 19th century associating inflammation with malignancy, clinical studies have identified inflammatory conditions (eg, colitis, gastritis, pancreatitis, hepatitis, and esophagitis) as key risk factors for the development of cancer.1 Inflammation marked by the recruitment of leukocytes, particularly neutrophils, macrophages, and eosinophils, is a near-universal hallmark of cancer that is directed by malignant cells, which seek to establish a microenvironment conducive to survival, growth, and metastasis. This inflammatory reaction can also act as a barrier to the efficacy of cytotoxic and immunotherapeutic interventions. As a result, increasing attention has been placed on strategies designed to ablate or redirect tumor-infiltrating inflammatory cells to improve clinical outcomes. In this review, we discuss the clinical implications of cancer inflammation and emerging interventions that disengage elements of therapeutic resistance imparted by inflammation in cancer.
PROGNOSIS: LINKING INFLAMMATION WITH ONCOLOGY OUTCOMES
Inflammation is a negative prognostic factor across multiple cancers and has been associated with cachexia, muscle wasting, fatigue, decreased cognitive ability, and a reduced quality of life.2,3 Given the link between inflammation and cancer, several large clinical studies have evaluated biomarkers of inflammation for their prognostic value. For example, C-reactive protein (CRP) has been extensively studied as a marker of inflammation. Similarly, albumin is an acute-phase reactant that decreases in the setting of a systemic inflammatory response. Together, elevated CRP level and hypoalbuminemia are defining measures of the modified Glasgow Prognostic Score (mGPS), which has been validated across more than 70 studies involving more than 31,000 patients with a wide-range of malignancies.4 Increased CRP concentrations (> 10 mg/L) and decreased albumin levels (< 35 g/L), and, consequently, a higher mGPS score, are associated with worse overall survival in patients with inoperable non–small-cell lung cancer, independent of tumor staging and performance status. Higher mGPS scores also correlate with poorer quality of life and decreased nutritional status in multiple malignancies.4
In addition, mGPS has recently been explored for its value in predicting therapeutic response. In patients with rectal cancer undergoing neoadjuvant chemotherapy, mGPS was strongly associated with pathologic response to treatment.5 A post hoc analysis of patients with gemcitabine-resistant metastatic pancreatic cancer, treated with capecitabine and ruxolitinib in a randomized phase II study, also demonstrated a positive correlation between overall survival and mGPS.6 However, a subsequent phase III study investigating this same treatment combination and incorporating mGPS as an inclusion criterion was terminated early because of a lack of data confirming a role for CRP in selecting patients most likely to respond to therapy.7 Thus, mGPS is a strong prognostic determinant for patients with cancer, but its role as a predictor of treatment response and in guiding anti-inflammatory interventions remains ill defined.
Another measure of cancer inflammation with clinical significance is the neutrophil-to-lymphocyte ratio (NLR).8 In patients with solid malignancies, a high NLR correlates with a poor prognosis. The NLR has also been shown to predict outcomes in patients with advanced colorectal cancer who are receiving chemotherapy, and it has been used as a predictor of tumor recurrence and progression.9,10 However, the prognostic potential of the NLR may not apply to all therapeutic scenarios. For example, in an unplanned secondary analysis of patients with muscle-invasive bladder treated in a randomized phase III study with or without neoadjuvant chemotherapy, the NLR was not found to be prognostic of overall survival benefit from neoadjuvant chemotherapy.11 In general, though, the NLR describes a balance between innate and adaptive immunity, wherein an accumulation of neutrophils and/or diminution in lymphocytes portends an immune state that is less effective in controlling tumor progression.
The quality of the immune response to cancer has also been explored as a prognostic marker. Among the many cell types observed in tumors, high densities of CD3+ CD8+ T cells displaying a CD45RO+ memory phenotype correlates with improved clinical outcomes.12,13 Taking advantage of this observation, the Immunoscore is a prognostic factor that can be incorporated into routine testing14 and involves immunohistochemical analysis to detect the density and location of CD3+ and CD8+ cells in tumor tissue. An increased density of CD3+ and CD8+ cells in the core versus the invasive margin of the tumor correlates with the best prognosis. Compared with the American Joint Committee on Cancer/Union for International Cancer Control TNM classification system, the Immunoscore has demonstrated improved prognostic significance.13 In addition, it was recently found to be a better predictor of overall survival than microsatellite instability.15 Moreover, CD3+ CD8+ T-cell infiltration in melanoma is associated with treatment response to programmed cell death-1 (PD-1)/programmed cell death ligand-1 (PD-L1) checkpoint immunotherapy.16 An ongoing international effort is underway to validate the Immunoscore metric for colorectal carcinoma.
Together, the laboratory-based metrics described in the previous paragraphs and summarized in Table 1 underscore the prognostic utility of the immune reaction to cancer, ranging from features of systemic inflammation detected in the peripheral blood to the quality of the cellular immune infiltrate detected in tumors. Efforts are underway that aim to apply molecular profiling and multiplex imaging analyses to identify unique features of the inflammatory reaction to cancer for guiding treatment and predicting subsequent clinical outcomes.
Table 1.
Inflammation-Based Prognostic Scoring Systems

INHIBITING INFLAMMATION FOR CANCER PREVENTION AND TREATMENT
The link between inflammatory markers and treatment resistance obviates the potential for targeting key inflammation pathways and improving patient outcomes. Strategies to intervene in cancer inflammation may (1) deplete elements of the inflammatory response or block its recruitment to tumors, 2) inhibit components of the protumorigenic and immunosuppressive phenotype of inflammation, or (3) redirect inflammation with properties that are tumor inhibitory, immune stimulatory, or both (Fig 1).17
FIG 1.

Strategies to target inflammation for cancer therapy. Inflammatory cells (eg, monocytes) can be targeted in cancer using therapeutics that (1) block their recruitment to tumors (eg, C-C chemokine receptor type 2 inhibitors), (2) deplete inflammatory cell subsets (eg, trabectedin and CSF1R antibodies), (3) inhibit protumorigenic signaling pathways (eg, inhibitors of Bruton tyrosine kinase, colony-stimulating factor 1 receptor, focal adhesion kinase, hypoxia inducible factor-2α, indoleamine 2,3 dioxygenase, Janus kinase, phosphatidylinositol-4,5-bisphosphate 3-kinase δ, transforming growth factor-β), and (4) redirect tumor-infiltrating inflammatory cells with antitumor properties (eg, CD40 agonists, CD47 antagonists, dectin-1 agonists, and Toll-like receptor agonists).
Targeting Inflammation for Cancer Prevention
Early evidence of clinical benefit achieved with targeting inflammation was seen in a prospective cohort study of aspirin use among patients with colon cancer, for whom low-dose aspirin use correlated with reduced fatality.18 Aspirin irreversibly inactivates the cyclooxygenase (COX) enzyme that is required for prostaglandin synthesis, an important mediator of inflammation. This prophylactic effect seen with aspirin in colon cancer has been extended to randomized controlled studies comparing aspirin versus placebo among carriers of Lynch syndrome, a hereditary condition with high predisposition for colorectal cancer. In the randomized CAPP2 trial, daily aspirin use for at least 2 years significantly reduced the incidence of colorectal cancer as well as other Lynch syndrome cancers after almost 5 years of follow-up.19 An observational study of patients with Lynch syndrome included in the Colon Cancer Family Registry also showed a reduced risk for colorectal cancer among patients taking aspirin.20 The ongoing CAPP3 randomized double-blinded trial is evaluating the dose of aspirin needed for chemoprevention in Lynch syndrome (ClinicalTrials.gov identifier: NCT02497820).
The prophylactic effect of suppressing inflammation may extend beyond colon cancer to other malignancies such as breast, prostate, and lung cancers.21 In a population-based case-control study, the frequency and duration of using nonsteroidal anti-inflammatory agents (ie, aspirin and ibuprofen) were associated with reduced risk for hormone receptor–positive breast cancer.22 However, the most pronounced beneficial effect of aspirin has been observed in gastrointestinal malignancies, including esophageal, stomach, and colon cancers.21 For colon cancer, National Comprehensive Cancer Network guidelines suggest considering low-dose aspirin for secondary chemoprevention, based on the strong data indicating that this therapy decreases risk of recurrence and death.23
Selective COX inhibitors may exhibit similar benefits as aspirin. For example, COX-2 is overexpressed in many cancers, including colorectal adenomatous polyps, precursors to the development of colorectal cancer. An inhibitor against COX-2, celecoxib, decreased the incidence of precancerous colorectal adenomas in phase III clinical trials.24,25 Celecoxib also reduced the risk of colon adenocarcinoma in patients with familial adenomatous polyposis.26 However, COX-2 inhibitors are not recommended routinely at this time for sporadic or familial adenomatous polyp prevention, because of their increased risk of cardiovascular events and current lack of phase IV data.27
Targeting Inflammation for Improving Cytotoxic Therapy
Multiple cues present within the tumor microenvironment, including hypoxia, extracellular matrix components, and soluble factors, can shape cancer cell biology and, in turn, ignite an inflammatory response that promotes chemoresistance. For example, the stiffness of the extracellular matrix surrounding malignant cells can stimulate focal adhesion kinase (FAK) activity, which drives cancer cell release of chemoattractants that are important for the recruitment of myeloid cells.28 Inhibition of FAK signaling can decrease myeloid cell infiltration into tumors, delay tumor progression, and sensitize tumors to chemotherapy. Thus, cross-talk between malignant cells and their surrounding microenvironment is critical to defining therapeutic resistance.
Malignant cells respond to cytotoxic stress by releasing chemoattractants (eg, C-C chemokine ligand 2 [CCL2]) and damage-associated molecular patterns, or danger signals, that attract myeloid cells to the tumor microenvironment where they support malignant cell survival, suppress immunosurveillance, and promote subsequent tumor outgrowth. Tumor-infiltrating myeloid cells can mediate resistance to cytotoxic therapies, including chemotherapy and radiation.29,30 Disrupting myeloid cell recruitment by blocking chemokine/chemokine receptor interactions (eg, CCL2/C-C chemokine receptor type 2 [CCR2]) or myeloid growth factors (eg, colony-stimulating factor [CSF]-1/CSF-1R) improves the efficacy of cytotoxic therapies in mouse models of cancer. Early promising results also support the merit of inhibiting myeloid cell recruitment for enhancing therapeutic efficacy in patients. In a phase I study of patients with borderline resectable or locally advanced pancreatic adenocarcinoma, treatment with chemotherapy in combination with a CCR2 antagonist produced encouraging objective responses in 16 of 33 evaluable patients (49%), with local tumor control seen in 32 patients (97%).31
Inflammatory cells may mediate chemoresistance through multiple pathways. By releasing soluble factors (eg, interleukin [IL]-6 and soluble IL-6 receptor), inflammatory cells activate signal transducer and activator of transcription (STAT) signaling in malignant cells.32 In models of spontaneously arising pancreatic ductal adenocarcinoma, inflammation-directed STAT signaling in cancer cells is critical for tumor development.32 Moreover, pharmacologic inhibition of STAT signaling sensitizes tumors to chemotherapy.33 Tumor-associated macrophages can also release insulin-like growth factors that activate insulin-like growth factor signaling in malignant cells and, in doing so, reduce the sensitivity of cancer cells to chemotherapy.34 Moreover, macrophages can be key orchestrators of revascularization after cytotoxic therapy, which promotes tumor regrowth.35 Thus, myeloid cells recruited to the tumor microenvironment can regulate cancer cell sensitivity to cytotoxic therapies.
On the basis of strong preclinical data supporting a role for inflammation in therapeutic resistance, multiple early-phase clinical trials are underway investigating strategies designed to manipulate inflammation with the intent of conditioning tumors for enhanced responsiveness to cytotoxic therapies. Targets being explored to disrupt inflammation include chemokine receptors (eg, CCR2, C-X-C motif chemokine receptor [CXCR]1/CXCR2), cytokines (eg, IL-6, IL-6R, tumor necrosis factor-α, IL-1α), and cellular signaling pathways (eg, FAK, Janus kinase 1/2, and colony-stimulating factor 1 receptor [CSF1R]).
Targeting Inflammation to Restore T-Cell Immunosurveillance in Cancer
Although chronic inflammation predisposes to cancer and can promote tumor growth and therapeutic resistance, the immune system is inherently pliable, such that under the appropriate conditions, it can also mediate strong antitumor activity. In recent years, strategies designed to harness the therapeutic potential of the immune system have emerged as new treatment options for some patients across a wide range of malignancies. The robustness and durability of treatment responses produced with immune-directed interventions have incited a revolution in cancer therapy. However, not all patients respond to immunotherapy and, in some cases, treatment responses can be transient owing to mechanisms of adaptive immune resistance, a process in which cancer cells adapt to immune pressure.36
Antitumor immunity induced with immunotherapy is dependent on the existence of tumor-specific T cells capable of infiltrating tumors and then recognizing and eliminating malignant cells.37 This highly regulated process relies on productive and coordinated interactions between innate and adaptive immunity. Specifically, innate immune cells (eg, dendritic cells) must capture and present tumor-associated antigens to prime antigen-specific T cells and stimulate their entry into tumor tissue and subsequent effector activity. However, chronic inflammation can impair both the priming of T cells and their subsequent effector activity by limiting effective antigen presentation and suppressing T-cell activation in tumor tissues, respectively.
Chronic inflammation can suppress the development and productivity of T-cell dependent immunosurveillance in cancer through multiple mechanisms, including the release of immunosuppressive molecules (eg, transforming growth factor-β, IL-10, and nitric oxide), expression of immune checkpoint molecules (eg, PD-L1), and metabolism of amino acids (eg, arginine and tryptophan) that are important for T-cell phenotype and survival.38-40 Inflammatory cells can also regulate T-cell entry into tumors by establishing a physical barrier and upregulating immune checkpoint molecules including indoleamine 2,3 dioxygenase and PD-L1, as has been seen in colorectal cancers with microsatellite instability.41 The coexpression of T-cell immune checkpoint molecules in tumor tissues at the interface between innate and adaptive immunity has suggested redundancy in mechanisms of peripheral tolerance established within the tumor microenvironment and represents the impetus for ongoing studies combining multiple immune checkpoint inhibitors (eg, indoleamine 2,3 dioxygenase inhibition plus PD-1/PD-L1 blockade and cytotoxic T-lymphocyte-associated protein-4 plus PD-1/PD-L1 blockade).
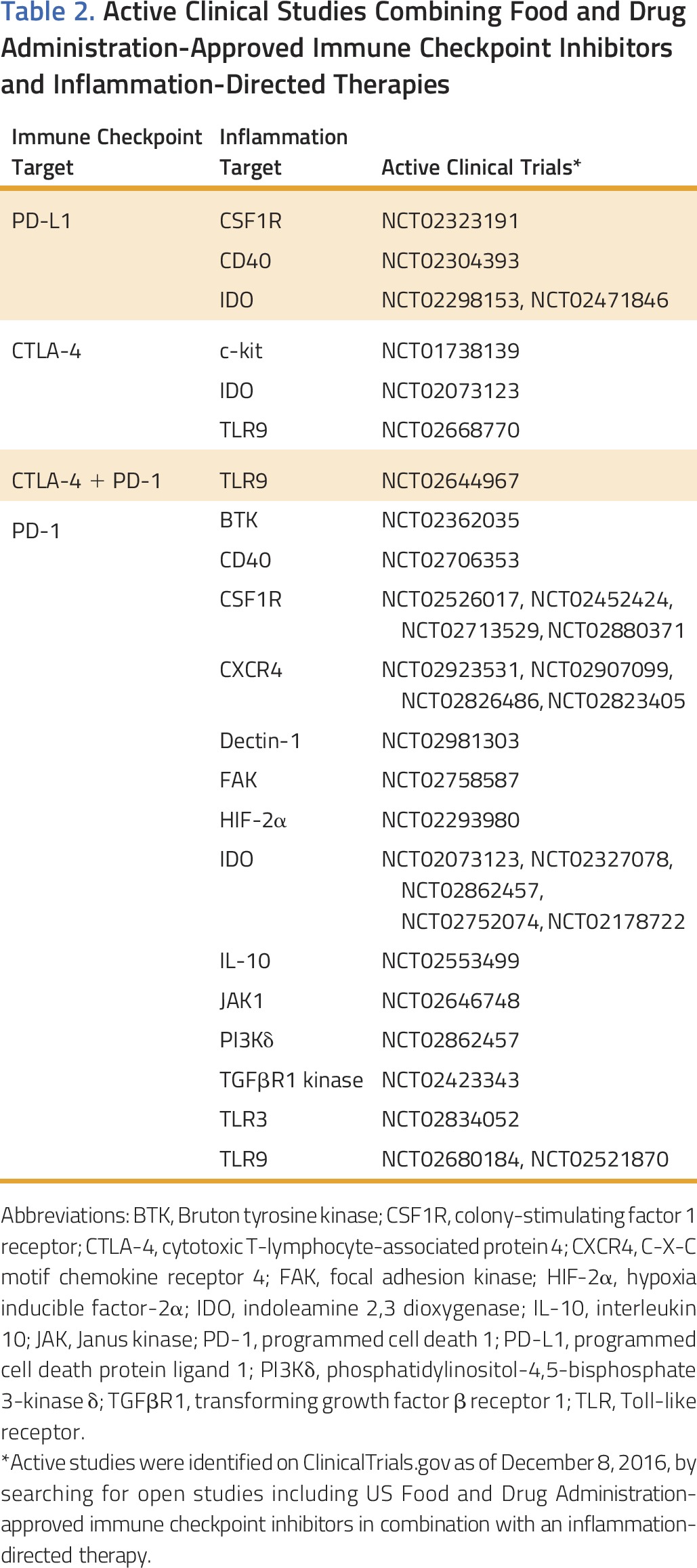
In preclinical mouse models of cancer, releasing immune suppression imposed by chronic inflammation can enhance the efficacy of T-cell directed immunotherapies, including adoptive T-cell therapy, immune checkpoint inhibitors, and vaccines.42-45 These findings form the basis for early-phase clinical trials evaluating immune checkpoint inhibitors in combination with strategies designed to inhibit the recruitment of myeloid cells (eg, inhibitors of FAK, CCR2, and CXCR2) or deplete myeloid cells (eg, CSF1R antagonists; Table 2). Combining multiple immune modulatory agents, though, poses challenges in determining the contribution of each therapeutic intervention to observed clinical responses. Determining the benefit of combination therapy over monotherapy is particularly critical in early therapeutic development and relies on effective biomarkers, identification of patients unlikely to respond to monotherapy, or both. However, biomarker development for immunotherapy has been particularly challenging in contrast to targeted therapies, which rely on the presence or absence of activating mutations in driver oncogenes. For example, immunologic biomarkers can vary with time of assessment, anatomic site, and even within a single lesion, owing to heterogeneity of the immune reaction. Overall, the dynamic nature of the immune reaction to cancer has made immunologic biomarkers a moving target; this is exemplified by the difficulties encountered with standardizing assays for PD-1/PD-L1 checkpoint therapy.46 Potential biomarkers for monitoring therapies targeting cancer inflammation include immunohistochemical and flow cytometry analyses that quantify (1) the presence and activation status of inflammatory cell subsets in tumor tissue and the peripheral blood, (2) the presence or absence of effector T cells in tumors, (3) activation of oncogenes (eg, KRAS47) and loss of tumor suppressor genes (eg, PTEN48) associated with increased inflammation, (4) signaling pathway activation in malignant and stromal cells (eg, phosphorylation of STAT349 and FAK molecules28), and (5) quantification of the matrix, including vascularity and fibrosis.50 In general, biomarker development for therapies targeting cancer inflammation will need to be informed by mechanisms of action of the pathways, molecules, or cell types targeted. However, benefit achieved with inflammation-directed therapies will ultimately rely on demonstrating improvement in standard clinical measures of response, including response rates, progression-free survival, and overall survival. Nonetheless, efficiently maximizing information acquired from tissues, peripheral blood, and clinical observations during early-phase studies will be important to the development of therapies targeting inflammation, especially given the unlikeliness that single agents targeting cancer inflammation will produce significant clinical benefit in the advanced-disease setting.
Table 2.
Active Clinical Studies Combining Food and Drug Administration-Approved Immune Checkpoint Inhibitors and Inflammation-Directed Therapies
REDIRECTING CANCER INFLAMMATION FOR CLINICAL BENEFIT
Although inflammation is most commonly considered an obstacle in cancer therapy, the ability of innate immune cells to also acquire antitumor properties has propelled the possibility that cancer inflammation may be an opportunity that can be harnessed and redirected for therapeutic benefit. Indeed, although T cells are touted as potent killers capable of seeking malignant cells with exquisite efficiency and specificity, actual tumor shrinkage requires removal of dead cells and matrix remodeling, which is a hallmark of innate immunity and, in particular, macrophages. Macrophages demonstrate remarkable plasticity, with their biology defined by signals received from their surrounding microenvironment.40 To this end, although macrophages can inhibit T-cell immunosurveillance in cancer, preclinical models have also demonstrated their importance for mediating antitumor activity directed by tumor-infiltrating effector T cells.51
Macrophages can be important effectors of cancer therapy through release of cytotoxins (eg, reactive oxygen species) and via engulfment of malignant cells.40 These mechanisms are tightly regulated by a balance of stimulatory and inhibitory signals. Similar to immune checkpoint molecules (eg, PD-L1) that are upregulated on malignant cells to evade T-cell immunosurveillance, macrophage inhibitory molecules (eg, CD47) can also be upregulated on malignant cells and thwart the potential of innate immunosurveillance in cancer.40,52 Multiple clinical-grade antagonists have been developed to disrupt one of these negative regulatory signals that involves CD47 interaction with signal regulatory protein α (SIRPα) on macrophages.53 SIRPα signaling inhibits macrophage engulfment of antibody-opsonized malignant cells; in preclinical models, disrupting CD47-SIRPα interactions enhances the therapeutic benefit of antibody-based therapies, such as anti-CD20 antibodies (ie, rituximab) used in the treatment of B-cell lymphomas.54
Macrophages can also be activated with antitumor activity using Toll-like receptor agonists and in response to systemic activation of the CD40 pathway.55,56 However, in general, strategies that merely redirect cancer inflammation have demonstrated minimal clinical benefit when translated from the laboratory to the clinic, perhaps due to a lack of immunologic memory associated with innate immunity. This does not preclude, though, the potential of these approaches, which may be particularly relevant for tumor debulking and stabilizing disease activity. For example, redirecting myeloid cells in cancer has recently been demonstrated to condition tumors for enhanced sensitivity to chemotherapy. This finding was dependent on cytokines and chemokines (ie, interferon [IFN]-γ and CCL2) released in response to treatment with a CD40 agonist that subsequently stimulated a subset of peripheral blood monocytes to rapidly infiltrate tumors and facilitate the degradation of the collagen-based extracellular matrix that surrounds malignant cells and can impede the efficacy of chemotherapy.50 Redirecting myeloid cell biology in cancer has also been found to enhance the efficacy of PD-1/PD-L1–blocking antibodies, illustrating the potential of redirecting myeloid cells for enhancing T-cell dependent antitumor immunity.57 Thus, redirecting cancer inflammation holds promise as an approach for remodeling tumors with enhanced sensitivity to chemotherapy and stimulating productive T-cell dependent antitumor immunity.
In conclusion, from Virchow’s observations to studies connecting inflammation with cancer, clinical evidence supports both permissive and inhibitory roles for the immune system in regulating cancer development, progression, and therapeutic resistance. The inflammatory reaction to cancer, characterized using routine laboratory-based protocols, has demonstrated prognostic utility. Moreover, clinical studies investigating daily aspirin use for cancer prevention have provided a strong rationale for therapeutic strategies that intervene in cancer inflammation. To this end, preclinical models of cancer have highlighted the inherent plasticity of the immune system and identified novel targets for depleting, inhibiting, and redirecting cancer inflammation. These therapeutic maneuvers have the potential to enhance the efficacy of cytotoxic therapies and to restore an effective communication between innate and adaptive immunity that is necessary for productive cancer immunosurveillance. The clinical translation of agents directed at manipulating cancer inflammation will be benefited by biomarkers on the basis of mechanisms of action for the signaling pathways and cellular subsets targeted. However, because cancer inflammation is only one element of a complex network of signals that can regulate tumor biology, combination studies will be paramount for effectively leveraging the potential of inflammation to serve as a therapeutic target in cancer.
ACKNOWLEDGMENT
Supported by National Institutes of Health Grants No. NIH R01 CA197916 (G.L.B.) and NIH F30 CA196124-01 (M.L.), and by Grant No. 2013107 from the Doris Duke Charitable Foundation (G.L.B.).
AUTHOR CONTRIBUTIONS
Conception and design: All authors
Collection and assembly of data: All authors
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Functio Laesa: Cancer Inflammation and Therapeutic Resistance
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/journal/jop/site/misc/ifc.xhtml.
Mingen Liu
Stock or Other Ownership: Genmab, Bristol-Myers Squibb
Anusha Kalbasi
No relationship to disclose
Gregory L. Beatty
Stock or Other Ownership: Johnson & Johnson, Advaxis
Honoraria: Janssen, Incyte
Consulting or Advisory Role: Incyte, Bristol-Myers Squibb, Array BioPharma, Vaccinex, Seattle Genetics, Janssen Research & Development, Opsona Therapeutics
Research Funding: Verastem, Incyte, Biothera, Halozyme, Bristol-Myers Squibb
Patents, Royalties, Other Intellectual Property: Human mesothelin chimeric antigen receptors and uses thereof (US Patent 15/105,082); Methods for b cell preconditioning in car therapy (US Patent 2015/063498)
REFERENCES
- 1.Ferrone C, Dranoff G. Dual roles for immunity in gastrointestinal cancers. J Clin Oncol. 2010;28:4045–4051. doi: 10.1200/JCO.2010.27.9992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141:39–51. doi: 10.1016/j.cell.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seruga B, Zhang H, Bernstein LJ, et al. Cytokines and their relationship to the symptoms and outcome of cancer. Nat Rev Cancer. 2008;8:887–899. doi: 10.1038/nrc2507. [DOI] [PubMed] [Google Scholar]
- 4.McMillan DC. The systemic inflammation-based Glasgow Prognostic Score: A decade of experience in patients with cancer. Cancer Treat Rev. 2013;39:534–540. doi: 10.1016/j.ctrv.2012.08.003. [DOI] [PubMed] [Google Scholar]
- 5.Dreyer SB, Powell AG, McSorley ST, et al. The pretreatment systemic inflammatory response is an important determinant of poor pathologic response for patients undergoing neoadjuvant therapy for rectal cancer. Ann Surg Oncol. doi: 10.1245/s10434-016-5684-3. 10.1245/s10434-016-5684-3 [epub ahead of print on November 21, 2016] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hurwitz HI, Uppal N, Wagner SA, et al. Randomized, double-blind, phase II study of ruxolitinib or placebo in combination with capecitabine in patients with metastatic pancreatic cancer for whom therapy with gemcitabine has failed. J Clin Oncol. 2015;33:4039–4047. doi: 10.1200/JCO.2015.61.4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hurwitz H, Garrett WM, Clark J, et al. JANUS 1: A phase 3, placebo-controlled study of ruxolitinib plus capecitabine in patients with advanced or metastatic pancreatic cancer (mPC) after failure or intolerance of first-line chemotherapy. J Clin Oncol. 2015;33 (suppl; abstr TPS4147) [Google Scholar]
- 8.Templeton AJ, McNamara MG, Šeruga B, et al. Prognostic role of neutrophil-to-lymphocyte ratio in solid tumors: A systematic review and meta-analysis. J Natl Cancer Inst. 2014;106:dju124. doi: 10.1093/jnci/dju124. [DOI] [PubMed] [Google Scholar]
- 9.Chua W, Charles KA, Baracos VE, et al. Neutrophil/lymphocyte ratio predicts chemotherapy outcomes in patients with advanced colorectal cancer. Br J Cancer. 2011;104:1288–1295. doi: 10.1038/bjc.2011.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaneko M, Nozawa H, Sasaki K, et al. Elevated neutrophil to lymphocyte ratio predicts poor prognosis in advanced colorectal cancer patients receiving oxaliplatin-based chemotherapy. Oncology. 2012;82:261–268. doi: 10.1159/000337228. [DOI] [PubMed] [Google Scholar]
- 11.Ojerholm E, Smith A, Hwang WT, et al. Neutrophil-to-lymphocyte ratio as a bladder cancer biomarker: Assessing prognostic and predictive value in SWOG 8710. Cancer. 2017;123:794–801. doi: 10.1002/cncr.30422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fridman WH, Pagès F, Sautès-Fridman C, et al. The immune contexture in human tumours: Impact on clinical outcome. Nat Rev Cancer. 2012;12:298–306. doi: 10.1038/nrc3245. [DOI] [PubMed] [Google Scholar]
- 13.Galon J, Costes A, Sanchez-Cabo F, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313:1960–1964. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- 14.Galon J, Pagès F, Marincola FM, et al. Cancer classification using the Immunoscore: A worldwide task force. J Transl Med. 2012;10:205. doi: 10.1186/1479-5876-10-205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mlecnik B, Bindea G, Angell HK, et al. Integrative analyses of colorectal cancer show Immunoscore is a stronger predictor of patient survival than microsatellite instability. Immunity. 2016;44:698–711. doi: 10.1016/j.immuni.2016.02.025. [DOI] [PubMed] [Google Scholar]
- 16.Tumeh PC, Harview CL, Yearley JH, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–571. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beatty GL. Overcoming therapeutic resistance by targeting cancer inflammation. Am Soc Clin Oncol Educ Book. 2016;35:e168–e173. doi: 10.1200/EDBK_158948. [DOI] [PubMed] [Google Scholar]
- 18.Thun MJ, Namboodiri MM, Heath CW., Jr Aspirin use and reduced risk of fatal colon cancer. N Engl J Med. 1991;325:1593–1596. doi: 10.1056/NEJM199112053252301. [DOI] [PubMed] [Google Scholar]
- 19.Burn J, Gerdes AM, Macrae F, et al. Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: An analysis from the CAPP2 randomised controlled trial. Lancet. 2011;378:2081–2087. doi: 10.1016/S0140-6736(11)61049-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ait Ouakrim D, Dashti SG, Chau R, et al. Aspirin, ibuprofen, and the risk of colorectal cancer in Lynch syndrome. J Natl Cancer Inst. 2015;107:dvj107. doi: 10.1093/jnci/djv170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rothwell PM, Fowkes FG, Belch JF, et al. Effect of daily aspirin on long-term risk of death due to cancer: Analysis of individual patient data from randomised trials. Lancet. 2011;377:31–41. doi: 10.1016/S0140-6736(10)62110-1. [DOI] [PubMed] [Google Scholar]
- 22.Terry MB, Gammon MD, Zhang FF, et al. Association of frequency and duration of aspirin use and hormone receptor status with breast cancer risk. JAMA. 2004;291:2433–2440. doi: 10.1001/jama.291.20.2433. [DOI] [PubMed] [Google Scholar]
- 23.Bains SJ, Mahic M, Myklebust TA, et al. Aspirin as secondary prevention in patients with colorectal cancer: An unselected population-based study. J Clin Oncol. 2016;34:2501–2508. doi: 10.1200/JCO.2015.65.3519. [DOI] [PubMed] [Google Scholar]
- 24.Arber N, Eagle CJ, Spicak J, et al. Celecoxib for the prevention of colorectal adenomatous polyps. N Engl J Med. 2006;355:885–895. doi: 10.1056/NEJMoa061652. [DOI] [PubMed] [Google Scholar]
- 25.Bertagnolli MM, Eagle CJ, Zauber AG, et al. Five-year efficacy and safety analysis of the Adenoma Prevention with Celecoxib Trial. Cancer Prev Res (Phila) 2009;2:310–321. doi: 10.1158/1940-6207.CAPR-08-0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Steinbach G, Lynch PM, Phillips RK, et al. The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N Engl J Med. 2000;342:1946–1952. doi: 10.1056/NEJM200006293422603. [DOI] [PubMed] [Google Scholar]
- 27. National Comprehensive Cancer Network: Genetic/familial high-risk assessment: Colorectal (version 2.2016). https://www.nccn.org/professionals/physician_gls/pdf/genetics_colon.pdf. [Google Scholar]
- 28.Jiang H, Hegde S, Knolhoff BL, et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med. 2016;22:851–860. doi: 10.1038/nm.4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kalbasi A, Komar CA, Tooker GM, et al. Tumor-derived CCL2 mediates resistance to radiotherapy in pancreatic ductal adenocarcinoma. Clin Cancer Res. 2016;23:137–148. doi: 10.1158/1078-0432.CCR-16-0870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DeNardo DG, Brennan DJ, Rexhepaj E, et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011;1:54–67. doi: 10.1158/2159-8274.CD-10-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nywening TM, Wang-Gillam A, Sanford DE, et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: A single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016;17:651–662. doi: 10.1016/S1470-2045(16)00078-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lesina M, Kurkowski MU, Ludes K, et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell. 2011;19:456–469. doi: 10.1016/j.ccr.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 33.Nagathihalli NS, Castellanos JA, Shi C, et al. Signal transducer and activator of transcription 3, mediated remodeling of the tumor microenvironment results in enhanced tumor drug delivery in a mouse model of pancreatic cancer. Gastroenterology. 2015;149:1932–1943.e9. doi: 10.1053/j.gastro.2015.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ireland L, Santos A, Ahmed MS, et al. Chemoresistance in pancreatic cancer is driven by stroma-derived insulin-like growth factors. Cancer Res. 2016;76:6851–6863. doi: 10.1158/0008-5472.CAN-16-1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stockmann C, Doedens A, Weidemann A, et al. Deletion of vascular endothelial growth factor in myeloid cells accelerates tumorigenesis. Nature. 2008;456:814–818. doi: 10.1038/nature07445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Beatty GL, Gladney WL. Immune escape mechanisms as a guide for cancer immunotherapy. Clin Cancer Res. 2015;21:687–692. doi: 10.1158/1078-0432.CCR-14-1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen DS, Mellman I. Oncology meets immunology: The cancer-immunity cycle. Immunity. 2013;39:1–10. doi: 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 38.Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell. 2015;27:450–461. doi: 10.1016/j.ccell.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Munn DH, Mellor AL. IDO in the tumor microenvironment: Inflammation, counter-regulation, and tolerance. Trends Immunol. 2016;37:193–207. doi: 10.1016/j.it.2016.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Long KB, Beatty GL. Harnessing the antitumor potential of macrophages for cancer immunotherapy. OncoImmunology. 2013;2:e26860. doi: 10.4161/onci.26860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Llosa NJ, Cruise M, Tam A, et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015;5:43–51. doi: 10.1158/2159-8290.CD-14-0863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kusmartsev S, Cheng F, Yu B, et al. All-trans-retinoic acid eliminates immature myeloid cells from tumor-bearing mice and improves the effect of vaccination. Cancer Res. 2003;63:4441–4449. [PubMed] [Google Scholar]
- 43.Ding ZC, Lu X, Yu M, et al. Immunosuppressive myeloid cells induced by chemotherapy attenuate antitumor CD4+ T-cell responses through the PD-1-PD-L1 axis. Cancer Res. 2014;74:3441–3453. doi: 10.1158/0008-5472.CAN-13-3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhu Y, Knolhoff BL, Meyer MA, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014;74:5057–5069. doi: 10.1158/0008-5472.CAN-13-3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kimura T, McKolanis JR, Dzubinski LA, et al. MUC1 vaccine for individuals with advanced adenoma of the colon: A cancer immunoprevention feasibility study. Cancer Prev Res (Phila) 2013;6:18–26. doi: 10.1158/1940-6207.CAPR-12-0275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Topalian SL, Taube JM, Anders RA, et al. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer. 2016;16:275–287. doi: 10.1038/nrc.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pylayeva-Gupta Y, Lee KE, Hajdu CH, et al. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell. 2012;21:836–847. doi: 10.1016/j.ccr.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ying H, Elpek KG, Vinjamoori A, et al. PTEN is a major tumor suppressor in pancreatic ductal adenocarcinoma and regulates an NF-κB-cytokine network. Cancer Discov. 2011;1:158–169. doi: 10.1158/2159-8290.CD-11-0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat Rev Cancer. 2009;9:798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Long KB, Gladney WL, Tooker GM, et al. IFNγ and CCL2 cooperate to redirect tumor-infiltrating monocytes to degrade fibrosis and enhance chemotherapy efficacy in pancreatic carcinoma. Cancer Discov. 2016;6:400–413. doi: 10.1158/2159-8290.CD-15-1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Spear P, Barber A, Rynda-Apple A, et al. Chimeric antigen receptor T cells shape myeloid cell function within the tumor microenvironment through IFN-γ and GM-CSF. J Immunol. 2012;188:6389–6398. doi: 10.4049/jimmunol.1103019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Crusz SM, Balkwill FR. Inflammation and cancer: Advances and new agents. Nat Rev Clin Oncol. 2015;12:584–596. doi: 10.1038/nrclinonc.2015.105. [DOI] [PubMed] [Google Scholar]
- 53.Chao MP, Majeti R, Weissman IL. Programmed cell removal: A new obstacle in the road to developing cancer. Nat Rev Cancer. 2011;12:58–67. doi: 10.1038/nrc3171. [DOI] [PubMed] [Google Scholar]
- 54.Weiskopf K, Ring AM, Ho CC, et al. Engineered SIRPα variants as immunotherapeutic adjuvants to anticancer antibodies. Science. 2013;341:88–91. doi: 10.1126/science.1238856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Feng M, Chen JY, Weissman-Tsukamoto R, et al. Macrophages eat cancer cells using their own calreticulin as a guide: Roles of TLR and Btk. Proc Natl Acad Sci USA. 2015;112:2145–2150. doi: 10.1073/pnas.1424907112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Beatty GL, Chiorean EG, Fishman MP, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331:1612–1616. doi: 10.1126/science.1198443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sockolosky JT, Dougan M, Ingram JR, et al. Durable antitumor responses to CD47 blockade require adaptive immune stimulation. Proc Natl Acad Sci USA. 2016;113:E2646–E2654. doi: 10.1073/pnas.1604268113. [DOI] [PMC free article] [PubMed] [Google Scholar]