

Abstract
Biofilms are structured communities found associated with a wide range of surfaces. Here we report the identification of a three-component regulatory system required for biofilm maturation by Pseudomonas aeruginosa strain PA14. A transposon mutation that altered biofilm formation in a 96-well dish assay originally defined this locus, which is comprised of genes for a putative sensor histidine kinase and two response regulators and has been designated sadARS. Nonpolar mutations in any of the sadARS genes result in biofilms with an altered mature structure but do not confer defects in growth or early biofilm formation, swimming, or twitching motility. After 2 days of growth under flowing conditions, biofilms formed by the mutants are indistinguishable from those formed by the wild-type (WT) strain. However, by 5 days, mutant biofilms appear to be more homogeneous than the WT in that they fail to form large and distinct macrocolonies and show a drastic reduction in water channels. We propose that the sadARS three-component system is required for later events in biofilm formation on an abiotic surface. Semiquantitative reverse transcription-PCR analysis showed that there is no detectable change in expression of the sadARS genes when cells are grown in a planktonic culture versus a biofilm, indicating that this locus is not itself induced during or in response to biofilm formation. DNA microarray studies were used to identify downstream targets of the SadARS system. Among the genes regulated by the SadARS system are those required for type III secretion. Mutations in type III secretion genes result in strains with enhanced biofilm formation. We propose a possible mechanism for the role that the SadARS system plays in biofilm formation.
Biofilms are surface-associated microbial communities found on a wide array of biotic and abiotic substrata. Such communities are ubiquitous in natural environments but can also be found in industrial and clinical settings. Biofilms contribute to nosocomial infections when they form on a variety of implants, such as catheter lines (11). Recent evidence suggests that biofilms may also contribute to some nonimplant infections in diseases such as cystic fibrosis, otitis media, and periodontitis (13, 19, 43, 69). Therefore, a greater understanding of biofilm formation is required to develop strategies to control the formation of these microbial communities.
Much of what is currently known about the molecular genetics of biofilm formation in gram-negative organisms comes from the study of Pseudomonas aeruginosa. Microscopic examination of early biofilm formation in combination with mutational studies showed that P. aeruginosa first adheres to a substratum as a monolayer, which is followed thereafter by the formation of microcolonies (56). Microcolonies are comprised of ∼30 to 100 cells and are thought to form largely as a consequence of cell division (40, 56). Microcolony formation is followed, at least under some conditions (40), by the appearance of macrocolonies which can attain heights approaching 100 μm or greater. Macrocolonies are typically separated by fluid-filled channels and surrounded by a matrix composed of polysaccharide, DNA, and proteins (1). Recently, genes proposed to be involved in the synthesis of the polysaccharide component of the matrix have been reported (12, 22, 23, 34, 48). The resulting elaborate architecture of the mature biofilm is thought to be important for the influx of nutrients and oxygen into the biofilm and for removal of metabolic waste products (12).
The formation of biofilms has been proposed to be a developmental process wherein planktonic (free-swimming) bacteria adapt to life on a surface (15). If biofilm formation is analogous to other developmental systems, there should be regulatory pathways that control the transition between planktonic and biofilm growth, and this predicted regulatory cascade should be controlled in response to environmental signals (15). For example, recent studies have shown that the availability of both iron and oxygen can have profound impacts on biofilm formation by P. aeruginosa (5, 68, 82). Furthermore, a number of regulatory systems that influence the early stages of biofilm formation by this organism have been described. A mutation in the global virulence factor regulator gacA results in a 10-fold decrease in biofilm formation, and this mutant fails to make microcolonies (59). The GacA-regulated genes required for biofilm formation have yet to be identified. Crc, the catabolite repression control protein originally identified for its role in catabolite repression of sugars by organic acids, has also been shown to play a role in biofilm formation by P. aeruginosa. A crc mutant fails to show high-level expression of at least a subset of type IV pilus biosynthetic genes, rendering these strains deficient in twitching motility (55). Like the gacA mutants, crc mutant strains fail to make microcolonies. AlgR, a response regulator protein required for synthesis of alginate, which is thought to be the major component of the matrix of biofilms in the cystic fibrosis lung (26), also appears to function in controlling assembly and/or disassembly of type IV pili (75, 78). Mutations in algR that prevent phosphorylation of the AlgR protein lead to defects in twitching motility and a decrease in biofilm formation but have no effect on alginate production. An algR null mutant fails to twitch but also shows more severe defects in biofilm initiation, suggesting that AlgR regulates additional functions required for attachment (76). These studies underscore the importance of regulators in early biofilm formation by P. aeruginosa.
Additional work indicates that regulatory systems are important for later stages of biofilm formation as well. Studies by Davies and colleagues suggest that the formation of mature biofilm architecture from microcolonies is not a stochastic process but is regulated by quorum sensing. Under some conditions, strains lacking a functional las quorum-sensing system initiate biofilms normally but are unable to establish the typical architecture of a mature biofilm. Instead, the las mutant strain makes a biofilm with a relatively thin layer of cells (5, 16, 38). Furthermore, recent evidence indicates that the complex architecture of the mature biofilm is actively maintained (14). Davey et al. provided evidence that rhamnolipid surfactants, produced by cells within the biofilm, maintain the open spaces or channels surrounding macrocolonies by inhibiting colonization by invading planktonic cells (14).
Other regulators may affect biofilm formation at multiple stages. RpoN, an alternative sigma factor for RNA polymerase, controls expression of a diverse set of genes, including those for flagella and pili (36, 67, 73), which affect early stages of biofilm formation. RpoN also positively regulates expression of rhlI (27), which is in turn required for rhamnolipid synthesis; these surfactants are important during later stages of biofilm formation, as described above.
Here we report the identification of a so-called three-component regulatory system involved in biofilm formation by P. aeruginosa strain PA14. This three-component regulatory system consists of a sensor histidine kinase and two response regulators. In flowing systems, strains carrying mutations in this three-component system appear to initiate biofilm formation identically to the wild-type (WT) strain but make mature biofilms with an altered structure lacking normal channels. We present DNA microarray data used to identify downstream targets of the SadARS system. Finally, a possible mechanism for the role played by this regulatory system in biofilm formation is proposed.
MATERIALS AND METHODS
Bacterial strains, plasmids, and culture conditions.
The parental strain for all studies is P. aeruginosa PA14 (63), unless otherwise stated. Strains were grown in lysogeny broth (LB) or minimal M63 medium (58) as indicated. For P. aeruginosa, cultures were supplemented with antibiotics as follows: ampicillin, 1,500 μg/ml; tetracycline, 150 μg/ml; or gentamicin, 100 μg/ml. For E. coli, cultures were supplemented with ampicillin at 150 μg/ml. Plasmid pSMC21 (green fluorescent protein positive; Apr Knr) constitutively expresses the gene for the green fluorescent protein and was used to label the strains for microscopy studies (4). The original sad-160::Tn5 strain was reconstructed by phage-mediated transduction as described previously (6) to produce strain SMC693. The strains and plasmids used in this study are described in Table 1.
TABLE 1.
Strains and plasmids
| Strain or plasmid | Relevant characteristics | Source or reference |
|---|---|---|
| P. aeruginosa strains | ||
| PA14 | Wild type | 63 |
| SMC241 | Wild type/pSMC21 | This study |
| SMC416 | sad-110 (pilA)::Tn5B21(Tcr, ′lacZ) | This study |
| SMC257 | sad-36 (flgK)::Tn5B30(Tc) | 56 |
| SMC693 | sad-160::Tn5B21(Tcr, ′lacZ) | This study |
| SMC707 | sad-160::Tn5B21(Tcr, ′lacZ)/pSMC21 | This study |
| SMC1022 | ΔsadRS::Gmr (ΔPA3946-PA3947) | This study |
| SMC1023 | ΔsadRS::Gmr/pSMC21 | This study |
| SMC1368 | ΔsadS (ΔPA3946) | This study |
| SMC1437 | ΔsadS/pSMC21 | This study |
| SMC1061 | ΔsadR (ΔPA3947) | This study |
| SMC1101 | ΔsadR/pSMC21 | This study |
| SMC1259 | ΔsadA (ΔPA3948) | This study |
| SMC1263 | ΔsadA/pSMC21 | This study |
| MID383 | pscI::TnphoA | F. Ausubela |
| MID241 | pcrD::TnphoA | F. Ausubela |
| PAO1 | Wild type | 35 |
| MID33913 | pcrV::ISphoA/hah | 35 |
| MID21792 | pcrH::ISphoA/hah | 35 |
| MID20540 | popB::ISphoA/hah | 35 |
| Plasmids | ||
| pACΩGM | Gmr | 65 |
| pCVD442 | Apr, sacB, suicide vector for generating chromosomal knockout mutations | 18 |
| pEXGW | Gateway modified pEX18Gm | Matt Wolfgang |
| pEX18Gm | Gmr, sacB, suicide vector for generating chromosomal knockout mutations | 30 |
| pSU21 | Cmr, cloning vector | 47 |
| pSMC21 | Apr, Kmr, GFP expression vector | This study |
| pSMC32 | Cloning vector, Kmr | This study |
| pSMC42 | KO plasmid for ΔsadRS::Gmr, in pCVD442 | This study |
| pSMC44 | KO plasmid for ΔsadR, in pEXGW | This study |
| pSMC57 | KO construct for ΔsadA in pGEM-T | This study |
| pSMC58 | KO construct for ΔsadS in pSU21 | This study |
| pSMC59 | KO plasmid for ΔsadA, in pEX18Gm | This study |
| pSMC65 | KO plasmid for ΔsadS, in pEX18Gm | This study |
| pGEM-T easy vector | Apr, cloning vector | Promega, Inc. |
P. aeruginosa PA14 strains were obtained from MGH-Parabiosys:NHLBI Program for Genomic applications, Massachusetts General Hospital and Harvard Medical School, Boston, Mass. (http://pga.mgh.harvard.edu/cgi-bin/pa14/mutants/retrieve.cgi) (February 2003).
Microscopy and image acquisition.
A DM IRB inverted microscope (Leica Microsystems, Wetzlar, Germany) equipped with a cooled charge-coupled-device digital camera and a 10×, 40×, or 63× PL FLOTAR objective lens was used for epifluorescence and phase-contrast microscopy. Digital images were captured with a G4 Macintosh computer with the OpenLab software package (Improvision, Coventry, England). The images were processed for publication with Photoshop software (Adobe, Mountain View, Calif.). Confocal scanning laser microscopy (CSLM) was performed with a Fluoview 300 confocal microscope in the IX70 inverted microscope platform (Olympus, Melville, N.Y.). Images were obtained with a 60× objective and captured on an IBM PC with the Fluoview software (Olympus). Image deconvolution and three-dimensional rendering were performed with the Volocity software package (Improvision).
Biofilm formation assays. (i) Ninety-six-well microtiter dish assay.
Initial biofilm formation was measured by using the microtiter dish assay system, as described previously (56, 57). Microtiter wells were inoculated from overnight LB-grown cultures diluted 1:50 in minimal M63 medium supplemented with glucose (0.2%), magnesium sulfate (1 mM), and Casamino Acids (CAA) (0.5%). Unless otherwise stated, cells were grown for 8 h before they were stained with crystal violet (CV) and quantified (57). Crystal violet-stained wells were digitally imaged with a Nikon (Melville, N.Y.) 990 digital camera. Biofilm formation was quantified by solubilization of the CV staining in 30% glacial acetic acid. The absorbance of the resulting solution was measured at 550 nm.
(ii) Microscopy of the air-liquid interface (ALI) of static biofilms.
ALI assays were performed as previously described, with modifications (7). Briefly, cultures were grown overnight in LB and diluted 1:50 into the minimal M63 glucose medium described above, and 350-μl aliquots were inoculated into 24-well, polystyrene, flat-bottomed plates (Corning, Inc., Corning, N.Y.). The plates were incubated at a 45° angle such that the air-liquid interface was located at the center of the well bottom for optimal microscopic visualization. After 6 h, wells were aspirated and 300 μl of M63 was added to each well. Bacteria attached to the well were then visualized by phase-contrast microscopy as described above, using the 40× objective.
(iii) Flow cells.
Biofilms were cultivated in flow chambers with channel dimensions of 5 by 1 by 30 mm. The flow system was assembled as described previously (9), using a modified EPRI medium lacking lactate, succinate, and trace metals. Modified EPRI medium is a phosphate-buffered minimal salts medium similar to M63 but containing nitrate as an alternative electron acceptor to promote growth of P. aeruginosa under oxygen-limiting conditions. The flow cell was inoculated from overnight LB-grown cultures diluted 10-fold in EPRI medium. The medium flow was turned off prior to inoculation and for 1 h after inoculation. Thereafter, medium was pumped through the flow cell at a constant rate (1.8 ml/h) for the duration of the experiment. The flow was controlled with a PumpPro MPL (Watson-Marlow, Cornwall, England). Quantitative analysis of epifluorescence microscopic images obtained from flow cell-grown biofilms was performed with COMSTAT image analysis software (28).
Construction of mutant strains. (i) sadS (PA3946).
A knockout mutation of sadS was constructed by amplifying 5′ flanking (primers S5Sac and S5SpX) and 3′ flanking (primers S3Spe and S3DnH) DNA fragments (primers are listed in Table 2). Genomic DNA used as the PCR template was isolated by the guanidium thiocyanate method (61). The 5′ and 3′ fragments were ligated (by using the SpeI sites engineered in primers S5SpX and S3Spe) and amplified with primers S3DnH and S5Sac. The resulting PCR product was cloned into the pGEM cloning vector (Promega, Inc., Madison, Wis.), which was then digested with HindIII and SstI. The insert fragment was cloned into plasmid pEX18Gm cut with the same enzymes to generate the knockout plasmid pSMC65. The plasmid was electroporated into Escherichia coli SM10-λpir. Conjugations were performed (see below), and integrants were confirmed by PCR from chromosomal DNA, using primers SHK-7 and RRI-10. The resulting ΔsadS strain was named SMC1368.
TABLE 2.
Primers used in these studies
| Primer | Sequence (5′ to 3′) |
|---|---|
| attB1 | GGGGACAAGTTTGTACAAAAAAGCAGGCT |
| attB2 | GGGGACCACTTTGTACAAGAAAGCTGGGT |
| omlAF2 | CATCCAGCAAGGCAACGTCGTAACGCAG |
| omlAR | TCATTGCGGGCTGGGGTCCAGAGGTTCG |
| pcrVF | AGTCAGAAACCTTAATGCCGCTCG |
| pcrVR | TGAAATAGGCCGACACCAGGAACT |
| pcrHF | TATGAACAGGCCCTGCAGAGCTAC |
| pcrHR | TGCTCGATCCTTTCTCGCGGTTAC |
| popBF | ATCATCGTCGGCGCAATCATGGTG |
| popBR | TTTCAGCGAGTGCGACAGCGATTG |
| pACR1 | TCTTGCCCGCCTGATGAATG |
| pACF1 | CATATCACCAGCTCACCGTC |
| rplU1 | CGCAGTGATTGTTACCGGTG |
| rplU2 | AGGCCTGAATGCCGGTGATC |
| RI-10 | CATCAGCAGTGTCGTGGCGTTG |
| R1-11 | AGCCCTGGGCTATGGAGCAAC |
| R1-12 | TGGTCGACAAGCTGTTCTGGC |
| R1-13 | GCCTGATCGAACTCGGTTGCTC |
| RR1-1 | ACATGCATGCATGTTGTAGGTGCTGACGGTCTTGTTG |
| RR1-2 | CGGGATCCCGCCACACAGGGTTTCCAGGAAATAC |
| RRI-10 | CATCAGCAGTGTCGTGGCGTTG |
| RR2-10 | ACGGACAATGGCGTGGATGCCATC |
| RR2-11 | CGAATAGCCCATGGCGAGATGCTG |
| RRAdU | AAAAAGCAGGCTCGAGCGATCCAGCGACTCTCCTGC |
| RRAdD | AGAAAGCTGGGTATCCGCCGACGCTCGTCGCGTGGC |
| RRD3′ | ATGAATGATTTGAATGTTCTGGTGAGATCTCTCGACTATTGCTCCGGATCCTGA |
| RRD5′ | TCAGGATCCGGAGCAATAGTCGAGAGATCTCACCAGAACATTCAAATCATTCAT |
| R5Sac | GGCGGAGCTCTACCCGTTGTTCGTCGCTCT |
| R5SpX | GGCGTCTAGAACTAGTACGATCAGGACGGTATGCAT |
| R3Spe | GGCGACTAGTGCAATTCGCTGATCCACTGA |
| SHK-7 | AATGCGCCTGGACGCGATCGAC |
| SHK-8 | CGGGATCCCGTCCGCGGAGCCTGACCAAAGATCGGAATAC |
| SHK-9 | ACATGCATGCATGGAAGTGCACGTCCATGGTCACGTCGAACAG |
| SK-10 | TTGAACACCGCCAATCGATCAC |
| SK-11 | TTCCTGCTGTTCTTCTGCCTG |
| SK-13 | TGAACAGCGTCAGGGACGGATG |
| SK-14 | GCAGCAGGCAGAAGAACAGCAG |
| S3DnH | GGCGAAGCTTCATTTCGATGATCGAGTGCCT |
| S5Sac | GGCGGAGCTCCACATCTCGGCGAACGCCTC |
| S5SpX | GGCGTCTAGAACTAGTGGAAGAGTGCACGGATTGAC |
| S3Spe | GGCGACTAGTGATCGCATGAAAGGTCTGTG |
| 3DHDIII | GGCGAAGCTTATCTGCTCGCGACGCCTGTT |
(ii) sadR (PA3947).
A knockout mutation of sadR was constructed by amplifying 5′ flanking (primers RRAdU and RRD3′) and 3′ flanking (primers RRAdD with RRD5′) DNA fragments. The products were then amplified in a mixed PCR with primers bearing att sites and regions that could anneal to the 5′ and 3′ ends of the knockout construct (attB1 and attB2). The cassette was integrated into the gene replacement vector pEXGW (a gift from Matt Wolfgang) by using the Gateway in vitro cloning system (Life Technologies, Carlsbad, Calif.) to generate plasmid pSMC44, which was electroporated into E. coli SM10-λpir. Conjugations were performed, and integrants were confirmed by PCR from chromosomal DNA, using primers RRAdU and RRAdD. The resulting ΔsadR strain was named SMC1061.
(iii) sadA (PA3948).
A knockout mutation of sadA was generated by amplifying 5′ flanking (primers R5Sac and R5SpX) and 3′ flanking (primers R3Spe and 3DHDIII) DNA fragments. The fragments were ligated (using the SpeI sites engineered in primers R5SpX and R3Spe) and amplified with primers R5Sac and 3DHDIII. The resulting PCR product was digested with SstI and HindIII and cloned into pEX18Gm cut with the same enzymes to generate the knockout plasmid pSMC59. The plasmid was electroporated into E. coli SM10-λpir. Conjugations were performed, and integrants were confirmed by PCR with primers RRAdU and RRAdD. The resulting ΔsadA strain was named SMC1259.
(iv) ΔsadRS::Gmr.
A double knockout mutation of the sadR and sadS genes was generated by amplifying a sadR-flanking PCR fragment (primers RR1-1 and RR1-2) and a sadS-flanking fragment (primers SHK-8 and SHK-9). The sadS fragment was cloned into the pGEM-T Easy vector (Promega, Inc.), excised by using BamHI and SpeI, and cloned adjacent to the sadR flanking sequence in the pGEM vector cut with BamHI and SpeI. The Gmr gene was amplified from pACΩGM by using primers pACF1 and pACR1, digested with BamHI, and cloned into the BamHI site between the sadR and sadS PCR fragments. The entire insert was then excised by using SphI and cloned into the SphI site of pCVD442 to generate plasmid pSMC42. Conjugations were performed, and integrants were confirmed by Southern blotting. The resulting strain, in which the sadRS region was replaced by the ΔsadRS::Gmr allele, was named SMC1022.
(v) Conjugation and sucrose selection.
For all mutant constructions, the knockout plasmid was transformed into SM10λpir and then transferred to PA14 by conjugation for gene replacement. Exconjugants harboring a chromosomal insertion of the knockout plasmid were selected on ampicillin and gentamicin (for pCVD422) or on gentamicin alone (for pEX18Gm and pEXGW). Selection on sucrose was used as described previously (18) to isolate Sucr colonies containing the targeted gene knockout. Mutations were confirmed by PCR and/or Southern blot analysis.
Growth assays.
Strains (WT, sad-160::Tn5, ΔsadRS, ΔsadR, ΔsadS, and ΔsadA) were grown overnight in LB and diluted 1:50 into either M63 minimal medium supplemented with MgSO4 (1 mM), glucose (0.2%), and CAA (0.5%) or modified EPRI medium supplemented with MgSO4 (1 mM) and glucose (0.2%). For these experiments, 0.2 ml of the freshly inoculated medium was added to the wells of a 96-well microtiter dish (four wells per strain). The dish was incubated at 37°C for 48 h. Readings of optical density at 650 nm (OD650) were taken at 2-h intervals for the first 8 h and then at 12 and 24 h, using a Vmax kinetic microplate reader (Molecular Devices Corp., Sunnyvale, Calif.). The plates were incubated for an additional 24 h to assess stationary-phase viability. At that time, CFU per milliliter were determined for all strains.
Motility assays.
Swimming and twitching experiments were performed in triplicate, and the averages and standard deviations of the data are presented.
(i) Swimming assay.
Bacteria were assayed for swimming motility as described previously (55). Briefly, a colony of the indicated strain was inoculated into M63 agar (0.3% agar) supplemented with MgSO4, glucose, and CAA and incubated at 37°C for 24 h. The diameter of the swim zone was measured in millimeters.
(ii) Twitching assay.
Bacteria were assayed for twitching motility as reported previously (56). Briefly, a colony of the indicated strain was inoculated into M63 agar (1.5% agar) supplemented with MgSO4, glucose, and CAA and incubated at 37°C for 24 h. The diameter of the twitching zone was measured in millimeters.
Rhamnolipid assays.
Rhamnolipid assays were performed as reported previously (66).
Arbitrary PCR and DNA sequencing.
Arbitrary PCR was performed as reported previously (57).
Expression analysis of individual genes. (i) Growth of bacteria.
For analysis of gene expression from planktonic batch cultures, strains were grown overnight in LB and then diluted 1:100 in fresh minimal M63 medium supplemented with glucose (0.2%), magnesium sulfate (1 mM), and CAA (0.5%). Strains were grown to late log phase (OD600 of 0.8), and experiments were run in triplicate.
For planktonic chemostat cultures, strains were diluted 1:100 from overnight LB cultures into 60 ml of the modified EPRI medium described for use in the flow cell analysis, supplemented with MgSO4 (1 mM), CAA (0.025%), and glucose (0.02%). Modified EPRI medium containing nitrate was the medium used in all chemostat studies comparing planktonic and biofilm populations, as it facilitates the growth of biofilm populations under oxygen-limiting (nonaerated) conditions. Planktonic cultures were grown in chemostats (two vessels per strain) with aeration for 4 days at 37°C and maintained in early log phase (OD600 of 0.25). Cell samples (10 ml) were harvested every 24 h for 3 days. Cells were pelleted by centrifugation at 4°C at 2,750 × g. Cell pellets were rapidly frozen in an ethanol-dry ice bath and stored at −80°C until RNA was isolated. Three samples for each strain were analyzed.
For growth of bacterial biofilms in chemostats, the method used was similar to that previously described (77). From overnight LB cultures, bacteria were diluted 1:20 into LB and grown for 6 h. Cultures were diluted such that 107 cells were added to 50 ml of a minimal EPRI medium supplemented with MgSO4 (1 mM), glucose (0.02%), and CAA (0.025%) in chemostat vessels (two vessels per strain). Each vessel was filled with 8.5 g of plastic filter devices (a gift of Colin Steven, University of Maryland Biotechnology Institute, Center for Marine Studies), and cells were given 24 h to attach before the flow of medium was initiated, followed by a washout period of 5 h at a high flow rate (150 ml h−1) to remove unattached planktonic bacteria. Growth of the biofilm was resumed at a dilution rate of 0.2 h−1 for 5 days. To harvest biofilm bacteria, the filters were sonicated (Fisher Scientific, Pittsburgh, Pa.) in EPRI medium for 5 min. Bacterial cells were centrifuged at 2,750 × g (4°C for 10 min) and stored at −80°C prior to RNA extraction.
(ii) Isolation of total RNA.
Total RNA was isolated by using the RNeasy kit from Qiagen (Valencia, Calif.), including the on-column DNase treatment. The protocol was modified such that the lysozyme digestion was performed for 5 min with a 1-mg/ml final concentration of lysozyme. A second DNase treatment using all 50 μl of RNA eluted from the column and 50 U of DNase I (Roche, Basel, Switzerland) was performed, and samples were incubated for 1 h at room temperature. Following this treatment, RNA was purified with the Qiagen columns according to the manufacturer's instructions, and the RNA was subsequently precipitated with 2.5 volumes of 100% ethanol and 0.1 volume of 3 M sodium acetate. RNA was pelleted by centrifugation at 13,000 × g, washed with 0.5 ml of 70% ethanol, and then centrifuged at 5,000 × g for 5 min at 4°C. The pellet was air dried for 5 min, resuspended in 0.02 ml of diethyl pyrocarbonate-treated water, and heated at 65°C for 5 min. The purity and concentration of the RNA were determined by spectrophotometry (Eppendorf, Hamburg, Germany). RNA samples were determined to be free from DNA contamination by PCR with 1 μl of RNA in a reaction with primers specific to the rplU gene.
(iii) Reverse transcription-PCR (RT-PCR).
For first-strand cDNA synthesis, 5 μg of total RNA was used in the first-strand cDNA synthesis kit (Amersham Biosciences, Piscataway, N.J.) according to the manufacturer's instructions. PCR was carried out with 1 μl of first-strand cDNA in a total volume of 50 μl containing a 0.2 mM concentration of each primer, 2.5 U of Taq DNA polymerase (Qiagen), 1× buffer, and 5% dimethyl sulfoxide. To quantify the PCR product, samples were run on a 5% polyacrylamide Ready Gel (Bio-Rad, Hercules, Calif.) in 1× Tris-borate-EDTA. Gels were stained in a 1× solution of SYBR gold (Molecular Probes, Eugene, Oreg.) in Tris-EDTA (pH 8) for 15 min and then scanned by using the blue fluorescence filter on the Molecular Dynamics Storm 860 and analyzed with Image Quant software (Amersham).
The following primer sets were used to quantify expression of individual sad genes: sadA, RR2-10 and RR2-11; sadR, R1-11 and R1-12; and sadS, SK-10 and SK-11 (Table 2). Expression levels were normalized to rplU or omlA by using primer set rplU1-rplU2 or omlAF2-omlAR, respectively. The expression of omlA has been shown to be unaltered under a variety of planktonic growth conditions (53). We have shown that the expression of neither rplU nor omlA changes detectably under any of the experimental conditions that we used in these studies. Three primer sets were used to confirm that the sadRS genes are cotranscribed: R1-10 and SK-13, R1-13 and SK-13, and R1-10 and SK-14.
Microarray Analysis. (i) Growth of bacteria. (a) WT versus ΔsadRS mutant, planktonic-grown.
Planktonic bacteria were grown in chemostats (two vessels per strain) as described above and maintained in early log phase (OD600 of 0.2) for 4 days. Three samples for each strain were used in the microarray analysis.
(b) WT versus ΔsadRS mutant, biofilm-grown.
Bacterial biofilms were grown in chemostats, as described above. Two samples for each strain were used in the microarray analysis.
WT versus ΔsadS mutant versus sad-160::Tn5 mutant.
Bacterial cultures were inoculated from single colonies on LB plates into 5 ml of LB and grown in batch culture to late log or early stationary phase (OD600 of 1). Cells from each strain were then diluted in triplicate 1:50 into 1× M63 supplemented with glucose (0.2%), MgSO4 (1 mM), and CAA (0.5%) and grown to early stationary phase (OD600 of 1.1). Cells from each of the replicates were harvested in 1-ml aliquots and spun to a pellet at 37°C for 1 min at 16,000 × g. The supernatant was removed, and the cell pellets were rapidly frozen in an ethanol-dry ice bath and stored at −80°C until RNA extractions were performed. Three samples for each strain were analyzed in these microarray studies.
(ii) RNA isolation.
Total RNA was isolated by using the Qiagen RNeasy miniprep kit as described above, with one modification. For the WT versus ΔsadS mutant versus sad-160::Tn5 arrays, the RNA samples did not require ethanol precipitation prior to cDNA synthesis.
(iii) cDNA probe generation and microarray hybridization.
cDNA synthesis, labeling, array hybridization, staining, and scanning were carried out according to the Affymetrix (Santa Clara, Calif.) Genechip P. aeruginosa Genome Array Expression Analysis Protocol. Briefly, cDNA was synthesized by using random hexamers as primers (Invitrogen Life Technologies, Carlsbad, Calif.) for reverse transcription. Primers were annealed with total RNA (10 μg) and with exogenous transcripts (130 pM; kindly provided by Steve Lory) that were added to each sample to monitor transcriptional efficiency and array performance. Reverse transcription proceeded with Superscript II reverse transcriptase (Invitrogen) (25°C for 10 min, 37°C for 60 min, and 42°C for 60 min, followed by enzyme inactivation at 70°C for 10 min). Residual RNA was removed by alkaline treatment followed by acid neutralization, and cDNA was purified with a QIAquick PCR purification kit. Purified cDNA was fragmented with DNase I (Amersham) and end labeled with biotin-ddUTP by using the Enzo BioArray terminal labeling kit. Target hybridization, array staining, and scanning were performed with the Affymetrix GeneChip system at the Dartmouth Microarray Shared Resource in the Norris Cotton Cancer Center at Dartmouth Hitchcock Medical Center (including a workstation with Microarray Suite software version 5.0, a hybridization oven, a fluidics workstation, and a Hewlitt-Packard GeneArray scanner).
(iv) Microarray data analysis.
Microarray data were initially analyzed by using protocols described by Affymetrix in Microarray Suite version 5.0. Raw data (.CEL files) were then uploaded into GeneTraffic version 3.0 (Iobion, La Jolla, Calif.) and processed with GeneTraffic by using the robust multiarray average analysis prior to statistical analysis. Statistical t tests were performed with the CyberT web interface (http://www.igb.uci.edu) as described previously (31, 33), and data are presented in Tables S1 (WT versus ΔsadRS mutant, chemostat-grown planktonic analysis), S2 (WT versus ΔsadRS mutant, chemostat-grown biofilm analysis), and S3 (WT versus ΔsadS mutant versus sad-160::Tn5 mutant, batch-grown planktonic analysis) in the supplemental material. The P values shown are those associated with the t test on log-transformed control and experimental data, using the Bayesian or regularized standard deviations.
(v) Array validation.
Differential gene expression was confirmed by using quantitative real-time PCR (QRT-PCR). QRT-PCR was performed with the original RNA samples from the array experiments and, where indicated, with RNA prepared from two independent sets of cultures grown as described for the microarray studies. cDNA synthesis was performed with the Invitrogen Superscript first-strand cDNA synthesis system according to the manufacturer's instructions. Real-time PCR was performed with the SYBR Green PCR master mix (Applied Biosystems, Foster City, Calif.) in an ABI Prism 7700 sequencing detection system, according to the manufacturer's instructions. Data were collected and analyzed with the ABI Sequence Detection System software, version 1.9.1. Expression levels were quantified in picograms of input cDNA by using a standard curve method for absolute quantification, and these values were then normalized to rplU expression. The following primer sets were used to quantify expression of individual type III secretion system (TTSS) genes: pcrV, pcrVF-pcrVR; pcrH, pvrHF-pvrHR; and popB, popBF-popBR (Table 2).
RESULTS
Identification and characterization of the sad-160 mutant, a mutant defective for biofilm formation.
In a previous report, we described the isolation of P. aeruginosa mutants that are defective for biofilm formation (56). A random Tn5 transposon library was screened for mutants that failed to form a biofilm in 96-well microtiter plates. In this assay, bacteria were inoculated in a minimal salts medium (M63) supplemented with 0.2% glucose and 0.5% CAA and grown at 37°C for 8 h. After 8 h, the cells were removed, the wells were rinsed, and biofilm formation was detected by CV staining (57). From this collection of mutants, we chose the sad-160::Tn5 mutant strain (SMC693) (sad, for surface attachment defective) for further study.
Figure 1A depicts the biofilm formation phenotype of the WT strain and the sad-160::Tn5 mutant strain at 8 h. WT cells form a CV-staining ring in the microtiter dish at the air-liquid interface (ALI). In contrast, sad-160::Tn5 mutant cells exhibit an altered pattern of biofilm formation, with a decrease in attachment at the air-liquid interface and a concomitant increase in staining below the interface.
FIG. 1.

Microtiter dish phenotypes of WT strain PA14 and isogenic mutant strains. (A) Biofilm formation phenotypes of the WT and sad-160::Tn5 mutant strains at 8 h in the microtiter dish assay. Cells were grown for 8 h in minimal M63 medium supplemented with MgSO4, glucose, and CAA. The arrowhead indicates crystal violet staining of the biofilm at the ALI. The bracket indicates the increased CV staining below the ALI in the Tn5 mutant. (B) Biofilm formation by the WT, the sad-160::Tn5 transposon mutant, and the ΔsadRS::Gmr mutant strain observed at 4 and 8 h. In panels A and B, the microtiter wells are inverted. (C) Quantification of the biofilm formed by the WT and the sad-160::Tn5 and ΔsadRS::Gmr mutants in microtiter dishes. Biofilms were quantified at 4, 8, and 24 h by solubilization in 30% glacial acetic acid, and the A550 of the resulting solution was measured. See the Materials and Methods for experimental details. Error bars indicate standard deviations.
When we examined biofilm formation over time, we observed a striking difference in the pattern of biofilm formation in the sad-160::Tn5 mutant relative to the WT. As shown in Fig. 1B and C, the sad-160::Tn5 mutant forms a dark ring at the air-liquid interface after incubation for 4 h in minimal medium supplemented with glucose and CAA. This phenotype is dependent upon the presence of amino acids in the medium (data not shown). In contrast, the biofilm ring formed by WT cells under these conditions is difficult to detect at this early time point. By 8 h, the intensity of the ring formed by the mutant at the air-liquid interface decreases as staining below the interface increases (Fig. 1B). In contrast, the intensity of the ring increases at the interface for the WT. Therefore, despite this qualitative difference in biofilm formation, quantification by CV assay revealed little difference in staining of the WT versus the sad-160::Tn5 mutant at later time points (Fig. 1C). The similarity in CV staining between the WT and the sad-160::Tn5 mutant is due to the increased CV staining at the bottom of the wells for the mutant. By 24 h, both the WT and the sad-160::Tn5 mutant form an equivalent ring at the air-liquid interface (data not shown). These results suggest that in a microtiter dish, the sad-160 locus may be involved in directing biofilm formation in both a temporal and a spatial manner, but eventually this mutant strain attaches to the surface to a degree comparable to that for the WT strain as measured by CV staining (Fig. 1C, 24-h time point).
The sad-160 locus encodes members of a three-component regulatory system.
The genomic DNA flanking the sad-160::Tn5 transposon insertion was isolated by using arbitrary PCR (57), and the sequence obtained was compared to the GenBank and P. aeruginosa PAO1 genome sequence (www.Pseudomonas.com) databases. Sequence analyses indicated that the transposon is located in an intergenic region between two divergently transcribed genes. The genomic arrangement of this locus is shown in Fig. 2A. The genes flanking the transposon insertion site code for polypeptides with similarity to response regulators (RRs) involved in two-component regulatory systems. One regulator gene is adjacent to and transcribed in the same orientation as a downstream gene encoding a polypeptide with high similarity to sensor histidine kinases (SHKs). Thus, we refer to this locus as a “three-component” system.
FIG. 2.
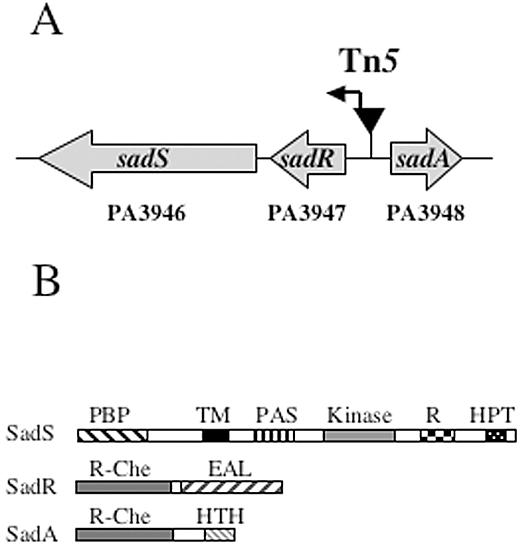
The sadARS locus. (A) The top of the panel shows the genomic organization of the sadARS region. The inverted triangle labeled Tn5 indicates the insertion site of the original sad-160::Tn5 mutant in the sadR-sadA intergenic region. The arrow within the Tn5 transposon indicates the direction of transcription of a putative promoter associated with the Tn5 insertion (see text for details). Each open reading frame is labeled with the annotation number (PA3946 to PA3948) from the P. aeruginosa genome project (www.Pseudomonas.com) and the corresponding sad gene designation. (B) Predicted structures of the SHK (SadS) and RRs (SadR and SadA). Abbreviations for the SHK are as follows: PBP, periplasmic binding protein domain; TM, transmembrane domain; PAS, PAS domain. The kinase, receiver (R), and HPT domains comprise the catalytic functions required for autophosphorylation and phosphotransfer activities. RR abbreviations: EAL, EAL family; R-Che, CheY-like phospho-receiver domain; HTH, HTH DNA binding domain.
The predicted protein structures for these genes are shown in Fig. 2B. The SHK protein (SadS, PA3946) shows a high degree of similarity to BvgS (57% amino acid identity and 40% similarity), an SHK required for virulence gene expression in Bordetella spp. (2, 45). The SHK also bears 25% amino acid identity and 42% similarity to the VieS protein of Vibrio cholerae. The VieS protein has been implicated as an in vivo regulator of virulence gene expression in the infant mouse model of cholera (44).
Of the two RR proteins identified, one protein (SadR, PA3947) contains an amino-terminal receiver domain with a high degree of similarity to CheY, the prototypical RR of the bacterial chemotaxis system (Fig. 2B). SadR also contains an effector domain that shows similarity to the EAL domain (named for the conserved amino acid residues) found in diverse bacterial signaling proteins. The VieA protein, a V. cholerae RR with an EAL domain, shares sequence similarity with SadR (29% amino acid identity and 53% similarity). Through its EAL domain, the VieA protein was shown to regulate intracellular levels of the unusual nucleotide signaling molecule cyclic di-GMP and, in turn, to control expression of the Vibrio exopolysaccharide synthesis (vps) genes, which are important for biofilm formation (72).
The second response regulator (SadA, PA3948) has a receiver domain with similarity to CheY and a predicted effector domain containing a helix-turn-helix (HTH) motif with similarity to HTH domains in the LuxR and GerE families of transcriptional regulators (Fig. 2B). SadA is highly similar over its length to BvgA (59% identity and 74% similarity), a positive transcriptional regulator of virulence gene expression in Bordetella spp. (45).
Construction of mutants.
To determine which of the genes at the sad-160 locus (hereafter referred to as the sadARS locus) are involved in biofilm formation, we generated a set of null mutations in each gene (see Materials and Methods for details). The ΔsadS, ΔsadR, and ΔsadA alleles are in-frame deletion mutations. A ΔsadRS::Gmr mutation was also generated by a drug replacement strategy and eliminates both the sadR (PA3947) and sadS (PA3946) genes and replaces them with a Gmr cassette. All knockout strains were confirmed by PCR and/or Southern blotting (data not shown). No difference in growth rate, stationary-phase viability, or rhamnolipid production was observed between the WT and any of the sadARS mutants (data not shown).
Mutations in the sadARS locus render cells defective for biofilm formation.
We tested the ability of the constructed mutants to form biofilms in the 96-well microtiter dish assay. We examined biofilm formation at 4, 8, and 24 h after inoculation into minimal M63 medium supplemented with glucose and CAA. In contrast to the sad-160::Tn5 mutant, neither the ΔsadRS::Gmr mutant (Fig. 1B) nor any of the other sadARS knockout mutants (data not shown) formed an intense ring of attached cells after 4 h. However, all of the knockout mutants showed slightly reduced biofilm formation at 8 h. Unlike the Tn5 mutant, the knockout mutants did not show an increase in CV staining below the ALI (Fig. 1B). Therefore, the quantification of CV staining in the knockout mutants more accurately reflects the amount of biofilm formation at the ALI relative to that for the WT. By 24 h, all strains attached to the surface of the microtiter wells to a degree comparable to that of the WT, as determined by visual inspection and CV quantification.
sadARS mutants exhibit normal motility.
Several studies have shown that bacterial motility is critical for early attachment events in P. aeruginosa biofilm formation (17, 42, 56, 62). We tested whether sadARS mutants were capable of swimming and twitching motility behaviors in minimal medium supplemented with glucose and CAA, as described previously (57). The results indicate that all of the mutants migrate from the point of inoculation on 0.3% motility agar and thus are capable of swimming motility. Measuring the zone of migration shows that all mutants except the sad-160::Tn5 mutant swim as well as the WT (Table 3). The sad-160::Tn5 mutant strain shows a less-than-twofold decrease in swimming motility relative to the WT on minimal medium. This deficiency is corrected on LB (data not shown). Thus, the sad-160::Tn5 mutant strain shows a small medium-dependent defect in swimming motility.
TABLE 3.
Motility assays
| Strain | Swim zone (mm)a | Twitch zone (mm)a |
|---|---|---|
| WT | 18.8 (0.2) | 10.0 (0.7) |
| sad-160::Tn5 | 11.6 (0.5) | 9.4 (0.5) |
| ΔsadRS::Gmr | 20.2 (2.9) | 10.0 (0.9) |
| ΔsadR | 19.4 (0.9) | 11.0 (0) |
| ΔsadA | 19.5 (0.3) | 7.2 (0.2) |
| pilA | NDb | 0 |
| flgK | 0 | ND |
Values are means of measurements of motility zones taken after 24 h at 37°C. The number in parentheses indicates the standard deviation for measurements of at least three plates per strain.
ND, not determined.
Twitching motility is assessed by using a similar plate assay, except that the agar is at a concentration of 1.5% instead of the 0.3% used to measure swimming motility. Twitch-positive strains of P. aeruginosa form a haze of growth surrounding the point of inoculation. Twitch-negative strains such as the pilA mutant (which cannot make type IV pili) fail to migrate from the inoculation point. In this assay, all sadARS mutants are capable of twitching motility (Table 3). Upon measurement of twitching zones (distance of migration from the inoculation point), we observed that the mutant strains twitch as well as the WT strain, with the exception of the ΔsadA strain, which shows slightly reduced twitching.
sadARS mutants exhibit defects in mature biofilm architecture.
We hypothesized that the early defect in biofilm formation displayed by the mutants in the microtiter dish would have an impact on the formation of mature biofilm architecture. To address this point, we examined biofilm maturation over the course of 5 days by using a once-through flow cell system in which a minimal medium was continuously supplied to the growing biofilm. To monitor the bacteria by epifluorescence microscopy or CSLM, the strains carried plasmid pSMC21, which constitutively expresses the green fluorescent protein (4).
We monitored biofilm formation at several time points during growth, as shown in Fig. 3. WT cells begin to attach to the glass slide within a few hours of incubation at 37°C (not shown). Under the conditions used in these studies, by day 2 (Fig. 3A), both the WT and mutant cells begin to cluster (and grow) and form microcolonies. At this time point, we quantified the number of cells attaching to the surface, using data from three independent experiments, and observed equivalent levels of attachment for the WT and the sadARS mutants (data not shown). Therefore, none of the sadARS mutants had any apparent defects in attachment or microcolony formation in the flow cell at day 2. This result is consistent with the subtle biofilm formation defect observed for the constructed sadARS mutants in the microtiter dish assay shown in Fig. 1B and C. By day 5, epifluorescence microscopy showed that the WT microcolonies had grown to form macrocolonies, and the macrocolonies appeared dispersed on the surface (Fig. 3B). These macrocolony structures have defined borders and are separated by a network of channels. In contrast, all of the sadARS mutant strains form a biofilm lacking distinct macrocolonies with defined borders. Furthermore, the network of channels seen in the WT is drastically reduced in all of the sadARS mutants. A similar striking phenotype was observed for the ΔsadRS::Gmr mutant compared to the WT when visualized by CSLM (Fig. 3C).
FIG. 3.

Biofilm formation under continuous flow. Biofilm formation at day 2 (A) and day 5 (B) is shown for modified EPRI-grown bacteria in a flow cell. (A) Top-down phase-contrast micrographs at a magnification of ×1,400 are shown for the WT and three representative mutants. (B) Top-down fluorescence images of 5-day-old biofilms at a magnification of ×630. (C) CSLM images of 5-day old biofilms, at a magnification of ×600, of the WT and the ΔsadRS::Gmr mutant, showing the xy and xz planes. Flow cell experiments were performed as described in Materials and Methods.
Quantitative analysis of biofilm structure.
Visual inspection of the biofilm formed by sadARS mutants relative to that formed by the WT indicated that the mutants are defective for mature biofilm architecture. To confirm our observations in the flow cell, we utilized the COMSTAT image analysis program to perform a quantitative analysis of biofilm architecture at the 5-day time point (28). As shown in Table 4, four variables were used to evaluate biofilm architecture. The WT and the sadARS mutants had the same maximum thickness; however, the mean thickness of the mutants was ∼2-fold greater than that of the WT. The increased mean thickness in this analysis reflects the decreased prevalence of channels (i.e., “low-height” regions) in the mutant strains. The increased total biomass and decreased roughness coefficient (a measure of biofilm heterogeneity) in the biofilms formed by the mutants is consistent with the small, poorly defined channels observed for these mutant strains (Fig. 3B and C). Taken together, these quantitative analyses support our visual observations.
TABLE 4.
COMSTAT: quantitative analysis of biofilm structurea
| Strain | Mean thickness (μm) | Maximum thickness (μm) | Total biomass (μm3/μm2) | Roughness coefficient |
|---|---|---|---|---|
| WT | 16.5 (6.4) | 76.8 (11) | 15.2 (6.3) | 1.3 (0.2) |
| sad-160::Tn5 | 34.8 (7.6) | 81.0 (8.2) | 32.0 (7.3) | 0.7 (0.1) |
| ΔsadRS::Gmr | 32.8 (4.5) | 81.3 (8.4) | 30.0 (3.8) | 0.8 (0.1) |
| ΔsadA | 25.1 (7.9) | 77.2 (6.4) | 21.3 (6.4) | 0.9 (0.2) |
Values are means of data from six z-series image stacks for each strain taken at day 5. The number in parentheses indicates the standard deviation.
Expression of the sadARS locus.
Semiquantitative RT-PCR was used to examine the relative steady-state levels of the sadARS transcripts, using primer pairs specific for each transcript (see Materials and Methods and Table 2). We found that the sad-160::Tn5 mutant strain expresses the sadR and sadS transcripts at much higher levels (∼20-fold) than the WT strain (Fig. 4A and B). Overexpression of sadR and sadS in the Tn5 mutant is likely due to an outwardly directed promoter activity associated with Tn5 (Fig. 2A). The activation of genes by insertion of Tn5-derived transposons has been documented previously (3, 37, 74). However, we cannot rule out an alternative possibility that the Tn5 insertion has disrupted a negative regulatory element in the intergenic region between the two RRs. This mutant also shows increased expression of the sadA transcript, but the increase is only twofold compared to that in the WT (Fig. 4B). We also quantified expression levels of the sadARS transcripts in each of the mutant strains relative to the WT. In the ΔsadRS::Gmr mutant, the sadR and sadS transcripts are undetectable, as expected. Similarly, in the ΔsadR mutant, sadR expression is undetectable; however, levels of sadS transcript are elevated (∼8-fold) relative to those in the WT. These data suggest that SadR may negatively regulate SadS and potentially autoregulate its own expression, since the sadRS genes appear to be in an operon (see below). In both the ΔsadRS::Gmr and ΔsadR mutant strains, the sadA mRNA is expressed at levels comparable to those in the WT. In the ΔsadA mutant, expression of sadR and sadS is comparable to that seen for the WT.
FIG. 4.

Expression of the sadARS locus. (A) Semiquantitative RT-PCR analysis of sadR expression in planktonically grown cells of WT strain PA14 and various isogenic sadARS mutants. (B) Relative expression levels of the sadR, sadA, and sadS genes in the WT and various sadARS mutants in planktonic cultures analyzed as for panel A. Expression levels are measured in fluorescence units (emitted at 537 nm), using Image Quant software and normalized to the rplU gene. (C) Semiquantitative RT-PCR analysis of sadR, sadA, and sadS gene expression in planktonic (P)- versus biofilm (B)-grown populations of the WT and the sad-160::Tn5 mutant. (D) Expression levels from panel C, measured in fluorescence units (as described above) and normalized to the rplU gene. Error bars indicate standard deviations.
The genomic arrangement of the sadR and sadS genes suggests that these genes comprise an operon. To test this prediction, we used RT-PCR to detect a product by using three different primer pairs that span the 100-bp region between the open reading frames. We were able to amplify products of the expected size from the WT strain, and these products were present at elevated levels in the sad-160::Tn5 mutant and were absent from the ΔsadRS::Gmr mutant (data not shown). Thus, the sadR and sadS genes appear to be cotranscribed.
Biofilm versus planktonic expression.
We investigated whether expression of the sadARS locus might be differentially regulated at the transcriptional level in planktonic- versus biofilm-grown populations. We used semiquantitative RT-PCR to assess the relative steady-state levels of the sadARS transcripts in planktonic- versus biofilm-grown cells in both the WT and sad-160::Tn5 mutant strains (Fig. 4C and D). In WT cells, expression of sadR and sadS is not significantly different in planktonic versus biofilm populations, but sadA gene expression is slightly decreased (∼2-fold) in a biofilm. In the sad-160::Tn5 mutant, all three genes show increased expression relative to the WT in both planktonic and biofilm samples. Therefore, this mutant exhibits a constitutive high-level expression of sadR and sadS, regardless of growth mode. The sadA transcript level shows a slight decrease (∼1.5-fold) in the sad-160::Tn5 mutant grown in a biofilm, as was also observed for the WT.
Identification of SadARS-regulated genes by using DNA microarrays.
In order to better understand the role of the SadARS regulatory system during biofilm formation, we used DNA microarray analysis to identify potential gene targets of this system. Ideally, such an analysis would be performed under conditions in which the regulatory system is known to be active. However, in the case of the SadARS system, we do not know when SadARS activity is required during the process of biofilm formation, nor do we know the specific signals required for SadARS activation. To address these potential limitations, we approached the microarray analysis in two different ways, as described below.
First, we compared gene expression in the WT strain with that in the ΔsadRS::Gmr mutant strain in both planktonic and biofilm modes of growth. Gene expression profiles of planktonic cultures of the WT and ΔsadRS::Gmr mutant were determined for cells grown in chemostats (maintained in early logarithmic phase [OD600 of 0.2]) in a minimal EPRI medium supplemented with glucose and CAA. RNA for comparison of gene expression profiles of the WT and ΔsadRS::Gmr mutant biofilms was obtained from cells grown on plastic filter devices for a period of 5 days in chemostats with the same minimal medium used for the planktonic studies. Analysis of the microarray data shows that for planktonically grown bacteria, 61 genes were differentially expressed to a statistical significance of P < 0.005 between the WT and the ΔsadRS::Gmr mutant (see Table S1 in the supplemental material), whereas for these same strains grown as biofilms, ∼200 genes showed differential expression at this same level of significance (see Table S2 in the supplemental material).
The second approach to identifying SadARS targets is based on several studies which have shown that overexpression of an RR could be used to identify valid targets of a two-component regulatory system (8, 29, 41, 52, 54). We reasoned that use of the sad-160::Tn5 mutant, in which the SadARS system is constitutively overexpressed, might allow us to identify candidate gene targets in the absence of activating signals. The RNA for gene expression profiles comparing the WT versus ΔsadS versus sad-160::Tn5 strains was obtained from planktonic, batch-grown, test-tube cultures in early stationary phase (OD600 of ∼1.1), using M63 minimal medium supplemented with glucose and CAA. Analysis of these data revealed that ∼740 genes were differentially expressed at a statistical significance of P < 0.005 in the sad-160::Tn5 mutant compared to the WT, whereas only ∼100 genes showed differential expression between the ΔsadS mutant and the WT at this same level of significance (see Table S3 in the supplemental material).
Upon closer inspection of the genes whose expression was significantly and consistently altered among these microarray studies, we noted differential expression of genes encoding the TTSS. The TTSS in P. aeruginosa is a contact-dependent protein secretion and delivery system that is responsible for the translocation of bacterial virulence effectors directly into the host cell cytoplasm. This system consists of at least 35 coordinately regulated gene products required for the secretion and translocation of effector proteins. Most of the TTSS genes are organized in a cluster of five operons on the P. aeruginosa chromosome. Genes encoding the effector proteins are located elsewhere on the chromosome (20, 21, 46, 79-81).
Expression of 35 TTSS genes was elevated (2.7-fold on average, ranging from 1.6- to 4.1-fold) in the ΔsadRS::Gmr mutant growing in a biofilm relative to a WT biofilm (Table 5). In addition, the genes exoT (PA0044) and exoY (PA2191), encoding two of the four known TTSS effector proteins, also showed elevated expression in this analysis. No change in expression was detected for these genes in the comparison of the WT and the ΔsadRS::Gmr mutant grown planktonically in chemostats (see Table S1 in the supplemental material).
TABLE 5.
TTSS genes regulated by SadARS as determined by DNA microarray analysis
| PA no. | Gene | Predicted functiona | Fold change in expressionb
|
|
|---|---|---|---|---|
| ΔsadRS::Gm/WT, biofilm | Tn5/WT | |||
| PA0044 | exoT | Toxin, ADP-ribosyltransferase | 3.4 | −3.8d |
| PA1690 | pscU | Translocation machinery | 2.0 | |
| PA1691 | pscT | Translocation machinery | 1.9 | |
| PA1692 | Translocation machinery | 3.7 | −2.0 | |
| PA1693 | pscR | Translocation machinery | 2.2 | |
| PA1694 | pscQ | Translocation machinery | 3.2 | −1.7 |
| PA1695 | pscP | Translocation machinery | 2.8 | |
| PA1696 | pscO | Translocation machinery | 4.1 | −1.3 |
| PA1697 | ATP synthase | 2.4 | ||
| PA1698 | popN | Outer membrane protein | 2.4 | |
| PA1699 | Unknown | 2.5 | ||
| PA1700 | Unknown | 2.8 | ||
| PA1701 | Unknown | 4.0 | −1.8 | |
| PA1702 | Unknown | 1.6 | ||
| PA1703 | pcrD | Unknown | 2.7 | |
| PA1704 | pcrR | Transcriptional regulator | 1.9 | |
| PA1705 | pcrG | Type III regulatory protein | 2.8 | −1.5 |
| PA1706 | pcrV | Translocation machinery | 2.9c | −3.3d |
| PA1707 | pcrH | Type III regulatory protein | 2.7c | −4.2d |
| PA1708 | popB | Pore formation | 2.9c | −4.0d |
| PA1709 | popD | Pore formation | 2.9 | −3.7 |
| PA1710 | exsC | TTSS regulator | 2.6 | −3.2 |
| PA1711 | Unknown | −1.7 | ||
| PA1712 | exsB | TTSS regulator | 1.6 | −1.6 |
| PA1713 | exsA | TTSS regulator | 2.0 | −1.3d |
| PA1714 | exsD | TTSS regulator | 2.8 | −2.0d |
| PA1715 | pscB | Translocation machinery | 3.1 | −1.5 |
| PA1716 | pscC | Outer membrane protein | 3.4 | −1.6 |
| PA1717 | pscD | Translocation machinery | 2.5 | −1.5 |
| PA1718 | pscE | Translocation machinery | 3.1 | −3.2 |
| PA1719 | pscF | Translocation machinery | 2.7c | −2.9d |
| PA1720 | pscG | Translocation machinery | 2.1 | −1.8 |
| PA1721 | pscH | Translocation machinery | 2.5 | −2.1 |
| PA1722 | pscI | Translocation machinery | 3.5c | −2.4d |
| PA1723 | pscJ | Translocation machinery | 2.6 | −1.3 |
| PA1724 | pscK | Translocation machinery | 2.4 | |
| PA1725 | pscL | Translocation machinery | 2.3 | |
| PA2191 | exoY | Toxin, adenylate cyclase | 3.2 | −2.0 |
Predicted functions based on data at www.Pseudomonas.com.
Fold changes determined using the program CyberT, at a P value of <0.005.
Confirmed by QRT-PCR on original array samples.
Confirmed by QRT-PCR on original array samples plus two independent samples.
We also found that 25 TTSS genes, including exoT and exoY, showed decreased expression (2-fold on average, ranging from 1.3- to 4.2-fold) in the planktonic batch-grown sad-160::Tn5 mutant relative to the WT. None of the TTSS genes were differentially expressed in the planktonic, batch-grown ΔsadS mutant relative to the WT (see Table S3 in the supplemental material), a result consistent with the lack of differential expression of the TTSS genes observed in the comparison of the chemostat-grown planktonic WT versus the ΔsadRS::Gmr mutant.
QRT-PCR analysis of TTSS gene expression in the WT and in sadARS mutants.
Observations from our microarray studies prompted us to further examine whether the SadARS system might regulate TTSS gene expression. To this end, we used QRT-PCR to assess the relative steady-state levels of TTSS gene expression in various sadARS mutant backgrounds. We chose eight TTSS genes whose expression was altered in the array data (PA0044, PA1706, PA1707, PA1708, PA1713, PA1714, PA1719, and PA1722). The expression patterns observed for each of these genes in the sadARS mutants, as measured by QRT-PCR, are nearly identical. The results for three representative genes are shown in Fig. 5A. As observed in the array experiments, expression of TTSS genes is reduced (∼5-fold on average) in the sad-160::Tn5 mutant relative to the WT. In the ΔsadRS::Gmr mutant (Fig. 5A) and the ΔsadS mutant (data not shown), levels are approximately WT, whereas in the ΔsadR mutant, TTSS transcript levels are less than the WT levels and are comparable to those seen for the sad-160::Tn5 mutant (3.4-fold reduced on average). In the ΔsadA mutant, expression of the TTSS genes is elevated (1.5-fold on average) relative to the WT level.
FIG. 5.

Characterization of TTSS gene expression and TTSS mutant biofilm formation. (A) QRT-PCR analysis of TTSS gene expression of WT strain PA14 and isogenic sadARS mutants. Relative expression levels of PA1706 (pcrV), PA1707 (pcrH), and PA1708 (popB) in the WT (grey bars), sad-160::Tn5 (white bars), ΔsadRS (black bars), ΔsadR (hatched bars), and ΔsadA (stippled bars) strains are shown. Expression levels were quantified as picograms of input cDNA and normalized to rplU levels. Relative expression levels are plotted, with WT levels set equal to 1. (B) Quantification of the biofilm formed by WT PAO1 and the pcrV::ISphoA/hah, pcrH::ISphoA/hah, and popB::ISphoA/hah mutants in microtiter dishes. Biofilms were quantified at 6 h after inoculation into minimal glucose medium plus MgSO4 and CAA. Error bars indicate standard deviations. (C) Biofilm formation at the ALI in 24-well flat-bottomed plates. Top-down phase-contrast micrographs at a magnification of ×400 are shown for the WT PAO1 strain and two representative TTSS mutants. Bar, 35 μm. The lower panels are centered at 140 μm below the ALI, as indicated. Strains were grown in minimal glucose medium plus MgSO4 and CAA for 6 h. ALI assays were performed as described in Materials and Methods.
Analysis of biofilm formation by TTSS mutants.
One regulatory role of the SadARS system might include modulating TTSS gene expression during biofilm formation. Hence, we speculated that mutants defective for TTSS expression and/or function might also show defects in biofilm formation. To test this hypothesis, we obtained several TTSS transposon mutants from the PAO1 and PA14 transposon mutant libraries (35; http://pga.mgh.harvard.edu/cgi-bin/pa14/mutants/retrieve.cgi) and examined these strains in the microtiter plate biofilm assay. Biofilm formation was quantified at 6 h after inoculation into minimal glucose medium plus CAA, and results for three representative PAO1 mutants are plotted in Fig. 5B. The data indicate that TTSS mutants show increased CV staining (although to slightly different degrees) and thus enhanced biofilm formation compared to the WT. We observed similar enhanced biofilms for the PA14-derived transposon mutants with mutations in the TTSS pscI and pcrD genes (data not shown).
We next examined biofilm formation using phase-contrast microscopy to visualize cell attachment after 6 h of incubation under static conditions in minimal glucose medium plus CAA (Fig. 5C). Shown in the top left panel, WT cells have attached (dark regions) at the ALI and have formed small clusters or microcolonies interspersed by the plastic surface devoid of bacteria (light gray areas). In the lower left panel (centered ∼140 μm below the ALI), such cell-free spaces are again observed, and cell clusters are relatively small. In contrast, pcrV mutant cells show almost complete coverage of the surface both at and below the ALI (middle panels). In addition, pcrV mutant cells formed a thicker layer than the WT and formed large cell clusters below the ALI (compare left and middle bottom panels). In the rightmost panels, popB mutant cells also show increased attachment, with fewer cell-free spaces than for the WT. Below the ALI, the layer of popB mutant cells appears to be thicker than the WT layer but not as dense as that of pcrV mutant cells. Thus, the microscopic data support the observations from quantification of CV staining in the microtiter plate assay; that is, TTSS mutants exhibit enhanced biofilm formation compared to the WT.
DISCUSSION
In this study we describe the identification and initial genetic characterization of a three-component regulatory system required for biofilm formation. This regulatory locus adds to a growing list of regulatory factors that have been shown to be involved in the regulation of biofilm formation (10, 16, 55, 59). SadARS, the three-component system described here, appears to play a role in biofilm maturation, in particular the formation of large macrocolonies and fluid-filled channels observed in some mature P. aeruginosa biofilms (39, 40). This flow cell phenotype is distinct from those previously described, suggesting that the sadARS system affects a step in biofilm maturation distinct from that affected by other regulatory loci.
The original sad-160::Tn5 mutant was isolated due to its reduced ability to form a biofilm in a 96-well microtiter dish relative to the WT. This static system is typically thought to be effective for monitoring early events in biofilm formation, and thus it was surprising that a mutant initially identified with a phenotype in a microtiter plate screen at 8 h displayed a late biofilm maturation defect in the flow cell. However, it appears that the particular allele isolated, a transposon insertion that up-regulates the sadS and sadR genes over 20-fold, leads to a defect in biofilm formation that is evident at as early as 4 h in the microtiter dish (Fig. 1B). The more subtle biofilm defect of the sadARS knockout mutants observed at 8 and 24 h in the microtiter dish is consistent with a later defect in biofilm formation (as observed after 5 days in the flow cell [Fig. 3B and C]) and may reflect differences in the kinetics of biofilm formation in the static microtiter plate assay versus the flow cell system.
Biofilm maturation appears to require all three sadARS genes, and all of the mutations in this locus appear to confer the same defect in biofilm maturation when observed in a flow cell system. The biofilms formed between days 0 and 2 are indistinguishable from the WT; however, strains lacking a functional sadARS system do not make a normally structured mature biofilm by day 5 (Fig. 3). Interestingly, it appears that overexpression of sadS and/or sadR also leads to a defect in biofilm maturation. Given that we observed a constitutive low-level expression of the sadARS genes in the WT in both planktonic and biofilm modes of growth, it may not be surprising that overexpression of sadARS in the sad-160::Tn5 mutant would disrupt what appears to be an otherwise carefully regulated system. Furthermore, there are several other examples in which overexpression or hyperactivity of two-component systems results in disruption of the signal transduction pathway (8, 29, 41, 52, 54).
We used DNA microarray analysis to identify candidate gene targets of the SadARS regulatory system that might be important for its role in biofilm formation. In one approach, we compared gene expression in the WT versus the ΔsadRS::Gmr mutant when grown in chemostats either planktonically or as biofilms. Statistical analysis of the data revealed that expression of 35 TTSS genes was elevated in the ΔsadRS::Gmr mutant relative to the WT, but only in the biofilm mode of growth. In a second approach to microarray analysis, we compared gene expression in planktonically grown batch cultures of the WT versus the ΔsadS mutant versus the sad-160::Tn5 mutant. The results from this analysis showed that expression of 25 TTSS genes was reduced in the sad-160::Tn5 mutant but unaffected in the ΔsadS mutant relative to the WT. The fact that expression of the TTSS genes is unaltered in batch-grown cells of the ΔsadS mutant is consistent with the results from chemostat-grown planktonic cells. However, it appears that the sad-160::Tn5 mutant, likely as a result of the overexpression of sadRS, does show altered expression of TTSS genes in planktonic, batch-grown cells.
While array studies showed that the ΔsadRS::Gmr mutant grown as a biofilm has elevated TTSS gene expression, these same genes are repressed in the planktonically grown sad-160::Tn5 mutant. SadARS regulation of TTSS gene expression was further examined in batch-grown planktonic cultures by QRT-PCR (Fig. 5A). As expected from the array studies, TTSS gene expression is repressed in the sad-160::Tn5 mutant relative to the WT under these conditions. We postulate that the overexpression of the sadRS genes in this mutant leads to aberrant activity of this system under planktonic growth conditions, making it difficult to draw any firm conclusions from these data regarding “normal” SadARS-mediated control of gene expression. Interestingly, the ΔsadR mutant also shows decreased expression of TTSS genes under these planktonic conditions. In contrast, loss of the other RR, SadA, resulted in a small derepression of the TTSS genes. Perhaps SadR and SadA act to balance activation and repression of the TTSS genes, respectively. Consistent with this idea, loss of either the sadRS genes (Fig. 5A) or sadS alone (not shown) leads to either a slight derepression or no change in expression of the TTSS genes. That is, in the absence of the appropriate input signal via SadS, and therefore loss of appropriate signal(s) to SadR and SadA, perhaps there is a return to a baseline level of expression of the TTSS genes that is similar to that in the WT.
The studies described here support recent findings from other laboratories showing a link between the regulation of pathogenesis and biofilm formation. For example, Irie and coworkers have recently shown that BvgAS, a two-component regulatory system controlling virulence gene expression in Bordetella spp., is also important in regulating biofilm formation (32). The BvgAS system regulates gene expression in at least three distinct phases: a virulent Bvg+ phase, a nonvirulent Bvg− phase, and an intermediate Bvgi phase (45, 49). In Bordetella bronchiseptica, Irie et al. reported that biofilm formation is maximal in the Bvgi phase and requires both expression of BvgAS-regulated filamentous hemagglutinin (FHA) adhesins and the concomitant repression of the toxin adenylate cyclase/hemolysin (ACY), which is also under BvgAS control.
An in-frame deletion of the cya gene (which codes for ACY) leads to increased biofilm formation in the Bvg+ phase, comparable to that observed in the Bvgi phase, suggesting that ACY expression is capable of suppressing biofilm formation in the Bvg+ phase. Irie et al. presented evidence to suggest that biofilm inhibition is mediated, at least in part, by direct interaction of ACY with FHA (32), a model consistent with data from Zaretzky et al. (83) showing that ACY and FHA interact directly in Bordetella pertussis.
The notion that the SadARS system might play a role in pathogenesis in addition to regulating biofilm formation is not surprising given the similarity between the SadS and SadA proteins and the BvgS and BvgA proteins, respectively. Evidence supporting a role for SadS in promoting pathogenesis comes from studies by Gallagher et al., using a nematode model system for P. aeruginosa virulence (25). In those studies, the authors identified SadS as a regulator of several quorum-sensing-controlled genes, including the rhlI gene and the RhlIR-regulated genes required for cyanide production (hcn) in P. aeruginosa PAO1 (25). Cyanide is required for rapid killing of the nematode Caenorhabditis elegans, and it was shown that a sadS mutant has reduced levels of cyanide production and a fivefold reduction in virulence relative to the WT in the C. elegans model system (24). We did not observe any effects of the ΔsadS mutation on rhlI or hcn gene expression in our microarray analysis; however, our studies were performed with a different strain and under different growth conditions. In addition to similarity to BvgS, the SadS protein also shows similarity to VieS, a V. cholerae sensor shown to be involved in cholera toxin expression in vitro (71) and required for full virulence in vivo (44).
We have identified gene targets of the SadARS regulatory system; however, the specific mechanism by which SadARS regulates expression of these genes is unclear. The QRT-PCR data suggest that SadS and SadA are important for controlling expression of TTSS genes. SadA contains an HTH motif and thus may regulate TTSS gene expression at the level of transcription. In Bordetella spp., BvgA also contains an HTH motif, and under activating conditions (in the Bvg+ phase), this protein binds to virulence gene promoters and activates transcription. BvgA is generally thought to be an activator of virulence gene expression; however, our data suggest that SadA may have a small negative effect on TTSS expression. In Bordetella spp. an additional class of genes is repressed under conditions where virulence factor genes are maximally expressed, and repression of these genes is also under bvg control. The bvgR gene, which is located immediately downstream of bvgAS, has been shown to be required for repression of all of the known bvg-repressed genes in B. pertussis (50, 51). In contrast to BvgR, SadR appears to be a positive regulatory factor in regard to the TTSS genes. While there may be sequence similarity and some functional analogies between the BvgARS and SadARS systems, many details of their regulatory pathways and impact on cell physiology are likely to differ.
The mechanism of BvgR-mediated repression is unknown; however, BvgR contains an EAL domain and was the founding member of the EAL protein family. Since its discovery, numerous examples of proteins containing EAL domains, including PleD (60), VieA (72), and SadR, have emerged. The EAL domain has been associated with a phospodiesterase activity that degrades the unusual nucleotide known as cyclic di-GMP, producing GMP (64, 70). Recent evidence shows that the VieA RR is involved in the regulation of biofilm formation via its ability to regulate the intracellular concentration of cyclic di-GMP (72). Whether and how the SadR EAL domain is involved in biofilm formation and/or pathogenesis remains to be determined.
The phenotypic analysis of sadARS mutants in the flow cell system along with data from our microarray and QRT-PCR studies implicate the SadARS system as a regulator of both biofilm formation and TTSS gene expression. Furthermore, the observation that TTSS mutants show enhanced biofilm formation relative to the WT (Fig. 5B and C) suggests that modulation of TTSS gene expression may be an important means of regulating biofilm formation. Taken together, these data suggest that the SadARS regulatory system may function to promote biofilm formation, possibly in part by repressing expression of the TTSS. Perhaps biofilm formation and contact-dependent secretion are incompatible. Given that the TTSS is a highly specialized virulence system utilized during host cell infection, it is also possible that the SadARS system functions at a more general level in controlling a switch between a biofilm mode and a pathogenic mode of existence. The studies presented here were conducted under conditions conducive to biofilm formation and not to TTSS expression and assembly. It is plausible that under different circumstances, the SadARS system may promote TTSS expression, favoring pathogenesis over biofilm formation. Thus, the SadARS system may function to control the switch from one developmental program, such as biofilm formation, to an alternate pathway, such as pathogenesis.
Supplementary Material
Acknowledgments
We thank S. Lory for providing the spike transcript for the array analyses.
This work was supported by grants from the Cystic Fibrosis Foundation (O'Toole01GO), the NIH (1-P20-RR01878-01 and 1 RO1 AI51360-01), and the Pew Charitable Trusts to G.A.O. G.A.O. is a Pew Scholar in the Biomedical Sciences. S.L.K. was supported by NIH training grant T32 DK07301.
Footnotes
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Allison, D. G. 2003. The biofilm matrix. Biofouling 19:139-150. [DOI] [PubMed] [Google Scholar]
- 2.Arico, B., V. Scarlato, D. M. Monack, S. Falkow, and R. Rappuoli. 1991. Structural and genetic analysis of the bvg locus in Bordetella species. Mol. Microbiol. 5:2481-2491. [DOI] [PubMed] [Google Scholar]
- 3.Berg, D. E., A. Weiss, and L. Crossland. 1980. Polarity of Tn5 insertion mutations in Escherichia coli. J. Bacteriol. 142:439-446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bloemberg, G. V., G. A. O'Toole, B. J. J. Lugtenberg, and R. Kolter. 1997. Green fluorescent protein as a marker for Pseudomonas spp. Appl. Environ. Microbiol. 63:4543-4551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bollinger, N., D. J. Hassett, B. H. Iglewski, J. W. Costerton, and T. R. McDermott. 2001. Gene expression in Pseudomonas aeruginosa: evidence of iron override effects on quorum sensing and biofilm-specific gene regulation. J. Bacteriol. 183:1990-1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Budzik, J. M., W. A. Rosche, A. Rietsch, and G. A. O'Toole. 2004. Isolation and characterization of a generalized transducing phage for Pseudomonas aeruginosa strains PAO1 and PA14. J. Bacteriol. 186:3270-3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Caiazza, N. C., and G. A. O'Toole. 2004. SadB is required for the transition from reversible to irreversible attachment during biofilm formation by P. aeruginosa PA14. J. Bacteriol. 186:4476-4485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chatterjee, A., Y. Cui, H. Yang, A. Collmer, J. R. Alfano, and A. K. Chatterjee. 2003. GacA, the response regulator of a two-component system, acts as a master regulator in Pseudomonas syringae pv. tomato DC3000 by controlling regulatory RNA, transcriptional activators, and alternate sigma factors. Mol. Plant-Microbe Interact. 16:1106-1117. [DOI] [PubMed] [Google Scholar]
- 9.Christensen, B. B., C. Sternberg, J. B. Andersen, R. J. Palmer, Jr., A. T. Nielsen, M. Givskov, and S. Molin. 1999. Molecular tools for study of biofilm physiology. Methods Enzymol. 310:20-42. [DOI] [PubMed] [Google Scholar]
- 10.Cochran, W. L., S. J. Suh, G. A. McFeters, and P. S. Stewart. 2000. Role of RpoS and AlgT in Pseudomonas aeruginosa biofilm resistance to hydrogen peroxide and monochloramine. J Appl. Microbiol. 88:546-553. [DOI] [PubMed] [Google Scholar]
- 11.Costerton, J. W., K.-J. Cheng, G. G. Geesey, T. I. Ladd, J. G. Nickel, M. Dasgupta, and T. J. Marrie. 1987. Bacterial biofilms in nature and disease. Annu. Rev. Microbiol. 41:435-464. [DOI] [PubMed] [Google Scholar]
- 12.Costerton, J. W., Z. Lewandowski, D. E. Caldwell, D. R. Korber, and H. M. Lappin-Scott. 1995. Microbial biofilms. Annu. Rev. Microbiol. 49:711-745. [DOI] [PubMed] [Google Scholar]
- 13.Costerton, J. W., P. S. Stewart, and E. P. Greenberg. 1999. Bacterial biofilms: a common cause of persistent infections. Science 284:318-322. [DOI] [PubMed] [Google Scholar]
- 14.Davey, M. E., N. C. Caiazza, and G. A. O'Toole. 2003. Rhamnolipid surfactant production affects biofilm architecture in Pseudomonas aeruginosa PAO1. J. Bacteriol. 185:1027-1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davey, M. E., and G. O. O'Toole. 2000. Microbial biofilms: from ecology to molecular genetics. Microbiol. Mol. Biol. Rev. 64:847-867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davies, D. G., M. R. Parsek, J. P. Pearson, B. H. Iglewski, J. W. Costerton, and E. P. Greenberg. 1998. The involvement of cell-to-cell signals in the development of a bacterial biofilm. Science 280:295-298. [DOI] [PubMed] [Google Scholar]
- 17.DeFlaun, M., F., B. M. Marshall, E.-P. Kulle, and S. B. Levy. 1994. Tn5 insertion mutants of Pseudomonas fluorescens defective in adhesion to soil and seeds. Appl. Environ. Microbiol. 60:2637-2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Donnenberg, M. S., and J. B. Kaper. 1991. Construction of an eae deletion mutant of enteropathogenic Escherichia coli by using a positive-selection suicide vector. Infect. Immun. 59:4310-4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ehrlich, G. D., R. Veeh, X. Wang, J. W. Costerton, J. D. Hayes, F. Z. Hu, B. J. Daigle, M. D. Ehrlich, and J. C. Post. 2002. Mucosal biofilm formation on middle-ear mucosa in the chinchilla model of otitis media. JAMA 287:1710-1715. [DOI] [PubMed] [Google Scholar]
- 20.Frank, D. W. 1997. The exoenzyme S regulon of Pseudomonas aeruginosa. Mol. Microbiol. 26:621-629. [DOI] [PubMed] [Google Scholar]
- 21.Frank, D. W., and B. H. Iglewski. 1991. Cloning and sequence analysis of a trans-regulatory locus required for exoenzyme S synthesis in Pseudomonas aeruginosa. J. Bacteriol. 173:6460-6468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Friedman, L., and R. Kolter. 2004. Genes involved in matrix formation in Pseudomonas aeruginosa PA14 biofilms. Mol. Microbiol. 51:675-690. [DOI] [PubMed] [Google Scholar]
- 23.Friedman, L., and R. Kolter. 2004. Two genetic loci produce distinct carbohydrate-rich structural components of the Pseudomonas aeruginosa biofilm matrix. J. Bacteriol. 186:4457-4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gallagher, L. A., and C. Manoil. 2001. Pseudomonas aeruginosa PAO1 kills Caenorhabditis elegans by cyanide poisoning. J. Bacteriol. 183:6207-6214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gallagher, L. A., S. L. McKnight, M. S. Kuznetsova, E. C. Pesci, and C. Manoil. 2002. Functions required for extracellular quinolone signaling by Pseudomonas aeruginosa. J. Bacteriol. 184:6472-6480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Govan, J. R. W., and V. Deretic. 1996. Microbial pathogenesis in cystic fibrosis: mucoid Pseudomonas aeruginosa and Burkholderia cepacia. Microbiol. Rev. 60:539-574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heurlier, K., V. Denervaud, G. Pessi, C. Reimmann, and D. Haas. 2003. Negative control of quorum sensing by RpoN (σ54) in Pseudomonas aeruginosa PAO1. J. Bacteriol. 185:2227-2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heydorn, A., B. K. Ersboll, M. Hentzer, M. R. Parsek, M. Givskov, and S. Molin. 2000. Experimental reproducibility in flow-chamber biofilms. Microbiology 146:2409-2415. [DOI] [PubMed] [Google Scholar]
- 29.Hirakawa, H., K. Nishino, T. Hirata, and A. Yamaguchi. 2003. Comprehensive studies of drug resistance mediated by overexpression of response regulators of two-component signal transduction systems in Escherichia coli. J. Bacteriol. 185:1851-1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hoang, T. T., R. R. Karkhoff-Schweizer, A. J. Kutchma, and H. P. Schweizer. 1998. A broad-host-range Flp-FRT recombination system for site specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212:77-86. [DOI] [PubMed] [Google Scholar]
- 31.Hung, S. P., P. Baldi, and G. W. Hatfield. 2002. Global gene expression profiling in Escherichia coli K12. The effects of leucine-responsive regulatory protein. J. Biol. Chem. 277:40309-40323. [DOI] [PubMed] [Google Scholar]
- 32.Irie, Y., S. Mattoo, and M. H. Yuk. 2004. The Bvg virulence control system regulates biofilm formation in Bordetella bronchiseptica. J. Bacteriol. 186:5692-5698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Irizarry, R. A., B. Hobbs, F. Collin, Y. D. Beazer-Barclay, K. J. Antonellis, U. Scherf, and T. P. Speed. 2003. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4:249-264. [DOI] [PubMed] [Google Scholar]
- 34.Jackson, K. D., M. Starkey, S. Kremer, M. R. Parsek, and D. J. Wozniak. 2004. Identification of psl, a locus encoding a potential exopolysaccharide that is essential for Pseudomonas aeruginosa PAO1 biofilm formation. J. Bacteriol. 186:4466-4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jacobs, M. A., A. Alwood, I. Thaipisuttikul, D. Spencer, E. Haugen, S. Ernst, O. Will, R. Kaul, C. Raymond, R. Levy, L. Chun-Rong, D. Guenthner, D. Bovee, M. V. Olson, and C. Manoil. 2003. Comprehensive transposon mutant library of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 100:14339-14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jyot, J., N. Dasgupta, and R. Ramphal. 2002. FleQ, the major flagellar gene regulator in Pseudomonas aeruginosa, binds to enhancer sites located either upstream or atypically downstream of the RpoN binding site. J. Bacteriol. 184:5251-5260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kendrick, K. E., and W. S. A. W. S. R. Reznikoff. 1988. Transposition of ISSOL activates downstream genes. J. Bacteriol. 170:1965-1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kjelleberg, S., and S. Molin. 2002. Is there a role for quorum sensing signals in bacterial biofilms? Curr. Opin. Microbiol. 5:254-258. [DOI] [PubMed] [Google Scholar]
- 39.Klausen, M., A. Aaes-Jorgensen, S. Molin, and T. Tolker-Nielsen. 2003. Involvement of bacterial migration in the development of complex multicellular structures in Pseudomonas aeruginosa biofilms. Mol. Microbiol. 50:61-68. [DOI] [PubMed] [Google Scholar]
- 40.Klausen, M., A. Heydorn, P. Ragas, L. Lambertsen, A. Aaes-Jorgensen, S. Molin, and T. Tolker-Nielsen. 2003. Biofilm formation by Pseudomonas aeruginosa wild type, flagella and type IV pili mutants. Mol. Microbiol. 48:1511-1524. [DOI] [PubMed] [Google Scholar]
- 41.Kobayashi, K., M. Ogura, H. Yamaguchi, K. Yoshida, N. Ogasawara, T. Tanaka, and Y. Fujita. 2001. Comprehensive DNA microarray analysis of Bacillus subtilis two-component regulatory systems. J. Bacteriol. 183:7365-7370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Korber, D. R., J. R. Lawrence, and D. E. Caldwell. 1994. Effect of motility on surface colonization and reproductive success of Pseudomonas fluorescens in dual-dilution continuous culture and batch culture systems. Appl. Environ. Microbiol. 60:1421-1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lamont, R. J., and H. F. Jenkinson. 1998. Life below the gum line: pathogenic mechanisms of Porphyromonas gingivalis. Microbiol. Mol. Biol. Rev. 62:1244-1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee, S. H., M. J. Angelichio, J. J. Mekalanos, and A. Camilli. 1998. Nucleotide sequence and spatiotemporal expression of the Vibrio cholerae vieSAB genes during infection. J. Bacteriol. 180:2298-2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Locht, C., R. Antoine, and F. Jacob-Dubuisson. 2001. Bordetella pertussis, molecular pathogenesis under multiple aspects. Curr. Opin. Microbiol. 4:82-89. [DOI] [PubMed] [Google Scholar]
- 46.Ma, Q., Y. Zhai, J. C. Schneider, T. M. Ramseier, and M. H. Saier, Jr. 2003. Protein secretion systems of Pseudomonas aeruginosa and P. fluorescens. Biochim. Biophys. Acta 1611:223-233. [DOI] [PubMed] [Google Scholar]
- 47.Martinez, E., B. Bartolome, and F. de la Cruz. 1988. pACYC184-derived cloning vectors containing the multiple cloning site and lacZα reporter gene of pUC8/9 and pUC18/19. Gene 68:159-162. [DOI] [PubMed] [Google Scholar]
- 48.Matsukawa, M., and E. P. Greenberg. 2004. Putative exopolysaccharide synthesis genes influence Pseudomonas aeruginosa biofilm development. J. Bacteriol. 186:4449-4456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mattoo, S., A. K. Foreman-Wykert, P. A. Cotter, and J. F. Miller. 2001. Mechanisms of Bordetella pathogenesis. Front. Biosci. 6:E168. [DOI] [PubMed] [Google Scholar]
- 50.Merkel, T. J., C. Barros, and S. Stibitz. 1998. Characterization of the bvgR locus of Bordetella pertussis. J. Bacteriol. 180:1682-1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Merkel, T. J., and S. Stibitz. 1995. Identification of a locus required for the regulation of bvg-repressed genes in Bordetella pertussis. J. Bacteriol. 177:2727-2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nishino, K., and A. Yamaguchi. 2001. Overexpression of the response regulator evgA of the two-component signal transduction system modulates multidrug resistance conferred by multidrug resistance transporters. J. Bacteriol. 183:1455-1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ochsner, U. A., A. I. Vasil, Z. Johnson, and M. L. Vasil. 1999. Pseudomonas aeruginosa fur overlaps with a gene encoding a novel outer membrane lipoprotein, OmlA. J. Bacteriol. 181:1099-1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ogura, M., H. Yamaguchi, K. Yoshida, Y. Fujita, and T. Tanaka. 2001. DNA microarray analysis of Bacillus subtilis DegU, ComA and PhoP regulons: an approach to comprehensive analysis of B. subtilis two-component regulatory systems. Nucleic Acids Res. 29:3804-3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.O'Toole, G. A., K. A. Gibbs, P. W. Hager, P. V. Phibbs, Jr., and R. Kolter. 2000. The global carbon metabolism regulator Crc is a component of a signal transduction pathway required for biofilm development by Pseudomonas aeruginosa. J. Bacteriol. 182:425-431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.O'Toole, G. A., and R. Kolter. 1998. Flagellar and twitching motility are necessary for Pseudomonas aeruginosa biofilm development. Mol. Microbiol. 30:295-304. [DOI] [PubMed] [Google Scholar]
- 57.O'Toole, G. A., L. A. Pratt, P. I. Watnick, D. K. Newman, V. B. Weaver, and R. Kolter. 1999. Genetic approaches to the study of biofilms. Methods Enzymol. 310:91-109. [DOI] [PubMed] [Google Scholar]
- 58.Pardee, A. B., F. Jacob, and J. Monod. 1959. The genetic control and cytoplasmic expression of “inducibility” in the synthesis of β-galactosidase in E. coli. J. Mol. Biol. 1:165-178. [Google Scholar]
- 59.Parkins, M. D., H. Ceri, and D. G. Storey. 2001. Pseudomonas aeruginosa GacA, a factor in multihost virulence, is also essential for biofilm formation. Mol. Microbiol. 40:1215-1226. [DOI] [PubMed] [Google Scholar]
- 60.Paul, R., S. Weiser, N. C. Amiot, C. Chan, T. Schirmer, B. Giese, and U. Jenal. 2004. Cell cycle-dependent dynamic localization of a bacterial response regulator with a novel di-guanylate cyclase output domain. Genes Dev. 18:715-727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pitcher, D. G., N. A. Saunders, and R. J. Owen. 1989. Rapid extraction of bacterial genomic DNA with guanidium thiocyanate. Lett. Appl. Microbiol. 8:151-156. [Google Scholar]
- 62.Pratt, L. A., and R. Kolter. 1998. Genetic analysis of Escherichia coli biofilm formation: roles of flagella, motility, chemotaxis and type I pili. Mol. Microbiol. 30:285-294. [DOI] [PubMed] [Google Scholar]
- 63.Rahme, L. G., E. J. Stevens, S. F. Wolfort, J. Shao, R. G. Tompkins, and F. M. Ausubel. 1995. Common virulence factors for bacterial pathogenicity in plants and animals. Science 268:1899-1902. [DOI] [PubMed] [Google Scholar]
- 64.Ross, P., R. Mayer, and M. Benziman. 1991. Cellulose biosynthesis and function in bacteria. Microbiol. Rev. 55:35-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schweizer, H. P. 1993. Small broad-host-range gentamycin resistance gene cassettes for site-specific insertion and deletion mutagenesis. BioTechniques 15:831-833. [PubMed] [Google Scholar]
- 66.Siegmund, I., and F. Wagner. 1991. New method for detecting rhamnolipids excreted by Pseudomonas aeruginosa species during growth on minimal agar. Biotechnol. Tech. 5:265-268. [Google Scholar]
- 67.Simpson, D. A., R. Ramphal, and S. Lory. 1992. Genetic analysis of Pseudomonas aeruginosa adherence: distinct genetic loci control attachment to epithelial cells and mucins. Infect. Immun. 60:3771-3779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Singh, P. K., M. R. Parsek, E. P. Greenberg, and M. J. Welsh. 2002. A component of innate immunity prevents bacterial biofilm development. Nature 417:552-555. [DOI] [PubMed] [Google Scholar]
- 69.Singh, P. K., A. L. Schaefer, M. R. Parsek, T. O. Moninger, M. J. Welsh, and E. P. Greenberg. 2000. Quorum-sensing signals indicate that cystic fibrosis lungs are infected with bacterial biofilms. Nature 407:762-764. [DOI] [PubMed] [Google Scholar]
- 70.Tal, R., H. C. Wong, R. Calhoon, D. Gelfand, A. L. Fear, G. Volman, R. Mayer, P. Ross, D. Amikam, H. Weinhouse, A. Cohen, S. Sapir, P. Ohana, and M. Benziman. 1998. Three cdg operons control cellular turnover of cyclic di-GMP in Acetobacter xylinum: genetic organization and occurrence of conserved domains in isoenzymes. J. Bacteriol. 180:4416-4425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tischler, A. D., S. H. Lee, and A. Camilli. 2002. The Vibrio cholerae vieSAB locus encodes a pathway contributing to cholera toxin production. J. Bacteriol. 184:4104-4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tischler, A. D., and A. Camilli. 2004. Cyclic diguanylate (c-di-GMP) regulates Vibrio cholerae biofilm formation. Mol. Microbiol. 53:857-869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Totten, P. A., J. C. Lara, and S. Lory. 1990. The rpoN gene product of Pseudomonas aeruginosa is required for expression of diverse genes, including the flagellin gene. J. Bacteriol. 172:389-396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang, A., and J. R. Roth. 1988. Activation of silent genes by transposons Tn5 and TnlO. Genetics 120:875-885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Whitchurch, C. B., R. A. Alm, and J. S. Mattick. 1996. The alginate regulator AlgR and an associated sensor FimS are required for twitching motility in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 93:9839-9843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Whitchurch, C. B., T. E. Erova, J. A. Emery, J. L. Sargent, J. M. Harris, A. B. Semmler, M. D. Young, J. S. Mattick, and D. J. Wozniak. 2002. Phosphorylation of the Pseudomonas aeruginosa response regulator AlgR is essential for type IV fimbria-mediated twitching motility. J. Bacteriol. 184:4544-4554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Whiteley, M., M. G. Bangera, R. E. Bumgarner, M. R. Parsek, G. M. Teitzel, S. Lory, and E. P. Greenberg. 2001. Gene expression in Pseudomonas aeruginosa biofilms. Nature 413:860-864. [DOI] [PubMed] [Google Scholar]
- 78.Wozniak, D. J., and D. E. Ohman. 1994. Transcriptional analysis of the Pseudomonas aeruginosa genes algR, algB, and algD reveals a hierarchy of alginate gene expression which is modulated by algT. J. Bacteriol. 176:6007-6014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yahr, T. L., and D. W. Frank. 1994. Transcriptional organization of the trans-regulatory locus which controls exoenzyme S synthesis in Pseudomonas aeruginosa. J. Bacteriol. 176:3832-3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yahr, T. L., J. Goranson, and D. W. Frank. 1996. Exoenzyme S of Pseudomonas aeruginosa is secreted by a type III pathway. Mol. Microbiol. 22:991-1003. [DOI] [PubMed] [Google Scholar]
- 81.Yahr, T. L., L. M. Mende-Mueller, M. B. Friese, and D. W. Frank. 1997. Identification of type III secreted proteins of the Pseudomonas exoenzyme S regulon. J. Bacteriol. 179:7165-7168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yoon, S. S., R. F. Hennigan, G. M. Hilliard, U. A. Ochsner, K. Parvatiyar, M. C. Kamani, H. L. Allen, T. R. DeKievit, P. R. Gardner, U. Schwab, J. J. Rowe, B. H. Iglewski, T. R. McDermott, R. P. Mason, D. J. Wozniak, R. E. Hancock, M. R. Parsek, T. L. Noah, R. C. Boucher, and D. J. Hassett. 2002. Pseudomonas aeruginosa anaerobic respiration in biofilms. Relationships to cystic fibrosis pathogenesis. Dev. Cell 3:593-603. [DOI] [PubMed] [Google Scholar]
- 83.Zaretzky, F. R., M. C. Gray, and E. L. Hewlett. 2002. Mechanism of association of adenylate cyclase toxin with the surface of Bordetella pertussis: a role for toxin-filamentous haemagglutinin interaction. Mol. Microbiol. 45:1589-1598. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.