

Abstract
Purpose
Adenoid cystic carcinomas (ACCs) represent a heterogeneous group of chemotherapy refractory tumors, with a subset demonstrating an aggressive phenotype. We investigated the molecular underpinnings of this phenotype and assessed the Notch1 pathway as a potential therapeutic target.
Methods
We genotyped 102 ACCs that had available pathologic and clinical data. Notch1 activation was assessed by immunohistochemistry for Notch1 intracellular domain. Luciferase reporter assays were used to confirm Notch1 target gene expression in vitro. The Notch1 inhibitor brontictuzumab was tested in patient-derived xenografts from patients with ACC and in a patient with ACC who was enrolled in a phase I study.
Results
NOTCH1 mutations occurred predominantly (14 of 15 patients) in the negative regulatory region and Pro-Glu-Ser-Thr–rich domains, the same two hotspots seen in T-cell acute lymphoblastic leukemias, and led to pathway activation in vitro. NOTCH1-mutant tumors demonstrated significantly higher levels of Notch1 pathway activation than wild-type tumors on the basis of Notch1 intracellular domain staining (P = .004). NOTCH1 mutations define a distinct aggressive ACC subgroup with a significantly higher likelihood of solid subtype (P < .001), advanced-stage disease at diagnosis (P = .02), higher rate of liver and bone metastasis (P ≤ .02), shorter relapse-free survival (median, 13 v 34 months; P = .01), and shorter overall survival (median 30 v 122 months; P = .001) when compared with NOTCH1 wild-type tumors. Significant tumor growth inhibition with brontictuzumab was observed exclusively in the ACC patient-derived xenograft model that harbored a NOTCH1 activating mutation. Furthermore, an index patient with NOTCH1-mutant ACC had a partial response to brontictuzumab.
Conclusion
NOTCH1 mutations define a distinct disease phenotype characterized by solid histology, liver and bone metastasis, poor prognosis, and potential responsiveness to Notch1 inhibitors. Clinical studies targeting Notch1 in a genotype-defined ACC subgroup are warranted.
INTRODUCTION
Adenoid cystic carcinoma (ACC) is a common malignant salivary gland tumor with a recurrence rate of 40% to 50% after curative intent treatment.1,2 Overall, ACC is chemotherapy refractory, and there is no standard of care treatment for patients with recurrent and/or metastatic disease.3
Whole exome sequencing (WES) of ACC samples has shed light on the genetic landscape of this disease and provides evidence for Notch pathway alterations in 11% to 29% of patients.4-6 The Notch pathway is involved in cancer-relevant functions, including maintenance of stem cells, cell fate specification, proliferation, and angiogenesis.7 There are four NOTCH genes that encode transmembrane receptors (NOTCH1, -2, -3, and -4) and five membrane-bound ligands: delta-like ligands (DLL1, -3, -4); and Jagged (JAG1, -2). Notch signaling is usually initiated by receptor-ligand interaction, which leads to consecutive receptor cleavages, the second cleavage by the gamma-secretase complex that frees the Notch intracellular domain (NICD) to enter the nucleus, displace corepressors such as SPEN,4 and form a transcriptional activation complex with the DNA-binding factor RBPJ and coactivators of the mastermind-like family.8 The generation and stability of NICD is regulated by the ubiquitin ligase complexes containing FBXW7.9
Deregulation of the Notch1 pathway occurs in multiple cancers, although its specific roles and potential value as a therapeutic target vary. NOTCH1 mutations are oncogenic drivers in 50% of T-cell acute lymphoblastic leukemias (T-ALLs).10 T-ALL–activating mutations concentrate in two hotspot regions: in-frame mutations in exons 25 to 28 that disrupt the negative regulatory region and lead to ligand-independent Notch1 activation and stop-codon or nonsense mutations in exon 34 that result in deletion of the C-terminal degron domain (eg, Pro-Glu-Ser-Thr–rich domain [PEST]) and NICD stabilization. Notch signaling can also be activated in T-ALL through translocations, duplication insertions in the vicinity of exon 28, or FBXW7 mutations.11,12 Notch1 can act as a tumor suppressor in other malignancies such as oral squamous cell carcinoma in which loss-of-function NOTCH1 mutations occur in the epidermal growth factor–like domain.13-15
In this article, we describe that NOTCH1 mutations in ACC occur predominately in the T-ALL hotspots, are activating, and define a subgroup of patients with solid subtype, advanced-stage disease, distinct pattern of metastasis, and worse prognosis. We also report in an index patient that the acquisition of mutations leading to further Notch1 pathway activation probably occurs as the tumor progresses. Furthermore, Notch1 inhibitor demonstrated antitumor activity in a NOTCH1-mutant ACC xenograft and in a patient with a NOTCH1 mutation, demonstrating that Notch1 is a potential therapeutic target in a subgroup of ACC.
METHODS
Patient Selection
The study population consisted of 102 patients with ACC: 70 patients with primary tumor available for WES (46 patients in addition to the 24 previously published4) and 32 patients who had their tumor genotyped by using target-sequencing platforms from January 1, 2013, to March 31, 2015, at the request of the treating oncologist. Patient samples were obtained by either an institutional review board–approved waiver of informed consent (for deceased patients) or informed consent (front-door consent). Pathologic and clinical data were retrospectively obtained from electronic medical records according to institutional review board–approved protocol PA14-0375. Data acquisition was locked on December 7, 2015. At the date of analysis, 46 patients were alive (33 with disease and 13 without disease), and 56 were deceased (44 as a result of disease, five without disease, and seven with unknown disease status).
Genomic Analysis
WES was performed by using DNA obtained from fresh-frozen samples, as previously described.4 Target exome sequencing or analysis of hotspot mutations in cancer-related genes was performed by using next-generation sequencing as described in the Data Supplement.
Immunohistochemistry
Rabbit monoclonal cleaved Notch1 antibody Val1744 (D3B8; #4147; Cell Signaling Technology, Danvers, MA) was used for NICD immunohistochemical (IHC) staining as previously described.16 Details are available in the Data Supplement.
Luciferase Reporter Assay
Luciferase reporter assay was performed by using 293T cells. NOTCH1 mutations identified in a patient were constructed by site-directed mutagenesis. Detail are provided in the Data Supplement.
Patient-Derived Xenograft Drug Screening
The antitumor activity of brontictuzumab was tested in previously established and genotyped ACC patient-derived xenografts (PDXs),17 as detailed in Data Supplement.
Statistical Analysis
Fisher’s exact test was used to determine the association between NOTCH1 mutation or NICD expression and clinicopathologic characteristics. An analysis evaluating the association between NOTCH1 mutational status and specific sites of disease recurrence was undertaken among patients with local or distant recurrence. Relapse-free survival (RFS) and overall survival (OS) were estimated by the Kaplan-Meier method. RFS was defined as the time from diagnosis to relapse or death, whichever occurred first. Observation for RFS was censored at the date of last contact for patients last known to be alive without relapse. OS was defined as the time from diagnosis to death as a result of any cause. Survivors or patients who were lost to follow-up were censored at the last contact date. Univariable and multivariable analyses that used Cox proportional hazards models were used to identify important prognostic factors for OS and RFS. All P values were two-sided. P < .05 was considered statistically significant.
RESULTS
NOTCH1 Mutations in ACC Occur in Hotspots and Are Associated With Pathway Activation
Expanding on our prior work of genetic sequencing for 24 patients with ACC,4 genomic profiling was conducted in a total of 102 tumors, WES in an additional 46 samples, and targeted sequencing for gene panel that included NOTCH1 in 32 samples. Eighteen NOTCH1 mutations were identified in 15 tumors, and two patients harbored more than one NOTCH1 mutation. Seventeen of these mutations in 14 patients (14 [13.7%] of 102; Appendix Table A1, online only) occurred in the T-ALL hotspots, suggesting that they are gain-of-function mutations (Fig 1A).
Table 1.
Baseline Patient and Tumor Characteristics
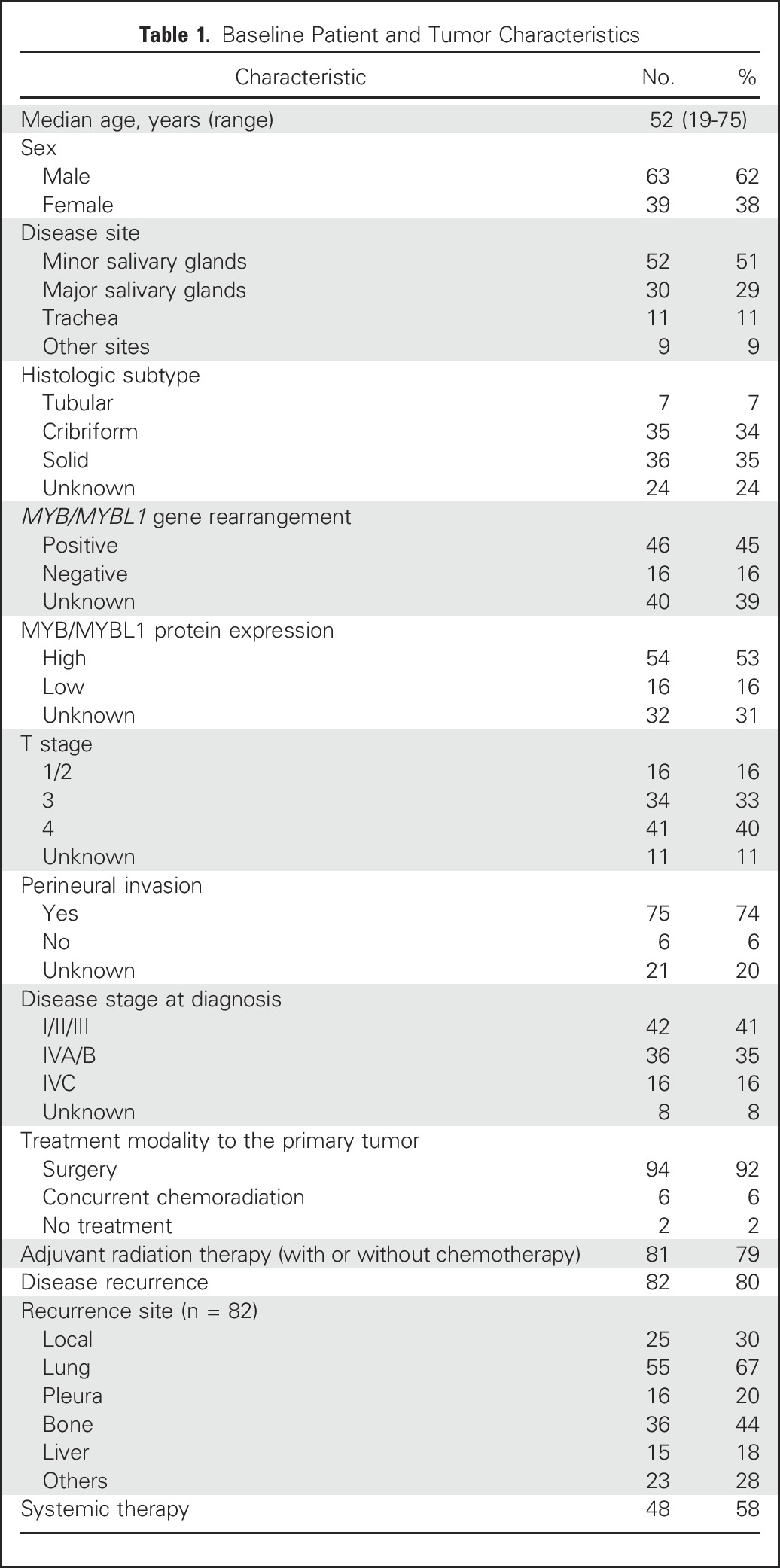
Fig 1.

(A) NOTCH1 mutations in patients with adenoid cystic carcinomas (ACCs) occurred predominantly in the same negative regulatory region and Pro-Glu-Ser-Thr–rich domain (PEST) hotspots as those observed in T-cell acute lymphoblastic leukemia and are predicted to be activating. (B) Notch1 intracellular domain (NICD) immunostaining in ACC. (Bi) Positive uniform nuclear expression of NICD in solid form of ACC. (Bii) ACC negative for NICD expression. (C) In vitro reporter assay assessing Notch1 pathway activation induced by individual mutations and the combination of both mutations observed in an index patient. 293T cells were cotransfected with NOTCH1-wild-type (WT) or NOTCH1-mutant (mut) constructs and HES1AB-responsive luciferase reporter, HES1AB-Δ luciferase mutant form, or Renilla luciferase control. Firefly/Renilla luciferase activity was measured in cell lysates after 48 hours. The NOTCH1 S2467fs* and L1600Q comutations led to a statistically significant 2.2-fold increase in reporter activity compared with wild-type NOTCH1. ANK, ankyrin repeat domain; DM, double mutation; LNR, Lin12/NOTCH repeats; M1, NOTCH1 S2467fs* mutation; M2, NOTCH1 L1600Q mutation; TM, transmembrane domain. (†) P < .001.
To evaluate whether these mutations were activating, we assessed the association between NOTCH1 mutations and NICD IHC staining (Fig 1B), an established marker for Notch1 pathway activation.16 Tumor tissues from 72 patients were available for NICD staining. There was a statistically significant association between NOTCH1 mutations and NICD positivity. All 10 tumors (100%) with NOTCH1 mutations predicted to be activating were NICD positive, whereas 30 (49%) of 61 NOTCH1 wild-type tumors stained positive (P = .004); the only tumor with a NOTCH1 mutation predicted to be inactivating (Y550fs*51) was NICD negative.
Double NOTCH1 Mutations Lead to Increased Pathway Activation
To further characterize the functional role of the NOTCH1 mutations observed in an index patient, we conducted in vitro analysis of pathway activation by using a luciferase reporter assay bearing the promoter of HES1, a Notch1 transcriptional target. 293T cells were cotransfected with an HES1-responsive luciferase reporter vector and constructs carrying the initially observed NOTCH1 mutation S2467fs* (M1), the acquired mutation L1600Q (M2), and the L1600Q/S2467fs* comutations (or double mutations). As expected, the cells cotransfected with the comutations increased luciferase activity irrespective of the presence of the ligand (Fig 1C) to a greater extent than either mutation alone or than wild-type NOTCH1, supporting the notion that the NOTCH1 mutations were transcriptionally activating. Although we detected a modest increase in pathway activation with each individual NOTCH1 mutation in the absence of ligand, these results were not statistically significant when compared with NOTCH1 wild-type. This could reflect the possibility that the Notch1 pathway activation may remain ligand-dependent for M1 or, alternatively, it may reflect a limitation of the standard transient cotransfection reporter system used.
Mutations in Other NOTCH-Related Genes
Mutations were also observed in other genes known to impact the Notch pathway. Mutations in SPEN were observed in six patients, including three concurrent with NOTCH1. Two patients had NOTCH2 mutations, one of them with a SPEN comutation. Interestingly, comutations in NOTCH1 and NOTCH4, NOTCH1 and JAG1, and NOTCH1 and FBXW7 were also identified (Data Supplement). In addition, one patient had a mutation in RBPJ, the main transcriptional effector of Notch signaling. In total, 21 patients (20.5%) had mutations in known Notch pathway-related genes.
Population Characteristics
The overall patient characteristics are provided in Table 1. The median age at diagnosis was 52 years, and the main primary tumor site was the minor salivary glands. MYB/MYBL1 rearrangement or overexpression was identified in 74% and 77% of the available samples, respectively. The majority of patients presented with stage I to III disease and were treated with surgery followed by adjuvant radiotherapy with or without concurrent cisplatin. Eighty percent of the patients relapsed, with lung being the most common site of recurrence. Metastasis to atypical sites such as brain, peritoneum, and subcutaneous tissue occurred in 23 patients. Fifty-eight percent of patients with recurrent disease received systemic therapy.
NOTCH1 Mutations Define a Distinct Biologic Phenotype
The correlation between clinicopathologic characteristics and NOTCH1 mutational status is presented in Table 2. Compared with patients who have NOTCH1 wild-type, those with mutations were more likely to have solid histology (P < .001), present with advanced-stage disease (P = .02), or both (solid subtype and stage IV v others; P = .01). In spite of lung being the most common site of metastasis among patients with recurrent ACC, patients with NOTCH1 mutations were less likely to develop lung metastasis (odds ratio [OR], 0.24; P = .02) but had a far higher likelihood of developing metastasis in the liver (OR, 8.5; P = .002), bone (OR, 5.4; P = .01), and atypical sites (OR, 3.8; P = .04; Fig 2A). Similar results were obtained when we included the 20 patients with mutations that were expected to activate the Notch pathway (Data Supplement). We also performed correlation analysis between NICD-positive (40) and NICD-negative (32) tumors. Patients with NICD-positive tumors were more likely to have solid histology (P = .02) and liver metastasis (P = .02).
Table 2.
Correlation Between Clinicopathologic Characteristics and NOTCH1 Mutational Status in Patients With and Without NOTCH1 Mutations
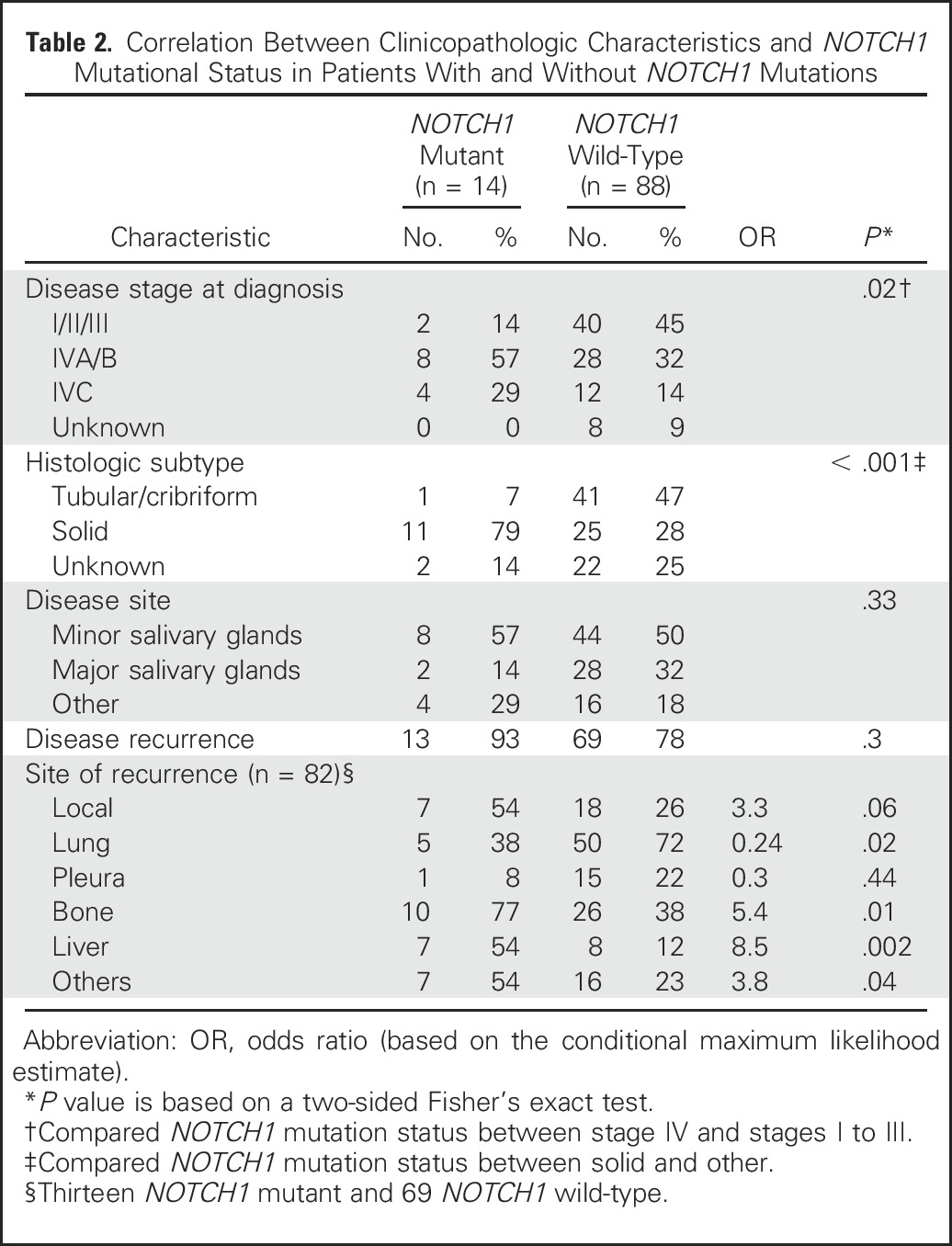
Fig 2.

(A) Odds ratio of metastasis to specific organs in patients with NOTCH1 mutations versus wild-type. The dashed yellow line represents the odds ratio of 1 (i.e., the incidence of metastasis in the NOTCH1 mutant group is the same as that of the NOTCH1 wild-type group). (B) Kaplan-Meier estimates of relapse-free survival (RFS) of patients with NOTCH1 mutations versus wild-type. (C) Kaplan-Meier estimates of overall survival (OS) of patients with NOTCH1 mutations versus wild-type.
NOTCH1 Mutation Is Prognostic in ACC But Is Not an Independent Prognostic Factor in the Presence of Histologic Subtype and Stage
The median RFS and OS in the overall population were 30 and 108 months, respectively. Median RFS was 12.5 versus 33.9 months for NOTCH1-mutant versus NOTCH1 wild-type (P = .01; Fig 2B). OS was significantly shorter in the patients with NOTCH1 mutations, with a median of 29.6 versus 121.9 months for NOTCH1 wild-type (P = .001; Fig 2C). MYB/MYBL1 rearrangement and/or overexpression did not influence RFS or OS irrespective of NOTCH1 mutational status (Data Supplement). The NICD-positive group showed a shorter RFS compared with the NICD-negative group (14.6 v 39 months; P = .03); however, OS did not significantly differ between the NICD-positive and the NICD-negative groups (44 v 108 months; P = .2).
Univariable and multivariable Cox models were performed for OS and RFS (Data Supplement). For OS, significant predictor variables by the univariable analysis were age, histology, disease stage, and NOTCH1 mutational status. Histologic subtype was the only significant predictor for OS in the multivariable analysis. For RFS, histology, disease stage, and NOTCH1 mutational status were significant predictors by univariable analysis. Both histologic subtype and stage remained significant in multivariable analysis. The results held when the nonsignificant predictors were removed from the model, and the results were consistent in the analysis that included mutation in genes predicted to activate the Notch pathway (Data Supplement). Hence, NOTCH1 mutation was not an independent prognostic factor when histology and stage were considered. The highly significant association between NOTCH1 mutation and advanced-stage disease (P = .02), solid histology (P < .001), or both (P = .01), together with additional factors such as tumor heterogeneity could account for dilution of the prognostic significance of NOTCH1 mutations in the multivariable analysis.
Notch1 Inhibitor Demonstrates Activity in NOTCH1-Mutant PDX
The PDX models ACCX9, ACCX11, ACCX5M1, and ACCX6 were screened against brontictuzumab, a humanized immunoglobulin G2 antibody that inhibits Notch1 signaling. ACCX9 harbors an HD NOTCH1 I1680N activating mutation, ACCX5M1 harbors a NOTCH1 S1004L inactivating mutation in the EGF-repeat domain, ACCX11 has a tandem duplication 3′ of NOTCH1, and ACCX6 is NOTCH1 wild-type. All four models have MYB rearrangements. NICD IHC staining was positive in the ACCX9 and ACCX11 models (Data Supplement). Brontictuzumab significantly inhibited tumor growth in ACCX9 (P < .05), but not in the models lacking activating NOTCH1 mutations (Fig 3), providing support for Notch1 as a therapeutic target in this NOTCH1-mutated PDX.
Fig 3.

The Notch1 inhibitor brontictuzumab led to significant tumor growth inhibition exclusively in the ACCX9 patient-derived xenograft model harboring a NOTCH1 activating mutation (I1680N). Mice were treated with brontictuzumab by intraperitoneal injection at 10 mg/kg once every 2 weeks for two total doses, and mean tumor volume was assessed. Error bars indicate SEM. mut, mutated; WT, wild-type. (*) P < .05.
Notch1 Inhibitor Led To Partial Response in a Patient With NOTCH1-Mutant ACC
A 28-year-old male presented with a tracheal ACC metastatic to mediastinal nodes, bone, and liver. He received palliative radiotherapy to the tracheal mass and osseous metastases, followed by two lines of chemotherapy. He had evidence of rapid disease progression and underwent biopsy and genotyping of a liver metastasis that revealed a NOTCH1 PEST domain mutation (S2467fs). He was then treated with third- and fourth-line targeted therapy. After further disease progression, a liver lesion biopsy revealed the original NOTCH1 mutation and an additional mutation in the HD (L1600Q; Fig 4A). All mutations were confirmed to be somatic and had similar variant allele frequency. As predicted, the co-occurrence of these mutations conferred greater ligand-independent NOTCH1 activation in vitro (Fig 1C). Notch1 pathway activation was also confirmed by NICD immunostaining (Fig 4B).
Fig 4.

(A) Tumor progression in an index patient with adenoid cystic carcinoma (ACC) was associated with the sequential identification of multiple mutations in the Notch1 pathway. The peripheral blood sample showed wild-type sequence at all NOTCH1 amplicons/codons covered by the assay. (B) Tumor from index patient with NOTCH1-mutant ACC was strongly positive for Notch1 intracellular domain by immunohistochemistry. (C) Patient with NOTCH1-mutant ACC achieved a partial response with a 38% reduction in the target lesion upon treatment with two doses of the anti-Notch1 monoclonal antibody brontictuzumab. CAP, cyclophosphamide, doxorubicin, and cisplatin; Carbo, carboplatin; ISIS482464, STAT3 inhibitor administered under a phase I clinical trial protocol; VAF, variant allele frequency. (†) No tumor available for genotyping. (‡) Genotyping performed in cell-free DNA.
The patient was treated with brontictuzumab and achieved a partial response (PR) after two doses (Fig 4C), which was accompanied by marked reduction in bone pain and lactate dehydrogenase levels. The patient unfortunately experienced a further increase in transaminases after cycle 2 that was questionably drug related, which led to brontictuzumab discontinuation and disease progression documented 44 days later. Liver toxicity was rarely observed in patients treated with the same drug.18 Sequencing of a new paraspinal metastasis confirmed the presence of the two NOTCH1 mutations and an additional mutation in FBXW7 (W606*). A third NOTCH1 mutation in the negative regulatory region (V1721G) was identified retrospectively in cell-free DNA. The patient received sunitinib but had rapid disease progression. He eventually succumbed as a consequence of his disease.
DISCUSSION
Recently, sequencing of ACC samples revealed genomic alterations in the Notch1 pathway in a subset of patients.4,5 In this study, we expanded our ACC WES efforts to include 46 patients in addition to the 24 previously published,4 which makes this the largest ACC series to be genotyped. In addition, we analyzed 32 patients who had their tumor tested for NOTCH1 mutations in at least exons 26, 27, and 34.
Our results show that the majority of NOTCH1 mutations in ACC (91%) are predicted to be activating. They occur mostly in the T-ALL hotspots and stain positive for NICD. As described in T-ALL,12 one patient with a juxtamembrane expansion mutation was identified. Furthermore, mutations in the HD and PEST domains co-occurred in two patients, including the reported patient. The double NOTCH1 mutations (S2467fs*/L1600Q) led to ligand-independent expression of the NOTCH1 target gene HES1. Tissue was scarce, which precluded us from establishing whether the NOTCH1 comutations in the index patient occurred in cis, limiting the extrapolation of the luciferase assay results to the clinical setting.
Although our data indicate that NOTCH1 mutations are associated with Notch1 pathway activation, they also suggest that pathway activation can occur by alternative routes. Canonical Notch signaling relies on nuclear translocation of NICD, and IHC NICD staining has been extensively validated in genotyped tumors.16 Our work demonstrates that NICD staining was quite sensitive (100%) in its ability to identify patients with NOTCH1 activating mutations; however, it lacks specificity, because 49% of NOTCH1 wild-type tumors were NICD positive. The specific drivers of Notch1 pathway activation independent of mutations are undetermined. The majority of patients with ACC overexpress Notch1 and its ligands, and receptor-ligand interaction is a known mechanism of pathway activation.19 Mutations in genes such as SPEN, FBXW7, or RBPJ can also activate the Notch1 pathway. Genes encoding chromatin-state regulators are frequently mutated in ACC, and epigenetic mechanisms may also have a role in Notch1 pathway activation.4,5,19,20
By using detailed histopathologic and clinical information from 102 patients, we demonstrated that NOTCH1 mutation defines a distinct ACC phenotype. Although the majority of patients with ACC have a protracted clinical course, patients with NOTCH1 mutations have an aggressive disease with a distinct pattern of metastasis and worse prognosis. The association between NOTCH1 mutation and the more dedifferentiated solid subtype, a poor prognostic factor in ACC,21 suggests that Notch1 drives this histologic prometastatic phenotype. Furthermore, the tendency of tumors with NOTCH1 mutations to metastasize to liver and bone is intriguing. Dysregulation of Notch signaling can cause developmental disorders characterized by defective bile duct formation, heart disease, and skeletal defects.22 The Notch pathway also plays a role in liver regeneration, osteoblastic maturation, and bone maintenance.23,24 Expression of JAG1 and DLL4 are seen in the normal liver, whereas JAG1 is overexpressed in bone marrow stromal cells.25,26 We hypothesize that the expression of Notch1 ligands in these organs may provide a permissive environment for growth; however, the mechanisms associated with the preferential homing of NOTCH1-mutant ACC to liver and bone are currently unknown and merit further investigation.
The potential oncogenic and prometastatic role of NOTCH1 mutations in ACC suggests that the pathway may be a therapeutic target. To test this directly, we used the Notch1-specific monoclonal antibody brontictuzumab and found that it significantly inhibited tumor growth exclusively in the ACCX9 NOTCH1-mutant model. The therapeutic potential of targeting Notch is also supported by a preclinical study in which a gamma-secretase inhibitor (GSI) led to tumor growth inhibition of the ACCX9 PDX.27 The lack of tumor growth inhibition in the NICD-positive ACCX11 model suggests that the mechanism by which the Notch1 pathway is activated may be important in predicting response from specific NOTCH1 inhibitors.
After validating the preclinical findings, we reported an index patient with NOTCH1-mutant ACC who achieved a PR after two doses of brontictuzumab administered during a clinical trial. This patient had at least two NOTCH1 mutations before starting treatment, with a third mutation detected in cell-free DNA, probably reflecting this patient’s tumor heterogeneity. Even though it was not possible to determine whether the NOTCH1 and/or FBXW7 mutations were present throughout the disease course or were acquired as the tumor progressed, the appearance of a detectable FBXW7 mutation in a new clinically evident mass is consistent with clonal evolution of the patients’ disease. The acquisition of additional mutations and progressive NOTCH1 oncogene addiction contributing to the clinical evolution of the disease has been described in T-ALL and chronic lymphocytic leukemia.10,28 Furthermore, in T-ALL, FBXW7 mutations were identified primarily in relapsed patients, and they predicted resistance to GSIs.11,29 Ultimately, irrespective of the timing in which the NOTCH1 and FBXW7 mutations occurred during the disease course, the presence of multiple alterations promoting Notch1 signaling supports its central role as an oncogenic driver in this cancer.
Preclinical studies in T-ALL lines demonstrated that GSIs induce growth suppression particularly in NOTCH1 double mutants; however, the GI toxicity associated with pan-Notch inhibitors has limited its clinical applicability.10 Although a biomarker predictive of response to Notch1 inhibitors remains to be determined, our findings suggest that mutations in Notch1 pathway genes and NICD staining may be used to select patients for clinical trials with potentially less toxic specific Notch1 inhibitors.30
In conclusion, our analysis integrating genomic, pathologic, and clinical outcomes data in ACC demonstrates that NOTCH1 mutations are activating and defines a subgroup of patients with an aggressive disease phenotype and distinct pattern of metastatic spread. Notch1 inhibition with a specific antibody demonstrated antitumor activity in preclinical models and an encouraging response in a NOTCH1-mutant patient. Further studies investigating the activity of Notch1 inhibitors in biomarker-selected patients with ACC are warranted.
ACKNOWLEDGMENT
We thank Kenna R. Shaw, Mark J. Routbort, and the Sheikh Khalifa Bin Zayed Al Nahyan Institute for personalized cancer therapy; The University of Texas MD Anderson Cancer Center for assistance with genomic profiling; all clinical investigators who participated in the clinical trial protocol NCT01778439; and Emily Roarty for editorial assistance.
Footnotes
Supported by the Ryan W. Smith Endowed Fund for Adenoid Cystic Carcinoma, the Adenoid Cystic Carcinoma Research Foundation, Cancer Center Support Grant No. P30 CA016672 from the National Cancer Institute, Grant No. U01DE019765 from the National Institutes of Health Office of Rare Diseases Research, and a Conquer Cancer Foundation Career Development Award (R.F.).
Presented as a poster at the 51st Annual Meeting of the American Society of Clinical Oncology, Chicago, IL, May 29-June 2, 2015.
AUTHOR CONTRIBUTIONS
Conception and design: Renata Ferrarotto, Yoshitsugu Mitani, Patrick Zweidler-McKay, Bonnie S. Glisson, Pamela Munster, Andrew Futreal, John V. Heymach
Financial support: John V. Heymach
Administrative support: John V. Heymach
Provision of study materials or patients: Adel K. El-Naggar, John V. Heymach
Collection and assembly of data: Renata Ferrarotto, Yoshitsugu Mitani, Irene Guijarro, Diana Bell, Michael J. Wick, Ann M. Kapoun, Amita Patnaik, Gail Eckhardt, Pamela Munster, Leonardo Faoro, Jakob Dupont, John V. Heymach
Data analysis and interpretation: Renata Ferrarotto, Yoshitsugu Mitani, Lixia Diao, Jing Wang, Diana Bell, William N. William Jr, J. Jack Lee, Andrew Futreal, Adel K. El-Naggar, John V. Heymach
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS’ DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Activating NOTCH1 Mutations Define a Distinct Subgroup of Patients With Adenoid Cystic Carcinoma Who Have Poor Prognosis, Propensity to Bone and Liver Metastasis, and Potential Responsiveness to Notch1 Inhibitors
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/site/ifc.
Renata Ferrarotto
Research Funding: OncoMed Pharmaceuticals (Inst), EMD Serono (Inst), AstraZeneca (Inst), G1 Therapeutics (Inst)
Yoshitsugu Mitani
No relationship to disclose
Lixia Diao
No relationship to disclose
Irene Guijarro
No relationship to disclose
Jing Wang
No relationship to disclose
Patrick Zweidler-McKay
No relationship to disclose
Diana Bell
No relationship to disclose
William N. William Jr
No relationship to disclose
Bonnie S. Glisson
Research Funding: OncoMed Pharmaceuticals (Inst)
Michael J. Wick
Employment: South Texas Accelerated Research Therapeutics
Stock or Other Ownership: South Texas Accelerated Research Therapeutics
Ann M. Kapoun
Employment: OncoMed Pharmaceuticals
Leadership: OncoMed Pharmaceuticals
Stock or Other Ownership: OncoMed Pharmaceuticals
Patents, Royalties, Other Intellectual Property: Co-author of patent: “Anti-Notch1 for the treatment of adenoid cystic carcinoma” (Inst)
Travel, Accommodations, Expenses: OncoMed Pharmaceuticals
Amita Patnaik
Research Funding: Merck (Inst), Pfizer (Inst), AVEO Pharmaceuticals (Inst), Eli Lilly (Inst), Plexxikon (Inst), Jiangsu Hengrui Medicine (Inst), Symphogen (Inst), Corvus Pharmaceuticals (Inst), TESARO (Inst), Bayer Healthcare Pharmaceuticals (Inst), Bayer Pharma AG (Inst)
Gail Eckhardt
Stock or Other Ownership: EntreMed
Honoraria: Kyowa Hakko Kirin, Eli Lilly/ImClone, Janssen Oncology, Boehringer Ingelheim
Consulting or Advisory Role: Sanofi, Taiho Pharmaceutical, CASI Pharmaceuticals, Pfizer, OnKure, SuviCa
Research Funding: Genentech (Inst), AstraZeneca (Inst), Rexahn Pharmaceuticals (Inst), OncoMed Pharmaceuticals (Inst), Cleave Biosciences (Inst)
Travel, Accommodations, Expenses: Boehringer Ingelheim, Kyowa Hakko Kirin, Sanofi, Eli Lilly/ImClone, Genentech
Pamela Munster
No relationship to disclose
Leonardo Faoro
Employment: OncoMed Pharmaceuticals
Stock or Other Ownership: OncoMed Pharmaceuticals
Jakob Dupont
Employment: OncoMed Pharmaceuticals
Leadership: OncoMed Pharmaceuticals
Stock or Other Ownership: OncoMed Pharmaceuticals
Patents, Royalties, Other Intellectual Property: Anti-Notch1 for the treatment of adenoid cystic carcinoma
Travel, Accommodations, Expenses: OncoMed Pharmaceuticals
J. Jack Lee
No relationship to disclose
Andrew Futreal
No relationship to disclose
Adel K. El-Naggar
No relationship to disclose
John V. Heymach
Stock or Other Ownership: Cardinal Spine and Pain Management, Bio-Tree Systems
Consulting or Advisory Role: AstraZeneca, AbbVie, Boehringer Ingelheim, Bristol-Myers Squibb, Medivation, ARIAD Pharmaceuticals, Synta Pharmaceuticals, Oncomed Pharmaceuticals, Novartis, Genentech, Calithera Biosciences
Research Funding: AstraZeneca (Inst)
REFERENCES
- 1.Fordice J, Kershaw C, El-Naggar A, et al. Adenoid cystic carcinoma of the head and neck: Predictors of morbidity and mortality. Arch Otolaryngol Head Neck Surg. 1999;125:149–152. doi: 10.1001/archotol.125.2.149. [DOI] [PubMed] [Google Scholar]
- 2.Spiro RH. Distant metastasis in adenoid cystic carcinoma of salivary origin. Am J Surg. 1997;174:495–498. doi: 10.1016/s0002-9610(97)00153-0. [DOI] [PubMed] [Google Scholar]
- 3.Laurie SA, Ho AL, Fury MG, et al. Systemic therapy in the management of metastatic or locally recurrent adenoid cystic carcinoma of the salivary glands: A systematic review. Lancet Oncol. 2011;12:815–824. doi: 10.1016/S1470-2045(10)70245-X. [DOI] [PubMed] [Google Scholar]
- 4.Stephens PJ, Davies HR, Mitani Y, et al. Whole exome sequencing of adenoid cystic carcinoma. J Clin Invest. 2013;123:2965–2968. doi: 10.1172/JCI67201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ho AS, Kannan K, Roy DM, et al. The mutational landscape of adenoid cystic carcinoma. Nat Genet. 2013;45:791–798. doi: 10.1038/ng.2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ross JS, Wang K, Rand JV, et al. Comprehensive genomic profiling of relapsed and metastatic adenoid cystic carcinomas by next-generation sequencing reveals potential new routes to targeted therapies. Am J Surg Pathol. 2014;38:235–238. doi: 10.1097/PAS.0000000000000102. [DOI] [PubMed] [Google Scholar]
- 7.Grego-Bessa J, Díez J, Timmerman L, et al. Notch and epithelial-mesenchyme transition in development and tumor progression: Another turn of the screw. Cell Cycle. 2004;3:718–721. [PubMed] [Google Scholar]
- 8.Lobry C, Oh P, Aifantis I. Oncogenic and tumor suppressor functions of Notch in cancer: It’s NOTCH what you think. J Exp Med. 2011;208:1931–1935. doi: 10.1084/jem.20111855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lai EC. Protein degradation: Four E3s for the notch pathway. Curr Biol. 2002;12:R74–R78. doi: 10.1016/s0960-9822(01)00679-0. [DOI] [PubMed] [Google Scholar]
- 10.Weng AP, Ferrando AA, Lee W, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306:269–271. doi: 10.1126/science.1102160. [DOI] [PubMed] [Google Scholar]
- 11.O’Neil J, Grim J, Strack P, et al. FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to gamma-secretase inhibitors. J Exp Med. 2007;204:1813–1824. doi: 10.1084/jem.20070876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sulis ML, Williams O, Palomero T, et al. NOTCH1 extracellular juxtamembrane expansion mutations in T-ALL. Blood. 2008;112:733–740. doi: 10.1182/blood-2007-12-130096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weng AP, Aster JC. Multiple niches for Notch in cancer: Context is everything. Curr Opin Genet Dev. 2004;14:48–54. doi: 10.1016/j.gde.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 14.Agrawal N, Frederick MJ, Pickering CR, et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science. 2011;333:1154–1157. doi: 10.1126/science.1206923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pickering CR, Zhang J, Yoo SY, et al. Integrative genomic characterization of oral squamous cell carcinoma identifies frequent somatic drivers. Cancer Discov. 2013;3:770–781. doi: 10.1158/2159-8290.CD-12-0537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kluk MJ, Ashworth T, Wang H, et al. Gauging NOTCH1 Activation in Cancer Using Immunohistochemistry. PLoS One. 2013;8:e67306. doi: 10.1371/journal.pone.0067306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moskaluk CA, Baras AS, Mancuso SA, et al. Development and characterization of xenograft model systems for adenoid cystic carcinoma. Lab Invest. 2011;91:1480–1490. doi: 10.1038/labinvest.2011.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Munster P, Eckhardt SG, Patnaik A, et al: Safety and preliminary efficacy results of a first-in-human phase I study of the novel cancer stem cell (CSC) targeting antibody brontictuzumab (OMP-52M51, anti-Notch1) administered intravenously to patients with certain advanced solid tumors. Mol Cancer Ther 14, 2015 (suppl 2; abstr C42) [Google Scholar]
- 19.Bell D, Hanna EY, Miele L, et al. Expression and significance of notch signaling pathway in salivary adenoid cystic carcinoma. Ann Diagn Pathol. 2014;18:10–13. doi: 10.1016/j.anndiagpath.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frierson HF, Jr, Moskaluk CA. Mutation signature of adenoid cystic carcinoma: Evidence for transcriptional and epigenetic reprogramming. J Clin Invest. 2013;123:2783–2785. doi: 10.1172/JCI69070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Weert S, van der Waal I, Witte BI, et al. Histopathological grading of adenoid cystic carcinoma of the head and neck: Analysis of currently used grading systems and proposal for a simplified grading scheme. Oral Oncol. 2015;51:71–76. doi: 10.1016/j.oraloncology.2014.10.007. [DOI] [PubMed] [Google Scholar]
- 22.Zanotti S, Canalis E. Notch and the skeleton. Mol Cell Biol. 2010;30:886–896. doi: 10.1128/MCB.01285-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morell CM, Strazzabosco M. Notch signaling and new therapeutic options in liver disease. J Hepatol. 2014;60:885–890. doi: 10.1016/j.jhep.2013.11.028. [DOI] [PubMed] [Google Scholar]
- 24.Weber JM, Calvi LM. Notch signaling and the bone marrow hematopoietic stem cell niche. Bone. 2010;46:281–285. doi: 10.1016/j.bone.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nijjar SS, Wallace L, Crosby HA, et al. Altered Notch ligand expression in human liver disease: Further evidence for a role of the Notch signaling pathway in hepatic neovascularization and biliary ductular defects. Am J Pathol. 2002;160:1695–1703. doi: 10.1016/S0002-9440(10)61116-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li L, Milner LA, Deng Y, et al. The human homolog of rat Jagged1 expressed by marrow stroma inhibits differentiation of 32D cells through interaction with Notch1. Immunity. 1998;8:43–55. doi: 10.1016/s1074-7613(00)80457-4. [DOI] [PubMed] [Google Scholar]
- 27.Stoeck A, Lejnine S, Truong A, et al. Discovery of biomarkers predictive of GSI response in triple-negative breast cancer and adenoid cystic carcinoma. Cancer Discov. 2014;4:1154–1167. doi: 10.1158/2159-8290.CD-13-0830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Puente XS, Pinyol M, Quesada V, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011;475:101–105. doi: 10.1038/nature10113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thompson BJ, Buonamici S, Sulis ML, et al. The SCFFBW7 ubiquitin ligase complex as a tumor suppressor in T cell leukemia. J Exp Med. 2007;204:1825–1835. doi: 10.1084/jem.20070872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu Y, Cain-Hom C, Choy L, et al. Therapeutic antibody targeting of individual Notch receptors. Nature. 2010;464:1052–1057. doi: 10.1038/nature08878. [DOI] [PubMed] [Google Scholar]